Identification of Cross Talk between FoxM1 and RASSF1A as a Therapeutic Target of Colon Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
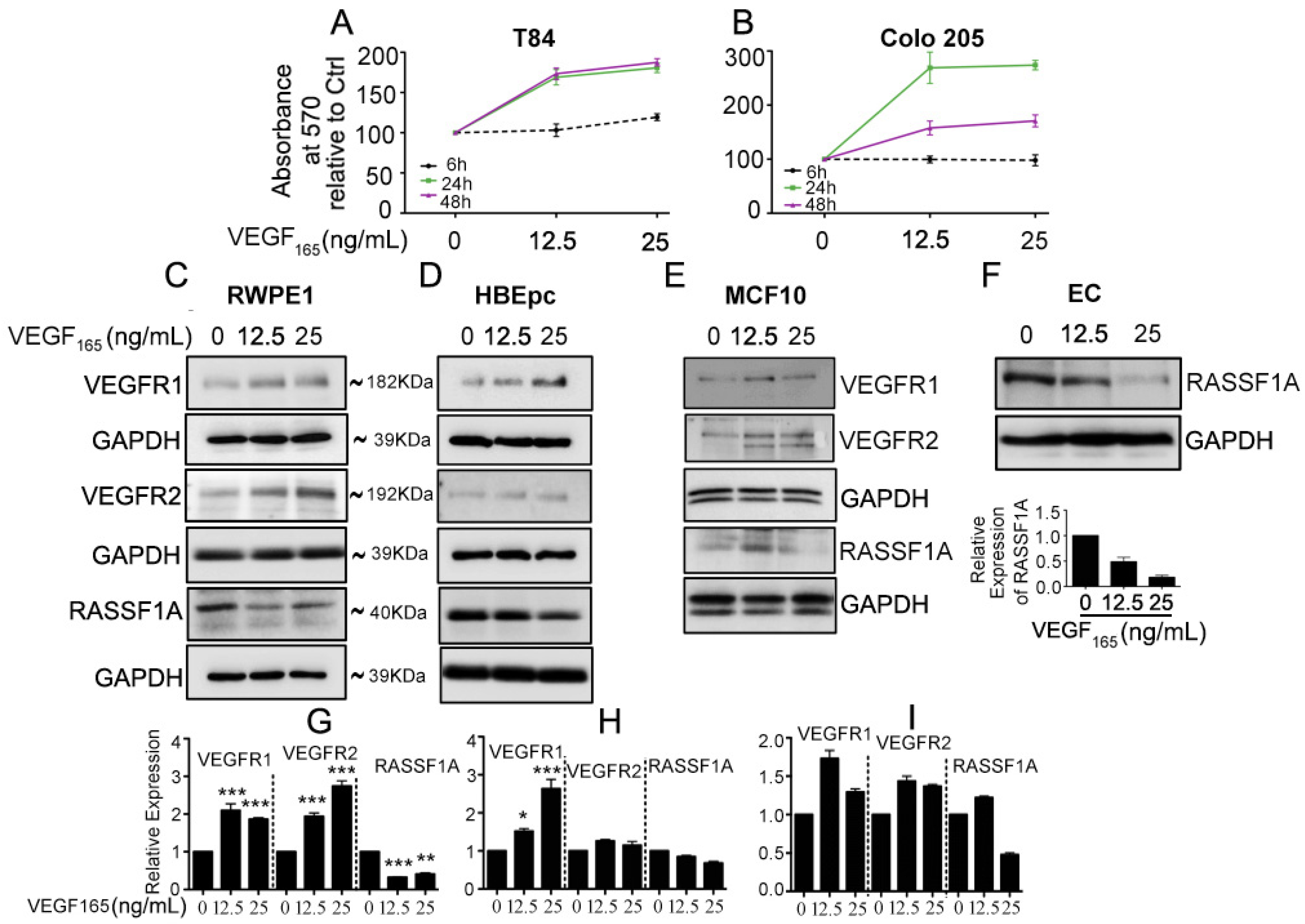
2.1. Exogenous VEGF Suppresses RASSF1A Expression in Normal Epithelial Cells
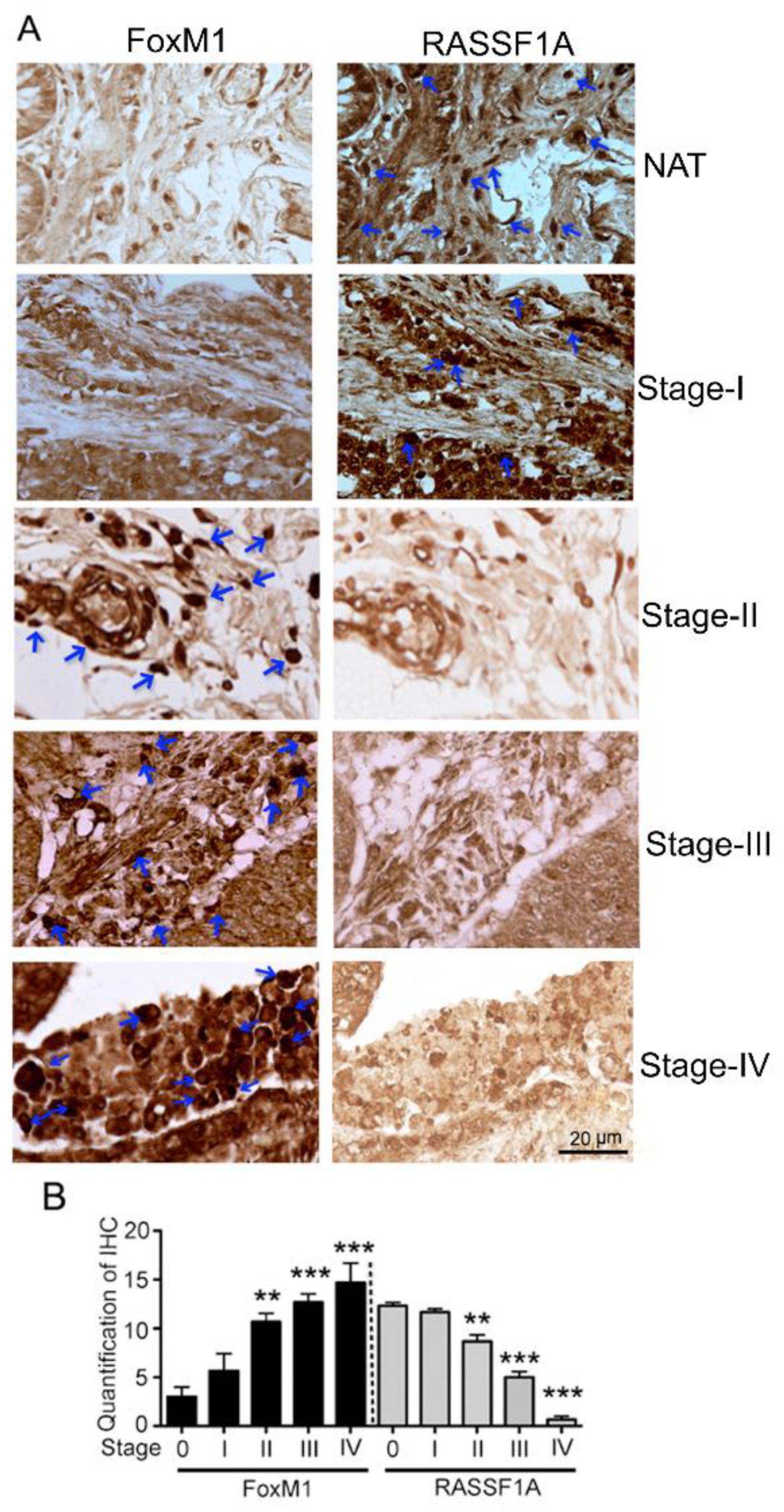
2.2. RASSF1A and FoxM1 Expression in Colon Cancer
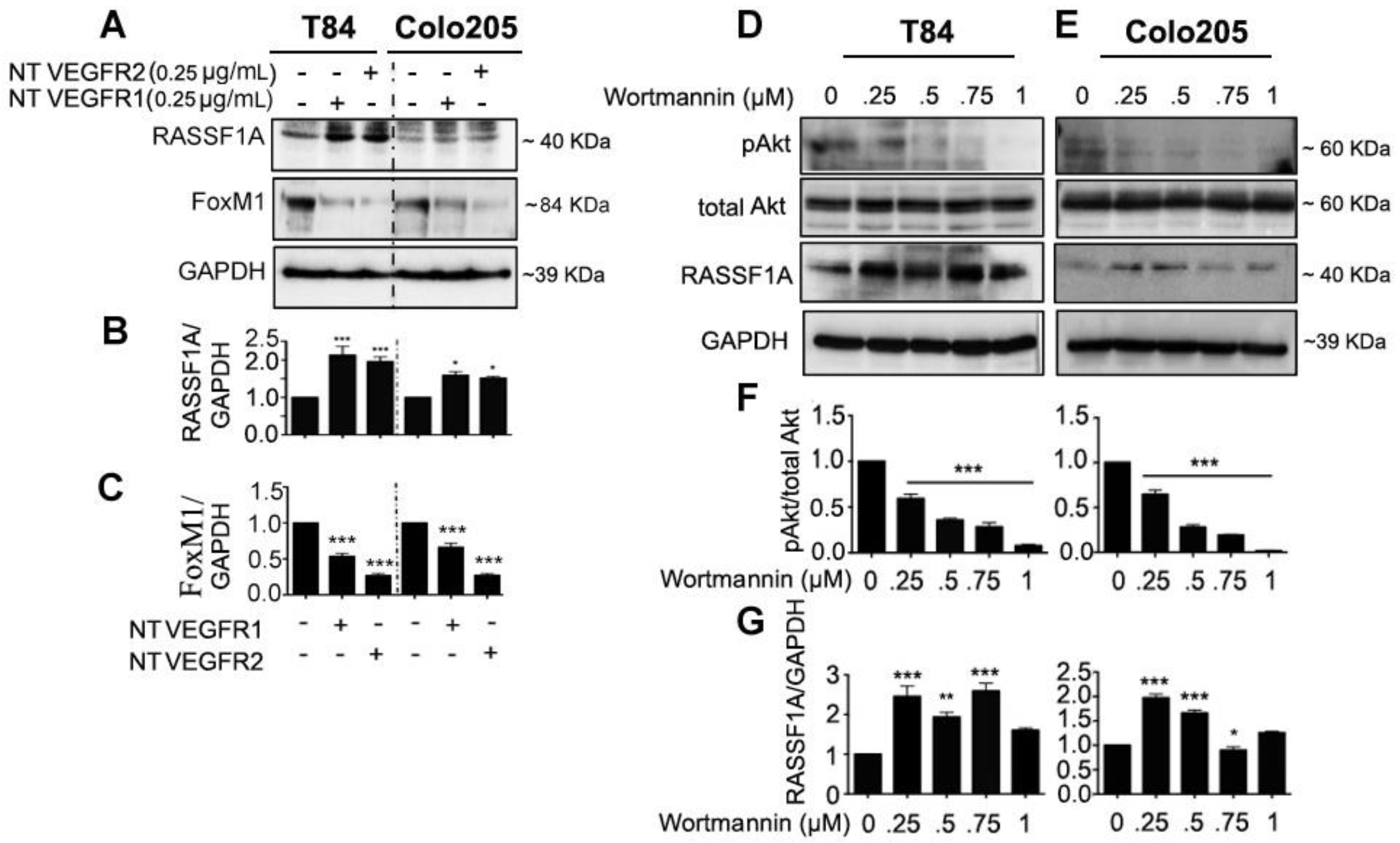
2.3. Neutralized VEGF Receptors and Akt Inhibition Upregulates RASSF1A Expression
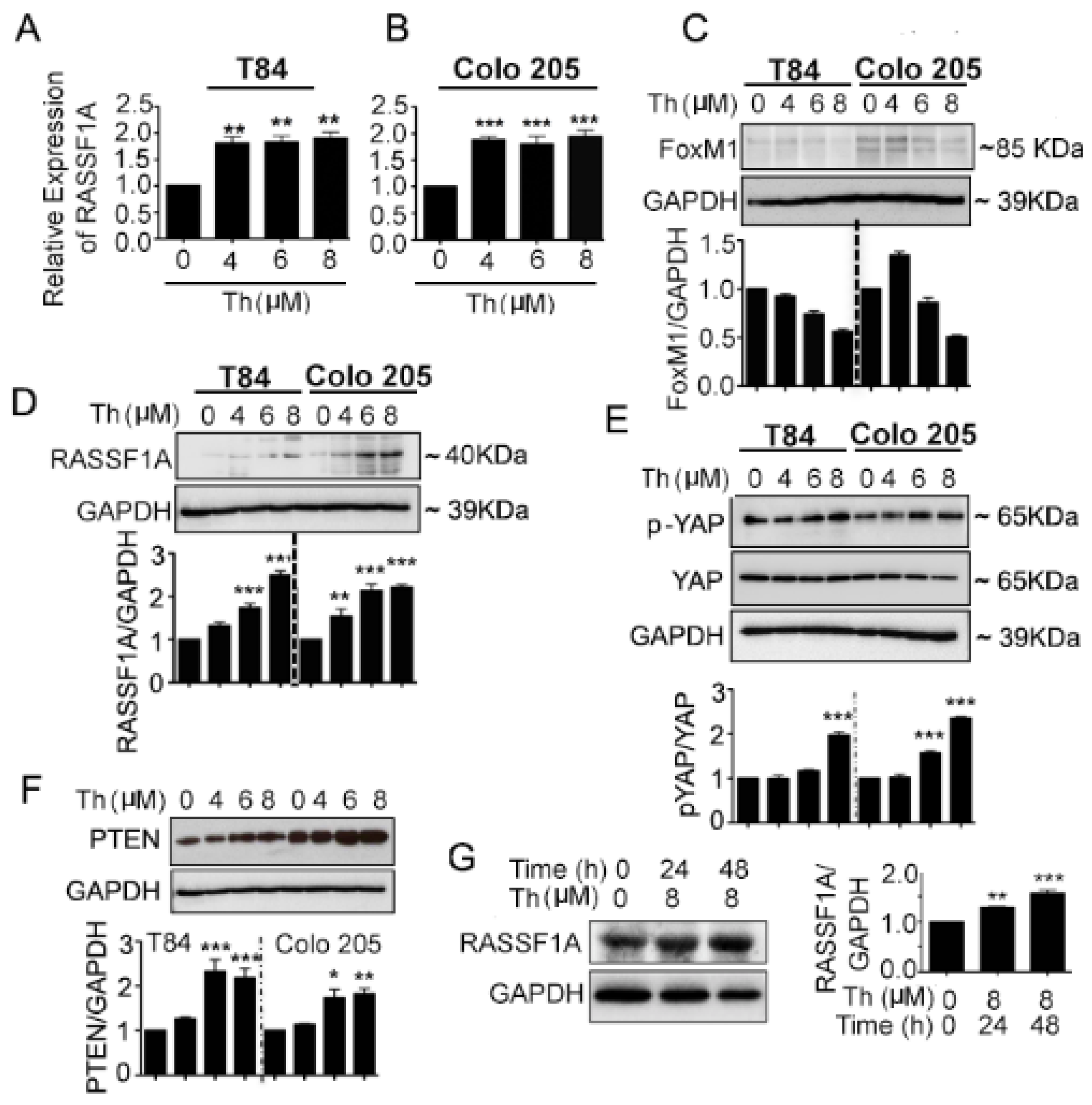
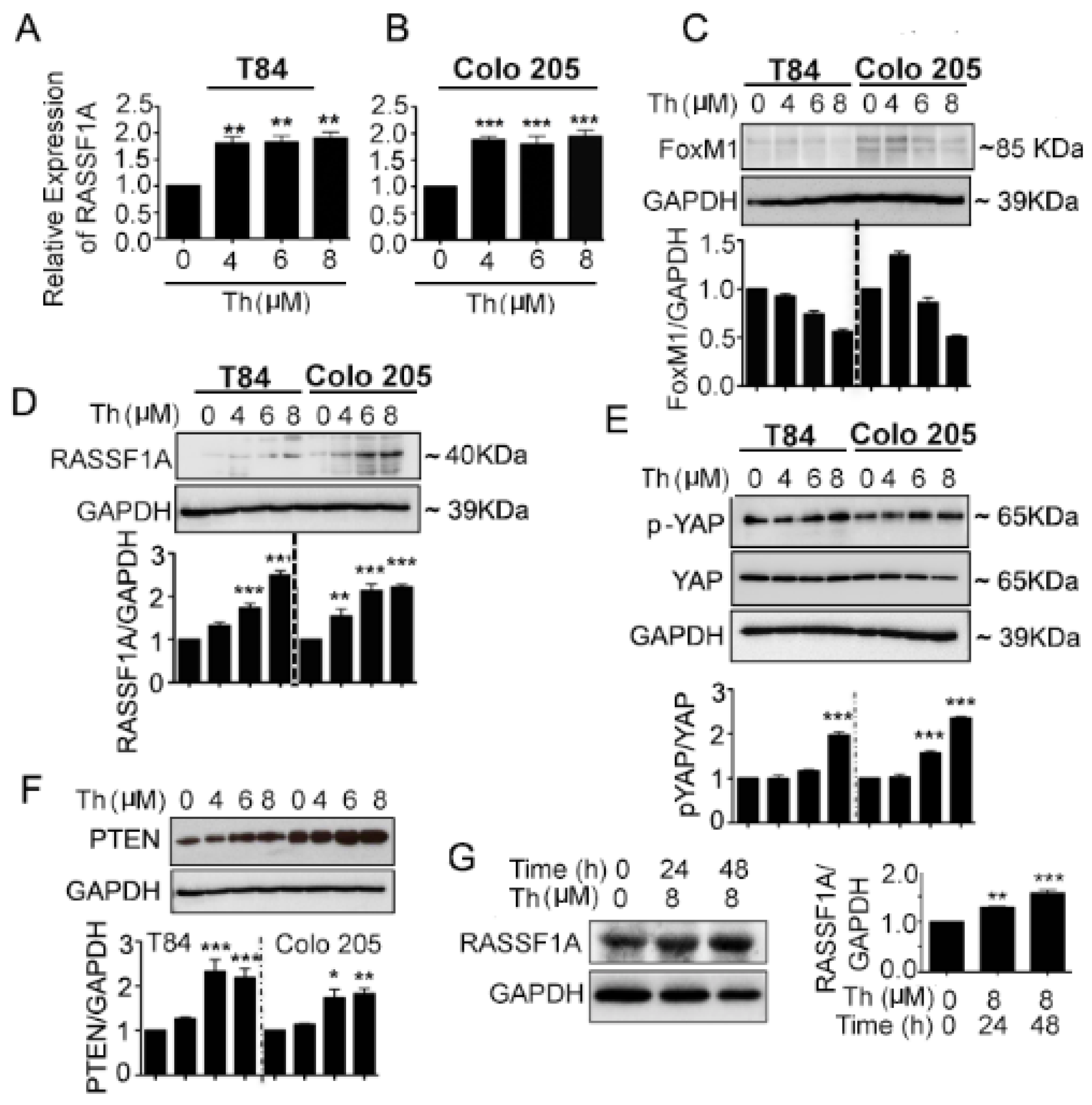
2.4. FoxM1 Inhibition Upregulates RASSF1A Expression
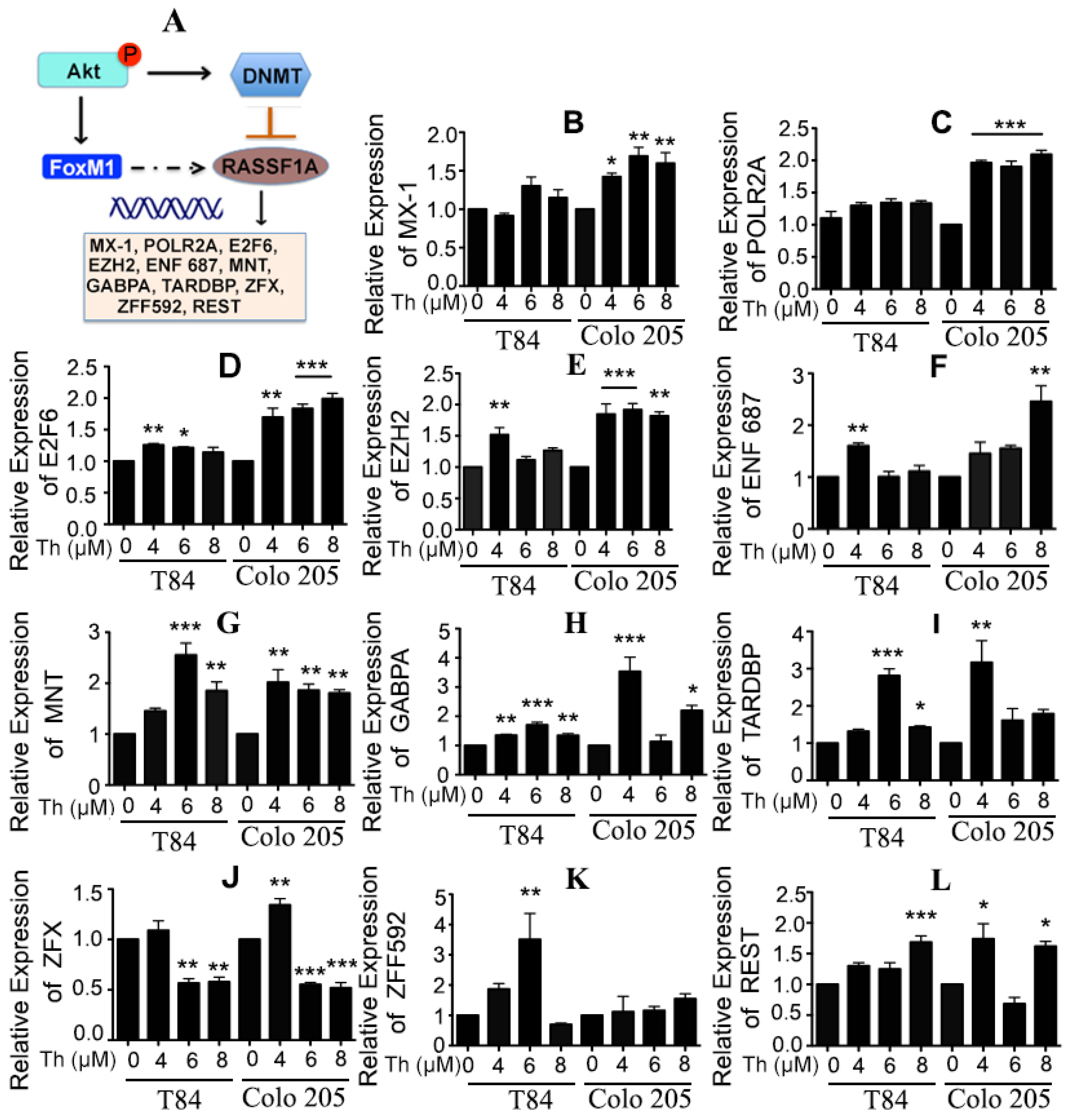
2.5. Potential Regulation of RASSF1A by FoxM1 through Regulation of Transcription Factor Activity
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Generation and Propagation of Patient-Derived Organoid Cell Cultures
4.3. Cell Culture
4.4. Viability Assay
4.5. Immunobloting
4.6. Immunohistochemistry
4.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.8. FoxM1 Overexpression and Depletion
