The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration

 , ,
, ,
Abstract
1. Introduction
2. Results
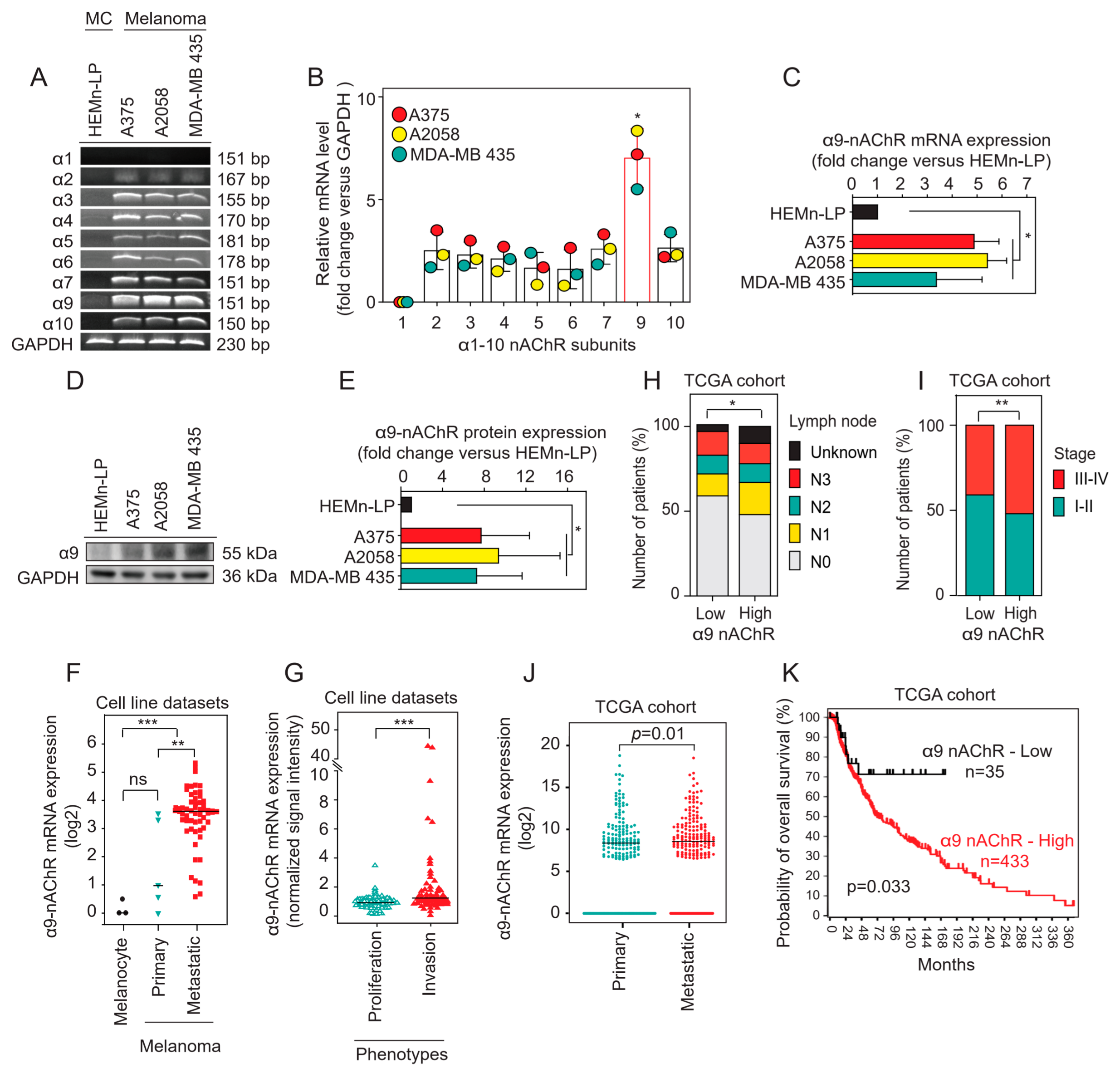
2.1. α9-nAChR Is Overexpressed in Melanoma
2.2. Correlation of α9-nAChR Levels with Clinicopathological Features in Melanoma
2.3. Correlation Between α9-nAChR and PD-L1 in Melanoma
2.4. α9-nAChR Induces PD-L1 Expression and Regulates in Proliferation and Migration
2.5. α9-nAChR Regulates Melanoma Cell Proliferation via the AKT and ERK Signaling Pathways
2.6. α9-nAChR Promotes Melanoma Cell Migration through EMT
2.7. α9-nAChR Regulates PD-L1 Expression via the STAT3 Signaling Pathway
2.8. Nicotine-Induced α9-nAChR Activity Upregulates PD-L1 Expression and Promotes Melanoma Cell Proliferation and Migration
2.9. Nicotine-Induced α9-nAChR Activity Significantly Increases Melanoma Cell Proliferation via the AKT and ERK Signaling Pathways
2.10. Nicotine-Induced α9-nAChR Activity Upregulates PD-L1 Expression via STAT3 Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Bioinformatics Analysis
4.2.1. Melanoma Cell Line Dataset Analysis
4.2.2. TCGA-SKCM Cohort Analysis
4.3. Kaplan-Meier Analysis
4.4. Nicotine Treatment
4.5. Inhibitor Treatment
4.6. Cell Proliferation and Viability Assays
4.6.1. Cell Counting Assay
4.6.2. MTT Assay
4.6.3. Soft-Agar Growth Assay
4.7. Wound healing Scratch Assay
4.8. Construction of Vectors
4.8.1. α9-nAChR siRNA
4.8.2. α9-nAChR Overexpression
4.9. Generation of Stable Cell Lines
4.10. RNA Isolation and RT-PCR Analysis
4.11. Protein Extraction, Western Blotting, and Antibody
4.12. ChIP Assay
4.13. IHC Staining and Scoring System
4.13.1. IHC Staining
4.13.2. Immunohistochemistry Score
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gershenwald, J.E.; Guy, G.P. Stemming the Rising Incidence of Melanoma: Calling Prevention to Action. J. Natl. Cancer Inst. 2016, 108, djv381. [Google Scholar] [CrossRef] [PubMed]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Hauschild, A.; Lindenblatt, N.; Pentheroudakis, G.; Keilholz, U.; on behalf of the ESMO Guidelines Committee. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Margolin, K.; Sullivan, R. Management of Metastatic Melanoma in 2018. JAMA Oncol. 2018, 4, 857–858. [Google Scholar] [CrossRef] [PubMed]
- Wolff, K.; Goldsmith, L.A.; Katz, S.I.; Gilchrest, B.A.; Paller, A.S.; Leffell, D.J. Melanoma. Fitzpatrick’s dermatology in general medicine, 2 volumes. Transplantation 2008, 85, 654. [Google Scholar]
