Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells

 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Results
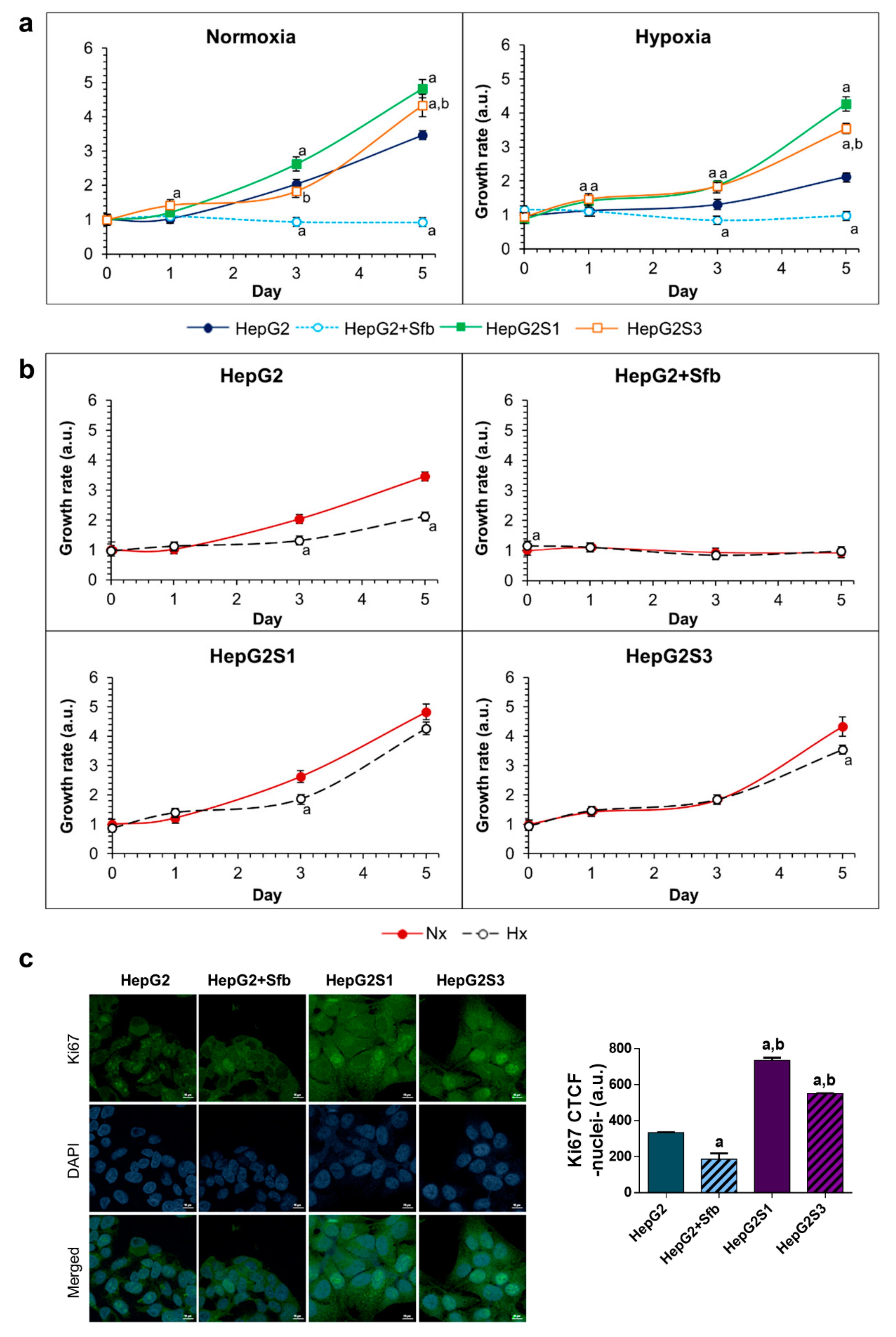
2.1. Resistant Hepatocarcinoma (HCC) Cell Lines Show a More Aggressive Growth than the HepG2 Parental Line under Both Normoxia and Hypoxia
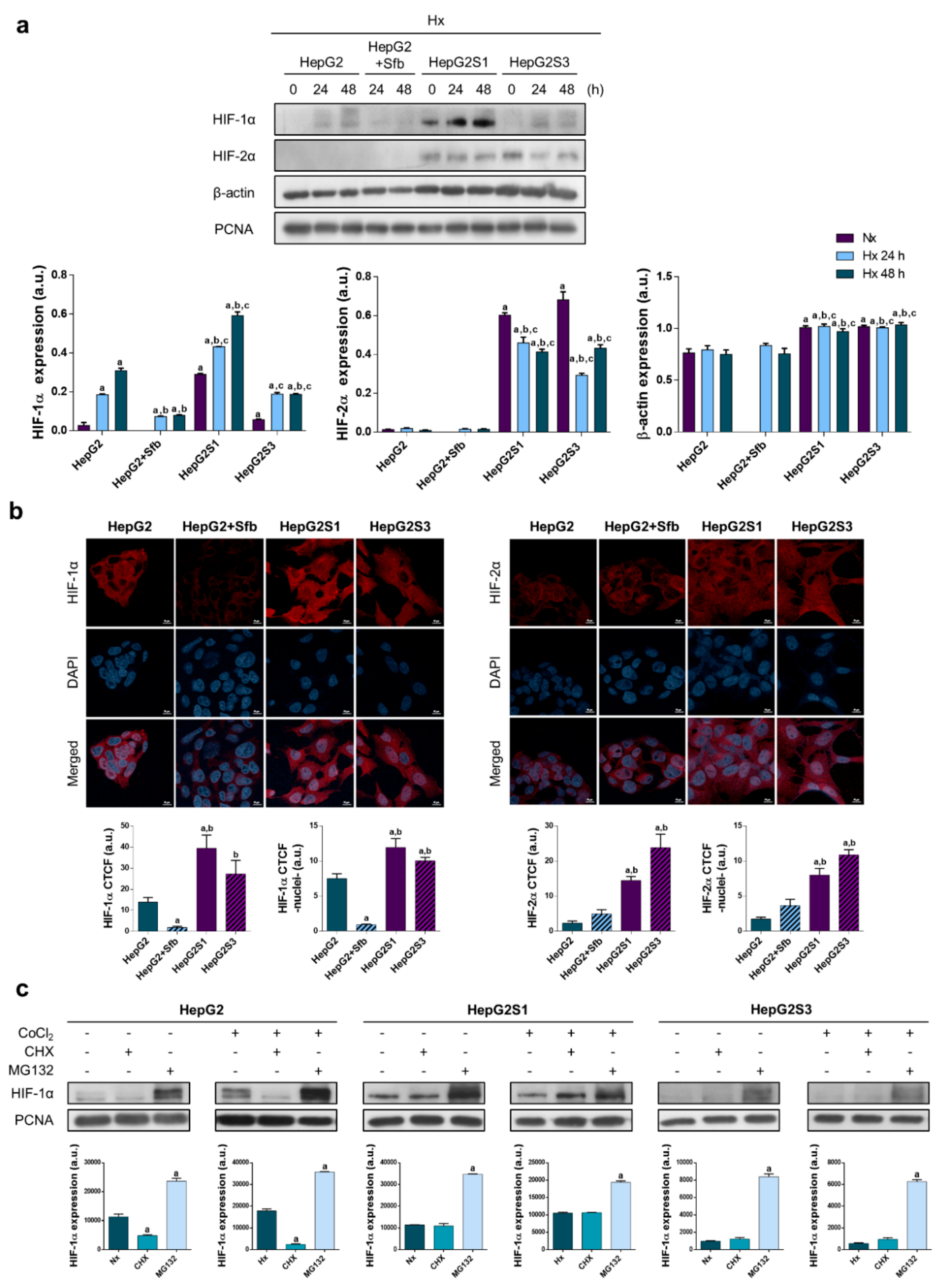
2.2. Sorafenib Resistant Cell Lines Overexpress Hypoxia-Inducible Factors (HIFs) and Display a Deregulation in the HIF-1α Degradation Mechanisms
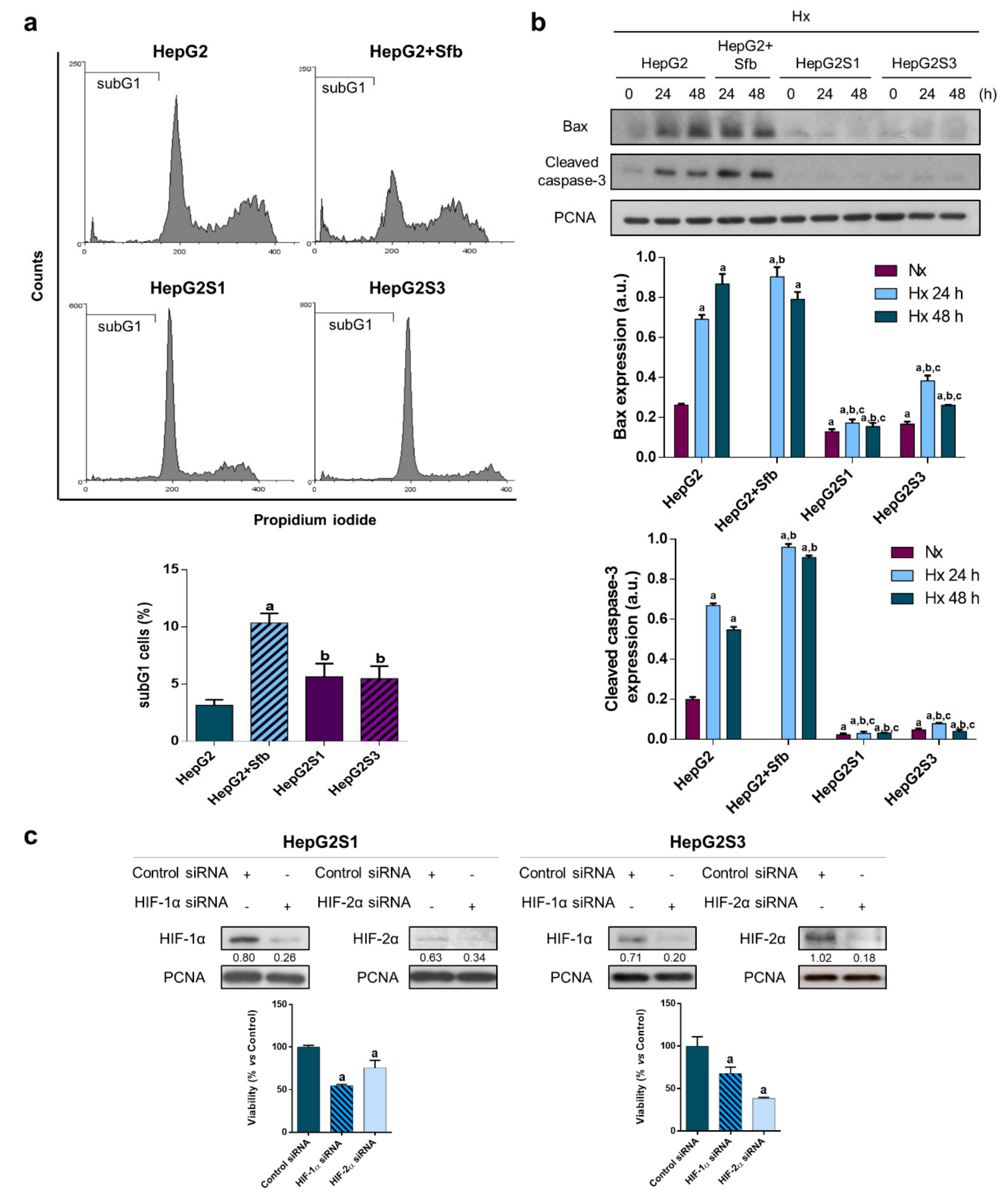
2.3. Resistant Cells Can Evade Sorafenib-Mediated Cell Death, Being HIFs Involved in This Lack of Cell Sensitivity to Sorafenib
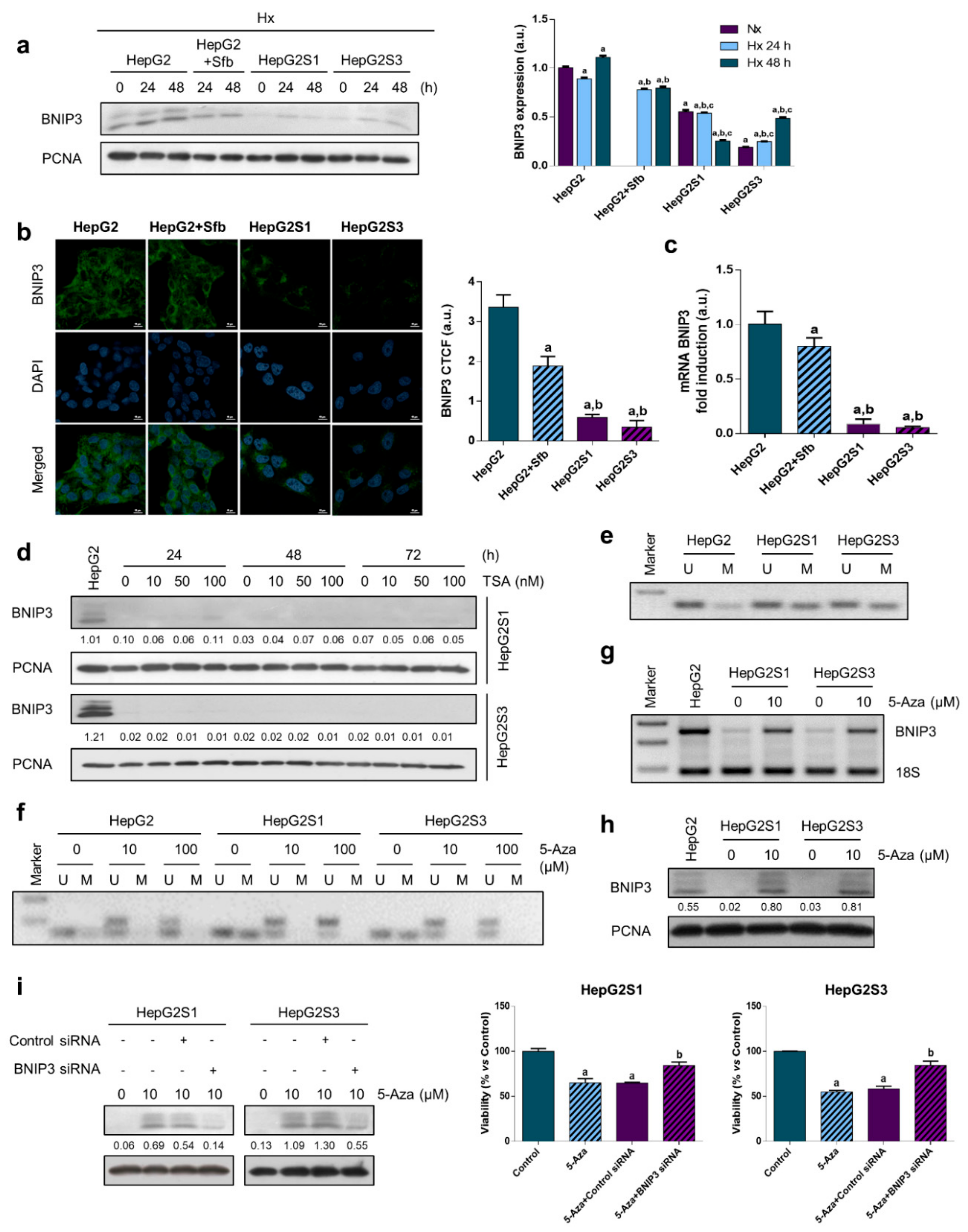
2.4. Methylation-Dependent Downregulation of Bcl-2 Interacting Protein 3 (BNIP3) Participates in Hypoxia-Mediated Sorafenib Resistance
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Growth Curve Based on Crystal Violet Staining
4.3. Immunofluorescence and Laser Confocal Imaging
4.4. Western Blot Assay
4.5. Microarray and Gene Expression Analysis
4.6. Flow Cytometry of SubG1 Cell Population
4.7. Gene Silencing
4.8. Cell Viability Assay
4.9. Real-Time (q) Reverse Transcriptase (RT)-Polymerase Chain Reaction (qRT-PCR), and RT-PCR
4.10. Methylation-Specific PCR (MSP)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Ahmed, T.S.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular carcinoma: Molecular mechanisms and targeted therapies. Medicina 2019, 55, 526. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zheng, B.; Wang, H.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, I.A.; Tsoulfas, G. Evolving role of Sorafenib in the management of hepatocellular carcinoma. World J. Clin. Oncol. 2017, 8, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, R.; Zhao, J.; Liu, J.; Ying, H.; Yan, H.; Zhou, S.; Liang, Y.; Huang, D.; Liang, X.; et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015, 367, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 134. [Google Scholar] [CrossRef]
- Ju, C.; Colgan, S.P.; Eltzschig, H.K. Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. 2016, 94, 613–627. [Google Scholar] [CrossRef]
- Xiong, X.X.; Qiu, X.Y.; Hu, D.X.; Chen, X.Q. Advances in hypoxia-mediated mechanisms in hepatocellular carcinoma. Mol. Pharmacol. 2017, 92, 246–255. [Google Scholar] [CrossRef]