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgements
Conflicts of Interest
References
- Wang, Y.; Wang, W.; Sanidad, K.Z.; Shih, P.A.; Zhao, X.; Zhang, G. Eicosanoid signaling in carcinogenesis of colorectal cancer. Cancer Metastasis Rev. 2018, 37, 257–267. [Google Scholar] [CrossRef]
- Society, A.C. Key Statistics for Colorectal Cancer. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html (accessed on 24 January 2019).
- Stenvang, J.; Andreassen, C.H.; Brünner, N. Screening of 129 FDA approved anti-cancer drugs in colorectal cancer cell lines resistant to oxaliplatin or irinotecan (SN38). J. Clin. Oncol. 2017, 35, 642. [Google Scholar] [CrossRef]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204–1225.e1212. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S.; et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, E3402–E3411. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Qi, H.X.; Hu, Z.M.; Chang, Y.N.; Shi, Z.M.; Han, X.H.; Han, Y.W.; Zhang, R.X.; Zhang, Z.; Chen, T.; et al. YAP and TAZ Take Center Stage in Cancer. Biochemistry 2015, 54, 6555–6566. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.; Sun, Z.; Li, S.; Li, G.; Li, X.; Ma, J. Dual roles of yes-associated protein (YAP) in colorectal cancer. Oncotarget 2017, 8, 75727–75741. [Google Scholar] [CrossRef]
- Jain, S.; Xie, L.; Boldbaatar, B.; Lin, S.Y.; Hamilton, J.P.; Meltzer, S.J.; Chen, S.H.; Hu, C.T.; Block, T.M.; Song, W.; et al. Differential methylation of the promoter and first exon of the RASSF1A gene in hepatocarcinogenesis. Hepatol. Res. 2015, 45, 1110–1123. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Yee, K.; Meissl, K.; Kucerova, L.; Piazzolla, D.; Baccarini, M.; Vass, J.K.; Kolch, W.; O’Neill, E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007, 27, 962–975. [Google Scholar] [CrossRef]
- Levy, D.; Reuven, N.; Shaul, Y. A regulatory circuit controlling Itch-mediated p73 degradation by Runx. J. Biol. Chem. 2008, 283, 27462–27468. [Google Scholar] [CrossRef]