- Rosko, A.J.; Vankoevering, K.K.; McLean, S.A.; Johnson, T.M.; Moyer, J.S. Contemporary management of early-stage melanoma: A systematic review. JAMA Facial Plast. Surg. 2017, 19, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Swetter, S.M.; Tsao, H.; Bichakjian, C.K.; Curiel-Lewandrowski, C.; Elder, D.E.; Gershenwald, J.E.; Guild, V.; Grant-Kels, J.M.; Halpern, A.C.; Johnson, T.M.; et al. Guidelines of care for the management of primary cutaneous melanoma. J. Am. Acad. Dermatol. 2019, 80, 208–250. [Google Scholar] [CrossRef]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Bressac-de-Paillerets, B.; Avril, M.-F.; Chompret, A.; Demenais, F. Genetic and environmental factors in cutaneous malignant melanoma. Biochimie 2002, 84, 67–74. [Google Scholar] [CrossRef]
- Wu, C.H.; Lee, C.H.; Ho, Y.S. Nicotinic acetylcholine receptor-based blockade: Applications of molecular targets for cancer therapy. Clin. Cancer Res. 2011, 17, 3533–3541. [Google Scholar] [CrossRef]
- Elgoyhen, A.B.; Johnson, D.S.; Boulter, J.; Vetter, D.E.; Heinemann, S. α9: An acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 1994, 79, 705–715. [Google Scholar] [CrossRef]
- Glowatzki, E.; Fuchs, P.A. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 2000, 288, 2366–2368. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Ndoye, A.; Grando, S.A. Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am. J. Pathol. 2000, 157, 1377–1391. [Google Scholar] [CrossRef]
- Liu, Q.; Whiteaker, P.; Morley, B.J.; Shi, F.-D.; Lukas, R.J. Distinctive Roles for α7*- and α9*-Nicotinic Acetylcholine Receptors in Inflammatory and Autoimmune Responses in the Murine Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis. Front. Cell. Neurosci. 2017, 11, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Romero, H.K.; Christensen, S.B.; Di Cesare Mannelli, L.; Gajewiak, J.; Ramachandra, R.; Elmslie, K.S.; Vetter, D.E.; Ghelardini, C.; Iadonato, S.P.; Mercado, J.L.; et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. USA 2017, 114, 1825–1832. [Google Scholar] [CrossRef] [PubMed]
- Bufalo, A.; Cesario, A.; Salinaro, G.; Fini, M.; Russo, P. Alpha9Alpha10 Nicotinic Acetylcholine Receptors as Target for the Treatment of Chronic Pain. Curr. Pharm. Des. 2014, 20, 6042–6047. [Google Scholar] [CrossRef] [PubMed]
- Luebke, A.E.; Makoni, P.D.; Guth, S.M.; Lysakowski, A. Alpha-9 nicotinic acetylcholine receptor immunoreactivity in the rodent vestibular labyrinth. J. Comp. Neurol. 2005, 492, 323–333. [Google Scholar] [CrossRef][Green Version]
- Colomer, C.; Olivos-Oré, L.A.; Vincent, A.; McIntosh, J.M.; Artalejo, A.R.; Guérineau, N.C. Functional characterization of α9-containing cholinergic nicotinic receptors in the rat adrenal medulla: Implication in stress-induced functional plasticity. J. Neurosci. 2010, 30, 6732–6742. [Google Scholar] [CrossRef]