- He, J.; Pei, L.; Jiang, H.; Yang, W.; Chen, J.; Liang, H. Chemoresistance of colorectal cancer to 5-fluorouracil is associated with silencing of the BNIP3 gene through aberrant methylation. J. Cancer 2017, 8, 1187–1196. [Google Scholar] [CrossRef]
- Vasagiri, N.; Kutala, V.K. Structure, function, and epigenetic regulation of BNIP3: A pathophysiological relevance. Mol. Biol. Rep. 2014, 41, 7705–7714. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Liu, Z.; Liu, J.; Wang, H.; Huang, L.; Lin, T.; Liu, J.; Wei, Q.; Zeng, H.; He, G.; et al. Expression and epigenetic regulatory mechanism of BNIP3 in clear cell renal cell carcinoma. Int. J. Oncol. 2019, 54, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Toyota, M.; Suzuki, H.; Murai, M.; Akino, K.; Ueno, M.; Nojima, M.; Yawata, A.; Miyakawa, H.; Suga, T.; et al. Upregulation of BNIP3 by 5-aza-2′-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J. Gastroenterol. 2005, 40, 504–510. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Esposito, I.; Giese, T.; Ketterer, K.; Büchler, M.W.; Giese, N.A.; Friess, H. Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 2005, 24, 4421–4432. [Google Scholar] [CrossRef] [PubMed]
- An, H.-J.; Lee, H.; Paik, S.-G. Silencing of BNIP3 results from promoter methylation by DNA methyltransferase 1 induced by the mitogen-activated protein kinase pathway. Mol. Cells 2011, 31, 579–583. [Google Scholar] [CrossRef]
- Tang, H.; Liu, Y.-J.; Liu, M.; Li, X. Establishment and gene analysis of an oxaliplatin-resistant colon cancer cell line THC8307/L-OHP. Anticancer Drugs 2007, 18, 633–639. [Google Scholar] [CrossRef]
- De Angelis, P.M.; Fjell, B.; Kravik, K.L.; Haug, T.; Tunheim, S.H.; Reichelt, W.; Beigi, M.; Clausen, O.P.; Galteland, E.; Stokke, T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int. J. Oncol. 2004, 24, 1279–1288. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Yang, J.; Zhang, Y.; Zhu, D.; Zhang, L.; Zhu, Y.; Li, D.; Zhou, J. Methylation of BNIP3 in pancreatic cancer inhibits the induction of mitochondrial-mediated tumor cell apoptosis. Oncotarget 2017, 8, 63208–63222. [Google Scholar] [CrossRef]