- Zheng, Y.; Guo, J.; Zhou, J.; Lu, J.; Chen, Q.; Zhang, C.; Qing, C.; Koeffler, H.P.; Tong, Y. FoxM1 transactivates PTTG1 and promotes colorectal cancer cell migration and invasion. BMC Med. Genomics 2015, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tong, Y.; Wawrowsky, K.; Melmed, S. PTTG acts as a STAT3 target gene for colorectal cancer cell growth and motility. Oncogene 2014, 33, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Dallol, A.; Agathanggelou, A.; Tommasi, S.; Pfeifer, G.P.; Maher, E.R.; Latif, F. Involvement of the RASSF1A tumor suppressor gene in controlling cell migration. Cancer Res. 2005, 65, 7653–7659. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhou, C.; Li, B.; Chen, Y.; Dai, J.; Mao, Y.; Huang, T.; Yu, H.; Chen, M.; Zhao, J.; et al. Diagnostic value of RASSF1A hypermethylation in colorectal cancer: a meta-analysis. Pathol. Res. Pract. 2018, 214, 1572–1578. [Google Scholar] [CrossRef]
- Wong, T.S.; Kwong, D.L.; Sham, J.S.; Tsao, S.W.; Wei, W.I.; Kwong, Y.L.; Yuen, A.P. Promoter hypermethylation of high-in-normal 1 gene in primary nasopharyngeal carcinoma. Clin. Cancer Res. 2003, 9, 3042–3046. [Google Scholar] [PubMed]
- Liu, L.; Broaddus, R.R.; Yao, J.C.; Xie, S.; White, J.A.; Wu, T.T.; Hamilton, S.R.; Rashid, A. Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod. Pathol. 2005, 18, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Sakano, S.; Suehiro, Y.; Okada, T.; Korenaga, Y.; Hara, T.; Naito, K.; Matsuyama, H.; Hinoda, Y. Methylation level of the RASSF1A promoter is an independent prognostic factor for clear-cell renal cell carcinoma. Ann. Oncol. 2010, 21, 1612–1617. [Google Scholar] [CrossRef]
- Lu, S.; Ahmed, T.; Du, P.; Wang, Y. Genomic Variations in Pancreatic Cancer and Potential Opportunities for Development of New Approaches for Diagnosis and Treatment. Int. J. Mol. Sci. 2017, 18, 1201. [Google Scholar] [CrossRef]