- Baumann, L.; Kauschke, V.; Vikman, A.; Dürselen, L.; Krasteva-Christ, G.; Kampschulte, M.; Heiss, C.; Yee, K.T.; Vetter, D.E.; Lips, K.S. Deletion of nicotinic acetylcholine receptor alpha9 in mice resulted in altered bone structure. Bone 2019, 120, 285–296. [Google Scholar] [CrossRef]
- Lee, C.H.; Huang, C.S.; Chen, C.S.; Tu, S.H.; Wang, Y.J.; Chang, Y.J.; Tam, K.W.; Wei, P.L.; Cheng, T.C.; Chu, J.S.; et al. Overexpression and activation of the α9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J. Natl. Cancer Inst. 2010, 102, 1322–1335. [Google Scholar] [CrossRef]
- Huang, L.C.; Lin, C.L.; Qiu, J.Z.; Lin, C.Y.; Hsu, K.W.; Tam, K.W.; Lee, J.Y.; Yang, J.M.; Lee, C.H. Nicotinic acetylcholine receptor subtype alpha-9 mediates triple-negative breast cancers based on a spontaneous pulmonary metastasis mouse model. Front. Cell. Neurosci. 2017, 11, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-negative breast cancer: Current understanding and future therapeutic breakthrough targeting cancer stemness. Cancers 2019, 11, 1334. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Lee, C.-H.; Chuang, Y.-H.; Lee, J.-Y.; Chiu, Y.-Y.; Wu Lee, Y.-H.; Jong, Y.-J.; Hwang, J.-K.; Huang, S.-H.; Chen, L.-C.; et al. Membrane protein-regulated networks across human cancers. Nat. Commun. 2019, 10, 3131–3148. [Google Scholar] [CrossRef] [PubMed]
- Balogh, E.P.; Dresler, C.; Fleury, M.E.; Gritz, E.R.; Kean, T.J.; Myers, M.L.; Nass, S.J.; Nevidjon, B.; Toll, B.A.; Warren, G.W. Reducing tobacco-related cancer incidence and mortality: Summary of an institute of medicine workshop. Oncologist 2014, 19, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The chemical components of tobacco and tobacco smoke. In The Chemical Components of Tobacco and Tobacco Smoke; CRC Press: Boca Raton, FL, USA, 2016; pp. 1–2332. [Google Scholar]
- Schane, R.E.; Ling, P.M.; Glantz, S.A. Health effects of light and intermittent smoking: A review. Circulation 2010, 121, 1518–1522. [Google Scholar] [CrossRef]
- Shaw, H.M.; Milton, G.W. Smoking and the development of metastases from malignant melanoma. Int. J. Cancer 1981, 28, 153–156. [Google Scholar] [CrossRef]
- Jones, M.S.; Jones, P.C.; Stern, S.L.; Elashoff, D.; Hoon, D.S.B.; Thompson, J.; Mozzillo, N.; Nieweg, O.E.; Noyes, D.; Hoekstra, H.J.; et al. The Impact of Smoking on Sentinel Node Metastasis of Primary Cutaneous Melanoma. Ann. Surg. Oncol. 2017, 24, 2089–2094. [Google Scholar] [CrossRef]
- Harris, C.C. Tobacco smoking, E-cigarettes, and nicotine harm. Proc. Natl. Acad. Sci. USA 2018, 115, 1406–1407. [Google Scholar] [CrossRef]
- Grando, S.A. Connections of nicotine to cancer. Nat. Rev. Cancer 2014, 14, 419–429. [Google Scholar] [CrossRef]