- van Malenstein, H.; Dekervel, J.; Verslype, C.; Van Cutsem, E.; Windmolders, P.; Nevens, F.; van Pelt, J. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013, 329, 74–83. [Google Scholar] [CrossRef]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; Méndez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; Mauriz, J.L.; González-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, M.A.; González, R.; de la Rosa, Á.J.; Gallego, P.; Ordóñez, R.; Navarro-Villarán, E.; Contreras, L.; Rodríguez-Arribas, M.; González-Gallego, J.; Álamo-Martínez, J.M.; et al. Molecular characterization of autophagic and apoptotic signaling induced by sorafenib in liver cancer cells. J. Cell. Physiol. 2018, 234, 692–708. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Domínguez, N.; Méndez-Blanco, C.; Carbajo-Pescador, S.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Melatonin enhances sorafenib actions in human hepatocarcinoma cells by inhibiting mTORC1/p70S6K/HIF-1α and hypoxia-mediated mitophagy. Oncotarget 2017, 8, 91402–91414. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, X.; Gan, L.; Yang, X.; Miao, Z. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol. Sin. 2017, 38, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Tak, E.; Lee, S.; Lee, J.; Rashid, M.A.; Kim, Y.W.; Park, J.-H.; Park, W.S.; Shokat, K.M.; Ha, J.; Kim, S.S. Human carbonyl reductase 1 upregulated by hypoxia renders resistance to apoptosis in hepatocellular carcinoma cells. J. Hepatol. 2011, 54, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Bielecka, Z.F.; Malinowska, A.; Brodaczewska, K.K.; Klemba, A.; Kieda, C.; Krasowski, P.; Grzesiuk, E.; Piwowarski, J.; Czarnecka, A.M.; Szczylik, C. Hypoxic 3D in vitro culture models reveal distinct resistance processes to TKIs in renal cancer cells. Cell Biosci. 2017, 7, 71. [Google Scholar] [CrossRef]
- Zhao, C.-X.; Luo, C.-L.; Wu, X.-H. Hypoxia promotes 786-O cells invasiveness and resistance to sorafenib via HIF-2α/COX-2. Med. Oncol. 2015, 32, 419. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, L.; Guo, D.; Wu, Z.; Chen, W. Co-delivery of hypoxia inducible factor-1α small interfering RNA and 5-fluorouracil to overcome drug resistance in gastric cancer SGC-7901 cells. J. Gene Med. 2017, 19, e2998. [Google Scholar] [CrossRef]