- Richter, A.M.; Walesch, S.K.; Dammann, R.H. Aberrant Promoter Methylation of the Tumour Suppressor RASSF10 and Its Growth Inhibitory Function in Breast Cancer. Cancers 2016, 8, 26. [Google Scholar] [CrossRef]
- Dammann, R.; Schagdarsurengin, U.; Strunnikova, M.; Rastetter, M.; Seidel, C.; Liu, L.; Tommasi, S.; Pfeifer, G.P. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol. Histopathol. 2003, 18, 665–677. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Yoon, J.H.; Liu, L.; Tommasi, S.; Wilczynski, S.P.; Dammann, R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002, 383, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.-w.; Zhao, L.-D.; Wang, G.-X.; Kang, X.-C.; Qin, L.; Ji, J.; Hao, S. Promoter methylation and expression of RASSF 1 A genes as predictors of disease progression in colorectal cancer. Biomed. Pharmacother. 2016, 80, 23–29. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Zumwalt, T.J.; Goel, A. DNA methylation patterns as noninvasive biomarkers and targets of epigenetic therapies in colorectal cancer. Epigenomics 2016, 8, 685–703. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, T.G.; Lapidus, R.; Banerjee, V.; Bafford, A.C.; Czinn, S.J.; Ahmed, H.; Banerjee, A. Upregulation of RASSF1A in Colon Cancer by Suppression of Angiogenesis Signaling and Akt Activation. Cell Physiol. Biochem. 2018, 48, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Banerjee, V.; Czinn, S.; Blanchard, T. Increased reactive oxygen species levels cause ER stress and cytotoxicity in andrographolide treated colon cancer cells. Oncotarget 2017, 8, 26142–26153. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, A.B.; Du, Y.; Fu, H.; Yang, V.W. Identification of novel small-molecule compounds that inhibit the proproliferative Kruppel-like factor 5 in colorectal cancer cells by high-throughput screening. Mol. Cancer Ther. 2009, 8, 563–570. [Google Scholar] [CrossRef]
- GenesCards Human Gene Database. RASSF1 Gene. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=RASSF1 (accessed on 11 July 2018).