- Lee, C.-H.; Chang, Y.-C.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chen, L.-C.; Chang, Y.-J.; Wei, P.-L.; Chang, H.-W.; Chang, C.-H.; et al. Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces α9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 2011, 129, 331–345. [Google Scholar] [CrossRef]
- Fararjeh, A.-F.S.; Tu, S.-H.; Chen, L.-C.; Cheng, T.-C.; Liu, Y.-R.; Chang, H.-L.; Chang, H.-W.; Huang, C.-C.; Wang, H.-C.R.; Hwang-Verslues, W.W.; et al. Long-term exposure to extremely low-dose of nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induce non-malignant breast epithelial cell transformation through activation of the a9-nicotinic acetylcholine receptor-mediated signaling pathway. Environ. Toxicol. 2019, 34, 73–82. [Google Scholar] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Teng, F.; Kong, L.; Yu, J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco. Targets. Ther. 2016, 9, 5023–5039. [Google Scholar] [PubMed]
- Sharpe, A.H.; Lafleur, M.W.; Muroyama, Y.; Drake, C.G. Therapy Inhibitors of the PD-1 Pathway in Tumor. J. Immunol. 2018, 200, 375–383. [Google Scholar]
- Arasanz, H.; Gato-Cañas, M.; Zuazo, M.; Ibañez-Vea, M.; Breckpot, K.; Kochan, G.; Escors, D. PD1 signal transduction pathways in T cells. Oncotarget 2017, 8, 51936–51945. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: Beyond immune evasion. Front. Oncol. 2018, 8, 386–394. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef]
- Lu, Y.; Robbins, P.F. Seminars in Immunology Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef]
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28–39. [Google Scholar] [CrossRef]
- Li, B.; Huang, X.; Fu, L. Impact of smoking on efficacy of PD-1/PD-l1 inhibitors in non-small cell lung cancer patients: A meta-analysis. Onco. Targets. Ther. 2018, 11, 3691–3696. [Google Scholar] [CrossRef]
- Cao, L.; Wang, X.; Li, S.; Zhi, Q.; Wang, Y.; Wang, L.; Li, K.; Jiang, R. PD-L1 is a prognostic biomarker in resected NSCLC patients with moderate/high smoking history and elevated serum SCCA level. J. Cancer 2017, 8, 3251–3260. [Google Scholar] [CrossRef]
- Calles, A.; Liao, X.; Sholl, L.M.; Rodig, S.J.; Freeman, G.J.; Butaney, M.; Lydon, C.; Dahlberg, S.E.; Hodi, F.S.; Oxnard, G.R.; et al. Expression of PD-1 and Its Ligands, PD-L1 and PD-L2, in Smokers and Never Smokers with KRAS-Mutant Lung Cancer. J. Thorac. Oncol. 2015, 10, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.Z.; Zhang, L.; Zhao, X.C.; Gao, S.H.; Qu, L.W.; Yu, H.; Fang, W.F.; Zhou, Y.C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Widmer, D.S.; Cheng, P.F.; Eichhoff, O.M.; Belloni, B.C.; Zipser, M.C.; Schlegel, N.C.; Javelaud, D.; Mauviel, A.; Dummer, R.; Hoek, K.S. Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment Cell Melanoma Res. 2012, 25, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 1337–1362. [Google Scholar] [CrossRef]