- van Oosterwijk, J.G.; Buelow, D.R.; Drenberg, C.D.; Vasilyeva, A.; Li, L.; Shi, L.; Wang, Y.-D.; Finkelstein, D.; Shurtleff, S.A.; Janke, L.J.; et al. Hypoxia-induced upregulation of BMX kinase mediates therapeutic resistance in acute myeloid leukemia. J. Clin. Investig. 2018, 128, 369–380. [Google Scholar] [CrossRef]
- Liu, L.; Ho, R.L.K.; Chen, G.G.; Lai, P.B.S. Sorafenib inhibits hypoxia-inducible factor-1α synthesis: Implications for antiangiogenic activity in hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 5662–5671. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, G.; Zhu, H.; Dong, X.; Zhao, D.; Jiang, X.; Li, J.; Qiao, H.; Ni, S.; Sun, X. 2-Methoxyestradiol synergizes with sorafenib to suppress hepatocellular carcinoma by simultaneously dysregulating hypoxia-inducible factor-1 and -2. Cancer Lett. 2014, 355, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Shan, W.; Yang, Y.; Jin, M.; Dai, Y.; Yang, H.; Jiao, R.; Xia, Y.; Liu, Q.; Ju, L.; et al. Reversal of sorafenib resistance in hepatocellular carcinoma: Epigenetically regulated disruption of 14-3-3η/hypoxia-inducible factor-1α. Cell Death Discov. 2019, 5, 120. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-C.; Hsu, C.-H.; Shao, Y.-Y.; Ho, W.-C.; Tsai, M.-H.; Feng, W.-C.; Chow, L.-P. Integrated Stable Isotope Labeling by Amino acids in cell Culture (SILAC) and isobaric Tags for Relative and Absolute Quantitation (iTRAQ) quantitative proteomic analysis identifies galectin-1 as a potential biomarker for predicting sorafenib resistance i. Mol. Cell. Proteom. 2015, 14, 1527–1545. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Dong, X.; Lv, H.; Xiu, P.; Li, T.; Wang, F.; Xu, Z.; Li, J. Targeting hypoxia-inducible factor-2α enhances sorafenib antitumor activity via β-catenin/C-Myc-dependent pathways in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 778–784. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wu, H.; Jiang, Q.; Liu, Y.; Han, L.; Yan, Y.; Wei, B.; Liu, F.; Deng, X.; Chen, H.; et al. Hypoxia-inducible factor-2α directly promotes BCRP expression and mediates the resistance of ovarian cancer stem cells to adriamycin. Mol. Oncol. 2019, 13, 403–421. [Google Scholar] [CrossRef]