- Banerjee, A.; Ahmed, H.; Yang, P.; Czinn, S.J.; Blanchard, T.G. Endoplasmic reticulum stress and IRE-1 signaling cause apoptosis in colon cancer cells in response to andrographolide treatment. Oncotarget 2016, 7, 41432–41444. [Google Scholar] [CrossRef]
- Pal, D.; Tyagi, A.; Chandrasekaran, B.; Alattasi, H.; Ankem, M.K.; Sharma, A.K.; Damodaran, C. Suppression of Notch1 and AKT mediated epithelial to mesenchymal transition by Verrucarin J in metastatic colon cancer. Cell Death Dis. 2018, 9, 798. [Google Scholar] [CrossRef]
- Agarwal, S.; Amin, K.S.; Jagadeesh, S.; Baishay, G.; Rao, P.G.; Barua, N.C.; Bhattacharya, S.; Banerjee, P.P. Mahanine restores RASSF1A expression by down-regulating DNMT1 and DNMT3B in prostate cancer cells. Mol. Cancer 2013, 12, 99. [Google Scholar] [CrossRef]
- Liu, Y.; Shreder, K.R.; Gai, W.; Corral, S.; Ferris, D.K.; Rosenblum, J.S. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem. Biol. 2005, 12, 99–107. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Linthicum, W.; Thanh, M.H.; Vitolo, M.I.; Wen, Q. Effects of PTEN Loss and Activated KRAS Overexpression on Mechanical Properties of Breast Epithelial Cells. Int. J. Mol. Sci. 2018, 19, 1613. [Google Scholar] [CrossRef] [PubMed]
- Mochin, M.T.; Underwood, K.F.; Cooper, B.; McLenithan, J.C.; Pierce, A.D.; Nalvarte, C.; Arbiser, J.; Karlsson, A.I.; Moise, A.R.; Moskovitz, J.; et al. Hyperglycemia and redox status regulate RUNX2 DNA-binding and an angiogenic phenotype in endothelial cells. Microvasc. Res. 2015, 97, 55–64. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Amin, M.N.; Frieman, M.B.; Chen, W.H.; Cross, A.S.; Wang, L.X.; Vasta, G.R. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol. Immunol. 2015, 65, 1–16. [Google Scholar] [CrossRef]
- Shiu, J.; Piazuelo, M.B.; Ding, H.; Czinn, S.J.; Drakes, M.L.; Banerjee, A.; Basappa, N.; Kobayashi, K.S.; Fricke, W.F.; Blanchard, T.G. Gastric LTi cells promote lymphoid follicle formation but are limited by IRAK-M and do not alter microbial growth. Mucosal. Immunol. 2015, 8, 1047–1059. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanchard, T.G.; Czinn, S.J.; Banerjee, V.; Sharda, N.; Bafford, A.C.; Mubariz, F.; Morozov, D.; Passaniti, A.; Ahmed, H.; Banerjee, A. Identification of Cross Talk between FoxM1 and RASSF1A as a Therapeutic Target of Colon Cancer. Cancers 2019, 11, 199. https://doi.org/10.3390/cancers11020199
Blanchard TG, Czinn SJ, Banerjee V, Sharda N, Bafford AC, Mubariz F, Morozov D, Passaniti A, Ahmed H, Banerjee A. Identification of Cross Talk between FoxM1 and RASSF1A as a Therapeutic Target of Colon Cancer. Cancers. 2019; 11(2):199. https://doi.org/10.3390/cancers11020199
Chicago/Turabian StyleBlanchard, Thomas G., Steven J. Czinn, Vivekjyoti Banerjee, Neha Sharda, Andrea C. Bafford, Fahad Mubariz, Dennis Morozov, Antonino Passaniti, Hafiz Ahmed, and Aditi Banerjee. 2019. "Identification of Cross Talk between FoxM1 and RASSF1A as a Therapeutic Target of Colon Cancer" Cancers 11, no. 2: 199. https://doi.org/10.3390/cancers11020199
APA StyleBlanchard, T. G., Czinn, S. J., Banerjee, V., Sharda, N., Bafford, A. C., Mubariz, F., Morozov, D., Passaniti, A., Ahmed, H., & Banerjee, A. (2019). Identification of Cross Talk between FoxM1 and RASSF1A as a Therapeutic Target of Colon Cancer. Cancers, 11(2), 199. https://doi.org/10.3390/cancers11020199