- Wang, W.B.; Yen, M.L.; Liu, K.J.; Hsu, P.J.; Lin, M.H.; Chen, P.M.; Sudhir, P.R.; Chen, C.H.; Chen, C.H.; Sytwu, H.K.; et al. Interleukin-25 Mediates Transcriptional Control of PD-L1 via STAT3 in Multipotent Human Mesenchymal Stromal Cells (hMSCs) to Suppress Th17 Responses. Stem Cell Rep. 2015, 5, 392–404. [Google Scholar] [CrossRef]
- Bergen, A.W.; Caporaso, N. Cigarette smoking. J. Natl. Cancer Inst. 1999, 91, 1365–1375. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Yao, H.; Li, C.; Fang, J.Y.; Xu, J. Regulation of PD-L1: Emerging routes for targeting tumor immune evasion. Front. Pharmacol. 2018, 9, 536–549. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Hu, D.X.; Chen, W.-Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Zerdes, I.; Matikas, A.; Bergh, J.; Rassidakis, G.Z.; Foukakis, T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene 2018, 37, 4639–4661. [Google Scholar] [CrossRef] [PubMed]
- Song, T.L.; Nairismägi, M.-L.; Laurensia, Y.; Lim, J.-Q.; Tan, J.; Li, Z.-M.; Pang, W.-L.; Kizhakeyil, A.; Wijaya, G.-C.; Huang, D.-C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Song, Z.; Wang, H.-B.; Lang, L.; Yang, Y.-Z.; Xiao, W.; Webster, D.E.; Wei, W.; Barta, S.K.; Kadin, M.E.; et al. A novel model of controlling PD-L1 expression in ALK + anaplastic large cell lymphoma revealed by CRISPR screening. Blood 2019, 134, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Shchepotin, I.B.; Galitovkiy, V.; Grando, S.A. Mechanisms of tumor-promoting activities of nicotine in lung cancer: Synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer 2015, 15, 152–164. [Google Scholar] [CrossRef]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.-A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef]
- Yang, L.; Huang, F.; Mei, J.; Wang, X.; Zhang, Q.; Wang, H.; Xi, M.; You, Z. Posttranscriptional Control of PD-L1 Expression by 17β-Estradiol via PI3K/Akt Signaling Pathway in ERα-Positive Cancer Cell Lines. Int. J. Gynecol. Cancer 2017, 27, 196–205. [Google Scholar] [CrossRef]
- García-Aranda, M.; Redondo, M. Targeting protein kinases to enhance the response to anti-PD-1/PD-l1 immunotherapy. Int. J. Mol. Sci. 2019, 20, 2296. [Google Scholar] [CrossRef]
- Kortylewski, M.; Jove, R.; Yu, H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005, 24, 315–327. [Google Scholar] [CrossRef]
- I Chernyavsky, A.; Arredondo, J.; Vetter, D.; Grando, S. Central role of α9 acetylcholine receptor in coordinating keratinocyte adhesion and motility at the initiation of epithelialization. Exp. Cell Res. 2007, 313, 3542–3555. [Google Scholar] [CrossRef]