- Wu, F.-Q.; Fang, T.; Yu, L.-X.; Lv, G.-S.; Lv, H.-W.; Liang, D.; Li, T.; Wang, C.-Z.; Tan, Y.-X.; Ding, J.; et al. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1α. J. Hepatol. 2016, 65, 314–324. [Google Scholar] [CrossRef]
- Wang, J.; Ma, Y.; Jiang, H.; Zhu, H.; Liu, L.; Sun, B.; Pan, S.; Krissansen, G.W.; Sun, X. Overexpression of von Hippel-Lindau protein synergizes with doxorubicin to suppress hepatocellular carcinoma in mice. J. Hepatol. 2011, 55, 359–368. [Google Scholar] [CrossRef]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef]
- Zhou, T.; Zhuang, L.; Hu, Y.; Zhou, Y.; Lin, W.; Wang, D.; Wan, Z.; Chang, L.; Chen, Y.; Ying, M.; et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance to sorafenib in hepatocellular carcinoma cells. Sci. Rep. 2016, 6, 30483. [Google Scholar] [CrossRef]
- Hajigholami, S.; Malekshahi, Z.V.; Bodaghabadi, N.; Najafi, F.; Shirzad, H.; Sadeghizadeh, M. Nano packaged tamoxifen and curcumin; effective formulation against sensitive and resistant MCF-7 cells. Iran. J. Pharm. Res. 2018, 17, 1–10. [Google Scholar] [PubMed]
- Fernando, J.; Sancho, P.; Fernández-Rodriguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gong, R.; Wang, M.; Yan, Z.; Yuan, B.; Wang, K.; Shi, L. Sorafenib down-regulates c-IAP expression post-transcriptionally in hepatic carcinoma cells to suppress apoptosis. Biochem. Biophys. Res. Commun. 2012, 418, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, Q.; Gong, Z.; Zhou, L.; You, N.; Wang, S.; Li, X.; Li, J.; An, J.; Wang, D.; et al. MicroRNA-34a targets Bcl-2 and sensitizes human hepatocellular carcinoma cells to sorafenib treatment. Technol. Cancer Res. Treat. 2014, 13, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; Noda, T.; Nagano, H.; et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J. Hepatol. 2010, 52, 698–704. [Google Scholar] [CrossRef]
- Hikita, H.; Takehara, T.; Shimizu, S.; Kodama, T.; Shigekawa, M.; Iwase, K.; Hosui, A.; Miyagi, T.; Tatsumi, T.; Ishida, H.; et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology 2010, 52, 1310–1321. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W.; et al. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef]
- Huang, S.; Qi, P.; Zhang, T.; Li, F.; He, X. The HIF-1α/miR-224-3p/ATG5 axis affects cell mobility and chemosensitivity by regulating hypoxia-induced protective autophagy in glioblastoma and astrocytoma. Oncol. Rep. 2019, 41, 1759–1768. [Google Scholar] [CrossRef]
- Long, Q.; Zou, X.; Song, Y.; Duan, Z.; Liu, L. PFKFB3/HIF-1α feedback loop modulates sorafenib resistance in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 513, 642–650. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, L.; Fang, Q.; Sun, J.; Zhang, S.; Zhan, C.; Liu, S.; Zhang, Y. MiR-338-3p inhibits hepatocarcinoma cells and sensitizes these cells to sorafenib by targeting hypoxia-induced factor 1α. PLoS ONE 2014, 9, e115565. [Google Scholar] [CrossRef] [PubMed]
- Krutilina, R.; Sun, W.; Sethuraman, A.; Brown, M.; Seagroves, T.N.; Pfeffer, L.M.; Ignatova, T.; Fan, M. MicroRNA-18a inhibits hypoxia-inducible factor 1α activity and lung metastasis in basal breast cancers. Breast Cancer Res. 2014, 16, R78. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, A.; Martínez-Ríos, J.; Castañeda-Patlán, M.C.; Sarabia-Sánchez, M.A.; Tejeda-Muñoz, N.; Chinney-Herrera, A.; Soldevila, G.; Benelli, R.; Santoyo-Ramos, P.; Poggi, A.; et al. Functional interaction of hypoxia-inducible factor 2-alpha and autophagy mediates drug resistance in colon cancer cells. Cancers 2019, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Cao, M.; Guo, Z.; Zuo, B.; Gao, J.; Zhou, H.; Li, H.; Cui, Y.; Fang, F.; Zhang, W.; et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J. Hematol. Oncol. 2016, 9, 20. [Google Scholar] [CrossRef]
- Jiang, W.; Li, G.; Li, W.; Wang, P.; Xiu, P.; Jiang, X.; Liu, B.; Sun, X.; Jiang, H. Sodium orthovanadate overcomes sorafenib resistance of hepatocellular carcinoma cells by inhibiting Na+/K+-ATPase activity and hypoxia-inducible pathways. Sci. Rep. 2018, 8, 9706. [Google Scholar] [CrossRef]
- Bacon, A.; Fox, S.; Turley, H.; Harris, A. Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene 2007, 26, 132–141. [Google Scholar] [CrossRef]
- Deng, Q.; Huang, C.; Chen, N.; Li, L.; Wang, X.; Zhang, W.; Bi, F.; Tang, Q.; Li, Z.; Wang, W. Chemotherapy and radiotherapy downregulate the activity and expression of DNA methyltransferase and enhance Bcl-2/E1B-19-kDa interacting protein-3-induced apoptosis in human colorectal cancer cells. Chemotherapy 2013, 58, 445–453. [Google Scholar] [CrossRef]