- Xu, R.; Won, J.Y.; Kim, C.H.; Kim, D.E.; Yim, H. Roles of the Phosphorylation of Transcriptional Factors in Epithelial-Mesenchymal Transition. J. Oncol. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Jin, L.; Datta, P. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J. Clin. Med. 2016, 5, 44. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CHRNA9 Low mRNA n (%) | CHRNA9 High mRNA n (%) | p-Value | |
|---|---|---|---|---|
| Age (year) | ≥40 | 147 (32%) | 257 (55%) | 0.33 |
| <40 | 18 (4%) | 42 (9%) | ||
| Sex | Female | 114 (24%) | 179 (38%) | 0.04 * |
| Male | 53 (11%) | 126 (27%) | ||
| Ulceration | Yes | 61 (19%) | 106 (34%) | 0.08 |
| No | 40 (13%) | 107 (34%) | ||
| Breslow depth (mm) | ≥2 | 76 (21%) | 145 (40%) | 0.8 |
| <2 | 46 (13%) | 93 (26%) | ||
| Clark level | I-III | 32 (10%) | 69 (21%) | 0.24 |
| IV-V | 85 (26%) | 136 (42%) | ||
| Tumor size | T1 | 14 (3%) | 27 (7%) | 0.87 |
| T2 | 27 (7%) | 52 (13%) | ||
| T3 | 32 (8%) | 59 (14%) | ||
| T4 | 60 (15%) | 93 (23%) | ||
| Unknown | 15 (4%) | 32 (8%) | ||
| Lymph node status | N0 | 94 (21%) | 141 (31%) | 0.035 * |
| N1 | 20 (4%) | 54 (12%) | ||
| N2 | 17 (4%) | 32 (7%) | ||
| N3 | 22 (5%) | 34 (8%) | ||
| Unknown | 6 (1%) | 30 (7%) | ||
| Distant metastasis status | M0 | 148 (33%) | 270 (61%) | 0.39 |
| M1 | 10 (2%) | 15 (3%) | ||
| Stage | I-II | 94 (25%) | 123 (30%) | 0.009 ** |
| III-IV | 64 (15%) | 131 (32%) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.D.; Liao, Y.-C.; Ho, Y.-S.; Chen, L.-C.; Chang, H.-W.; Cheng, T.-C.; Liu, D.; Lee, W.-R.; Shen, S.-C.; Wu, C.-H.; et al. The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers 2019, 11, 1991. https://doi.org/10.3390/cancers11121991
Nguyen HD, Liao Y-C, Ho Y-S, Chen L-C, Chang H-W, Cheng T-C, Liu D, Lee W-R, Shen S-C, Wu C-H, et al. The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers. 2019; 11(12):1991. https://doi.org/10.3390/cancers11121991
Chicago/Turabian StyleNguyen, Hai Duong, You-Cheng Liao, Yuan-Soon Ho, Li-Ching Chen, Hui-Wen Chang, Tzu-Chun Cheng, Donald Liu, Woan-Ruoh Lee, Shing-Chuan Shen, Chih-Hsiung Wu, and et al. 2019. "The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration" Cancers 11, no. 12: 1991. https://doi.org/10.3390/cancers11121991
APA StyleNguyen, H. D., Liao, Y.-C., Ho, Y.-S., Chen, L.-C., Chang, H.-W., Cheng, T.-C., Liu, D., Lee, W.-R., Shen, S.-C., Wu, C.-H., & Tu, S.-H. (2019). The α9 Nicotinic Acetylcholine Receptor Mediates Nicotine-Induced PD-L1 Expression and Regulates Melanoma Cell Proliferation and Migration. Cancers, 11(12), 1991. https://doi.org/10.3390/cancers11121991