- Liu, F.; Liu, Q.; Yang, D.; Bollag, W.B.; Robertson, K.; Wu, P.; Liu, K. Verticilin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [Google Scholar] [CrossRef]
- Murai, M.; Toyota, M.; Suzuki, H.; Satoh, A.; Sasaki, Y.; Akino, K.; Ueno, M.; Takahashi, F.; Kusano, M.; Mita, H.; et al. Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. 2005, 11, 1021–1027. [Google Scholar]
- Valdez, B.C.; Li, Y.; Murray, D.; Corn, P.; Champlin, R.E.; Andersson, B.S. 5-Aza-2’-deoxycytidine sensitizes busulfan-resistant myeloid leukemia cells by regulating expression of genes involved in cell cycle checkpoint and apoptosis. Leuk. Resist. 2010, 34, 364–372. [Google Scholar] [CrossRef]
- Ishiguro, M.; Iida, S.; Uetake, H.; Morita, S.; Makino, H.; Kato, K.; Takagi, Y.; Enomoto, M.; Sugihara, K. Effect of combined therapy with low-dose 5-aza-2′-deoxycytidine and irinotecan on colon cancer cell line HCT-15. Ann. Surg. Oncol. 2007, 14, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Miguel, B.S.; Laliena, A.; Álvarez, M.; Culebras, J.M.; González-Gallego, J.; Tuñón, M.J. Melatonin prevents the decreased activity of antioxidant enzymes and activates nuclear erythroid 2-related factor 2 signaling in an animal model of fulminant hepatic failure of viral origin. J. Pineal Res. 2010, 49, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Schiller, I.; Huat Lu, Z.; Vaughan, L.; Weilenmann, R.; Koundrioukoff, S.; Pospischil, A. Establishment of proliferative cell nuclear antigen gene as an internal reference gene for polymerase chain reaction of a wide range of archival and fresh mammalian tissues. J. Vet. Diagn. Investig. 2003, 15, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Mian, O.Y.; Khattab, M.H.; Hedayati, M.; Coulter, J.; Abubaker-Sharif, B.; Schwaninger, J.M.; Veeraswamy, R.K.; Brooks, J.D.; Hopkins, L.; Shinohara, D.B.; et al. GSTP1 loss results in accumulation of oxidative DNA base damage and promotes prostate cancer cell survival following exposure to protracted oxidative stress. Prostate 2016, 76, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Sakwe, A.M.; Nguyen, T.; Frappier, L. The MCM-associated protein MCM-BP is important for human nuclear morphology. J. Cell Sci. 2012, 125, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; San-Miguel, B.; Prause, C.; Marroni, N.; Cuevas, M.J.; González-Gallego, J.; Tuñón, M.J. Glutamine treatment attenuates endoplasmic reticulum stress and apoptosis in TNBS-induced colitis. PLoS ONE 2012, 7, e50407. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Full Name | Regulation | 2logFC HepG2S1/HepG2 | Corrected p |
|---|---|---|---|---|
| BAX | BCL2 associated X apoptosis regulator | Down | −0.93 | 5.74 × 10−9 |
| BCL2 | BCL2 apoptosis regulator | Up | +1.25 | 1.43 × 10−8 |
| BCL2L1 | BCL2 like 1 | Down | −1.21 | 1.59 × 10−10 |
| BIRC3 | Baculoviral IAP repeat containing 3 | Up | +5.06 | 1.32 × 10−15 |
| CASP2 | Caspase 2 | Up | +0.95 | 1.18 × 10−8 |
| CASP3 | Caspase 3 | Down | −2.06 | 4.59 × 10−11 |
| CASP8 | Caspase 8 | Down | −0.81 | 7.98 × 10−9 |
| CASP9 | Caspase 9 | Up | +1.21 | 1.76 × 10−9 |
| CASP10 | Caspase 10 | Down | −1.23 | 6.76 × 10−8 |
| HRK | Harakiri BCL2 interacting protein | Up | +1.34 | 1.17 × 10−7 |
| PMAIP1 | Phorbol-12-myristate-13-acetate-induced protein 1 | Up | +2.45 | 9.17 × 10−13 |
| PTPN13 | Protein tyrosine phosphatase non-receptor type 13 | Up | +1.97 | 5.59 × 10−10 |
| TNFRSF10B | TNF receptor superfamily member 10b | Down | −1.23 | 5.04 × 10−11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Méndez-Blanco, C.; Fondevila, F.; Fernández-Palanca, P.; García-Palomo, A.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers 2019, 11, 1984. https://doi.org/10.3390/cancers11121984
Méndez-Blanco C, Fondevila F, Fernández-Palanca P, García-Palomo A, van Pelt J, Verslype C, González-Gallego J, Mauriz JL. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers. 2019; 11(12):1984. https://doi.org/10.3390/cancers11121984
Chicago/Turabian StyleMéndez-Blanco, Carolina, Flavia Fondevila, Paula Fernández-Palanca, Andrés García-Palomo, Jos van Pelt, Chris Verslype, Javier González-Gallego, and José L. Mauriz. 2019. "Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells" Cancers 11, no. 12: 1984. https://doi.org/10.3390/cancers11121984
APA StyleMéndez-Blanco, C., Fondevila, F., Fernández-Palanca, P., García-Palomo, A., van Pelt, J., Verslype, C., González-Gallego, J., & Mauriz, J. L. (2019). Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers, 11(12), 1984. https://doi.org/10.3390/cancers11121984