ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Real-Time RT-PCR
2.3. Immunoblotting
2.4. siRNA-Mediated Silencing
2.5. Measurement of Cytosolic Free Ca2+
2.6. Cell Migration Assay
2.7. Development of MDA-MB-468-GCaMP6m Cell Line
2.8. Assessment of Spontaneous Ca2+ Transients
2.9. ORAI3 Promoter Analysis
2.10. Analysis of ORAI1 and ORAI3 Expression in Breast Tumors
2.11. Patient Survival Analysis
2.12. RNA Sequencing Analysis
2.13. Statistical Analysis
3. Results
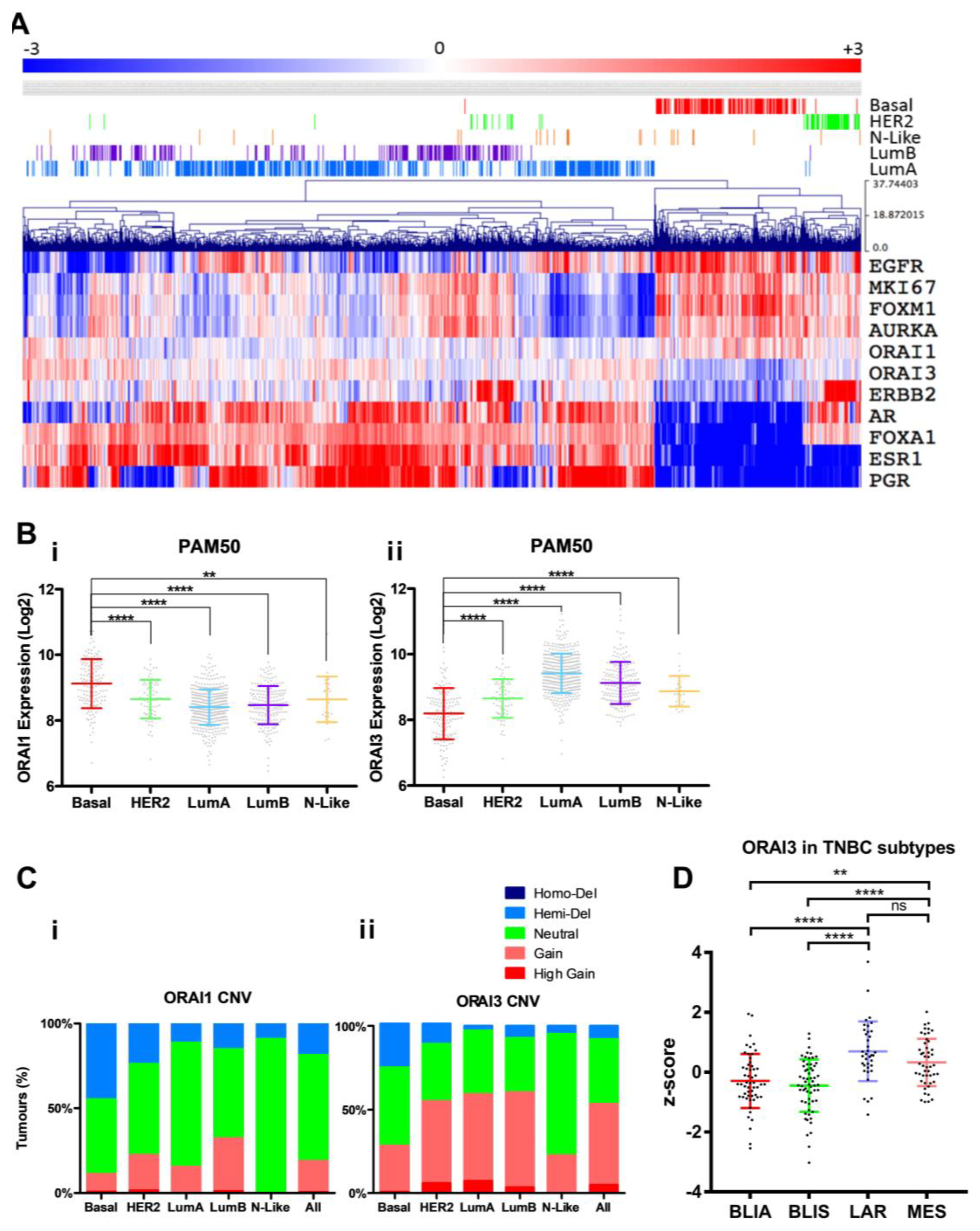
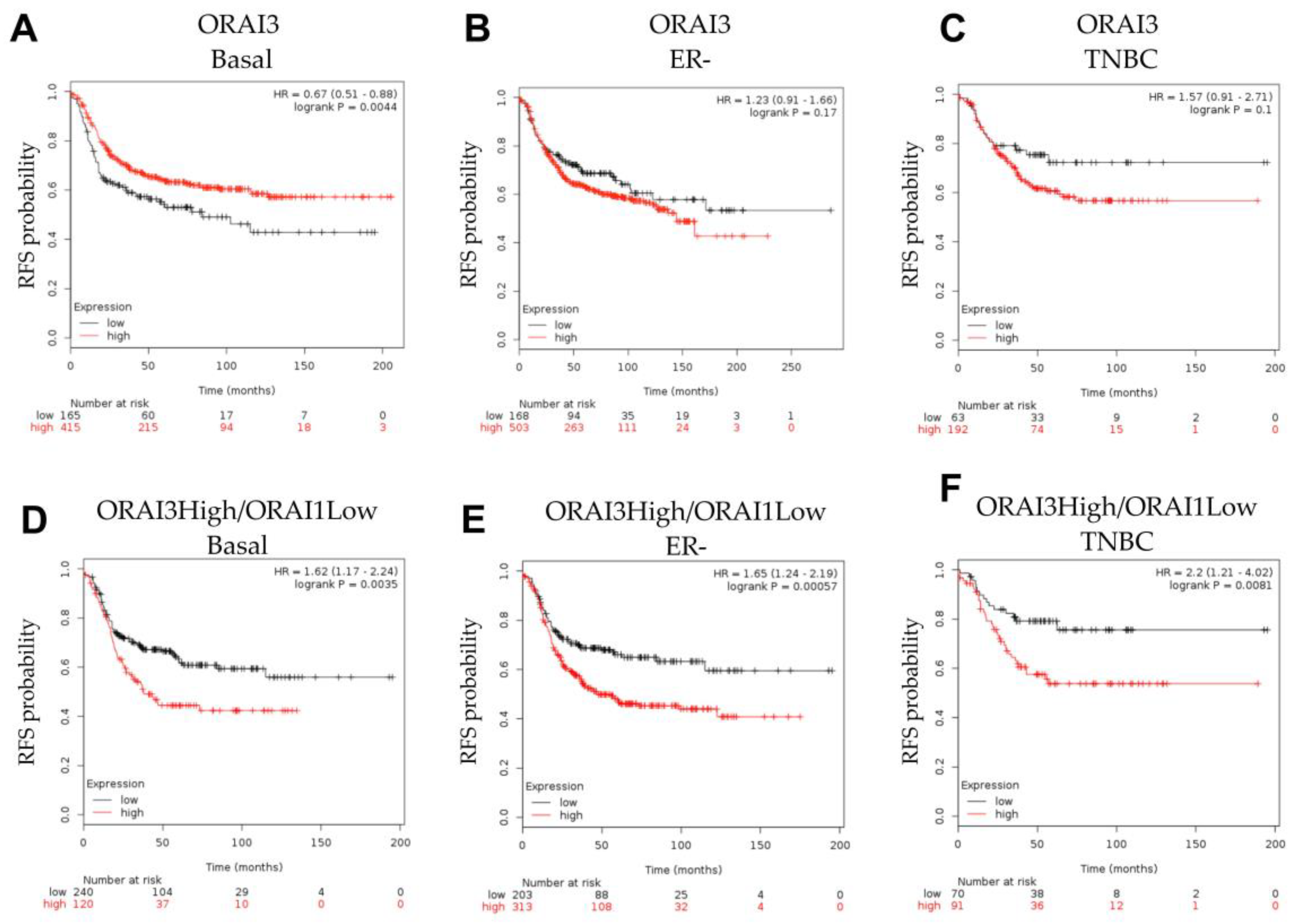
3.1. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes
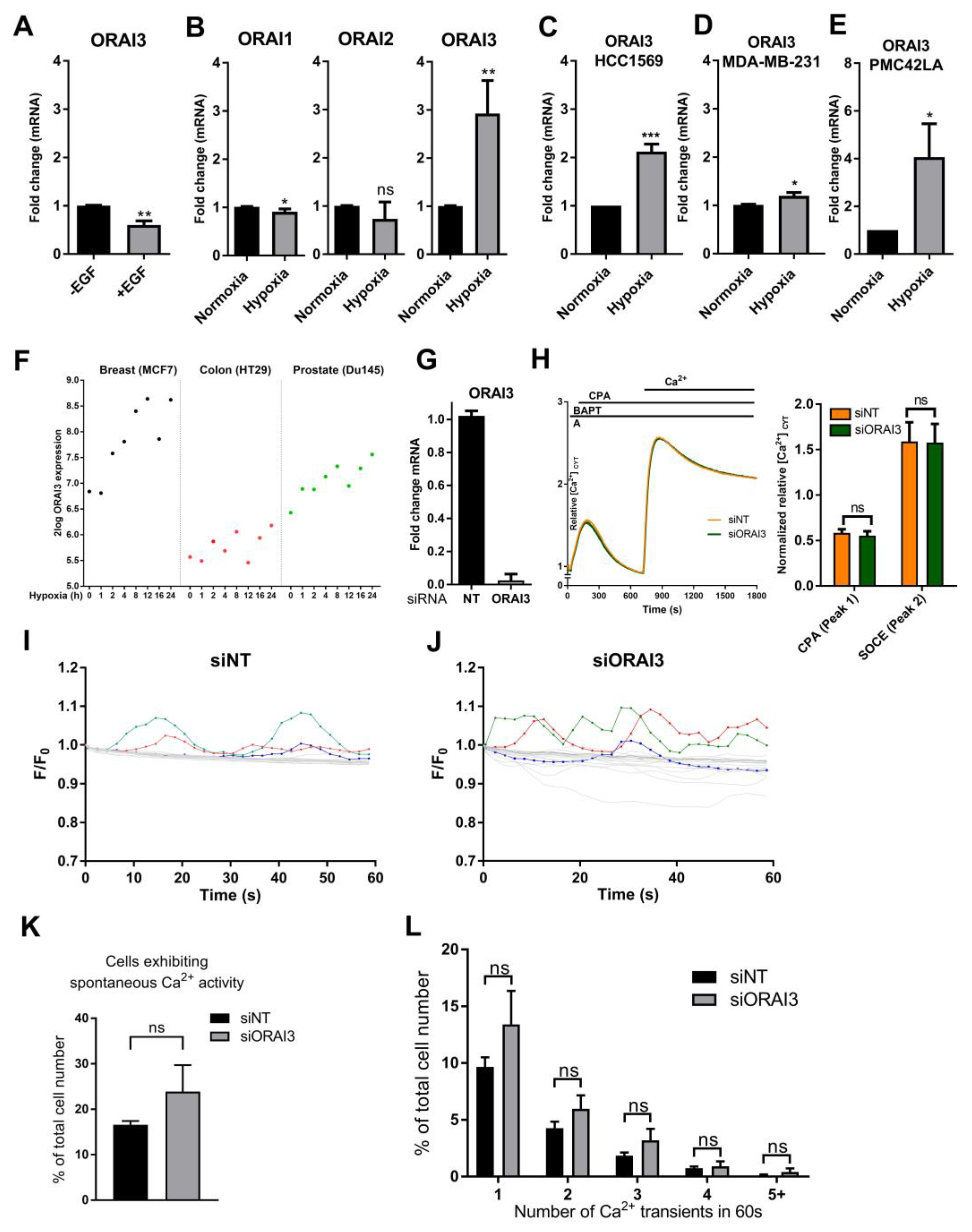
3.2. Hypoxia Induces ORAI3 Expression
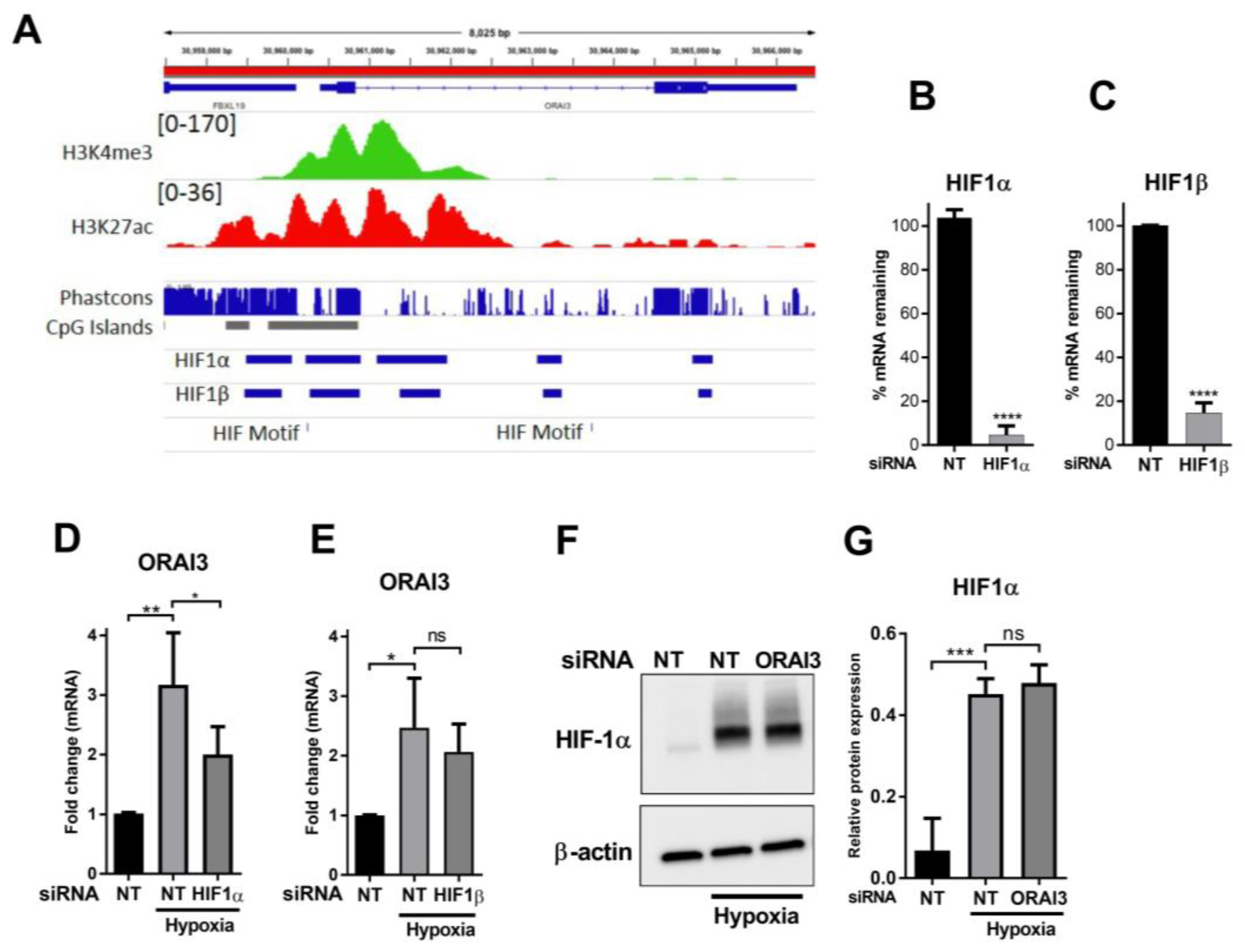
3.3. HIF1α Controls the Hypoxic Expression of ORAI3
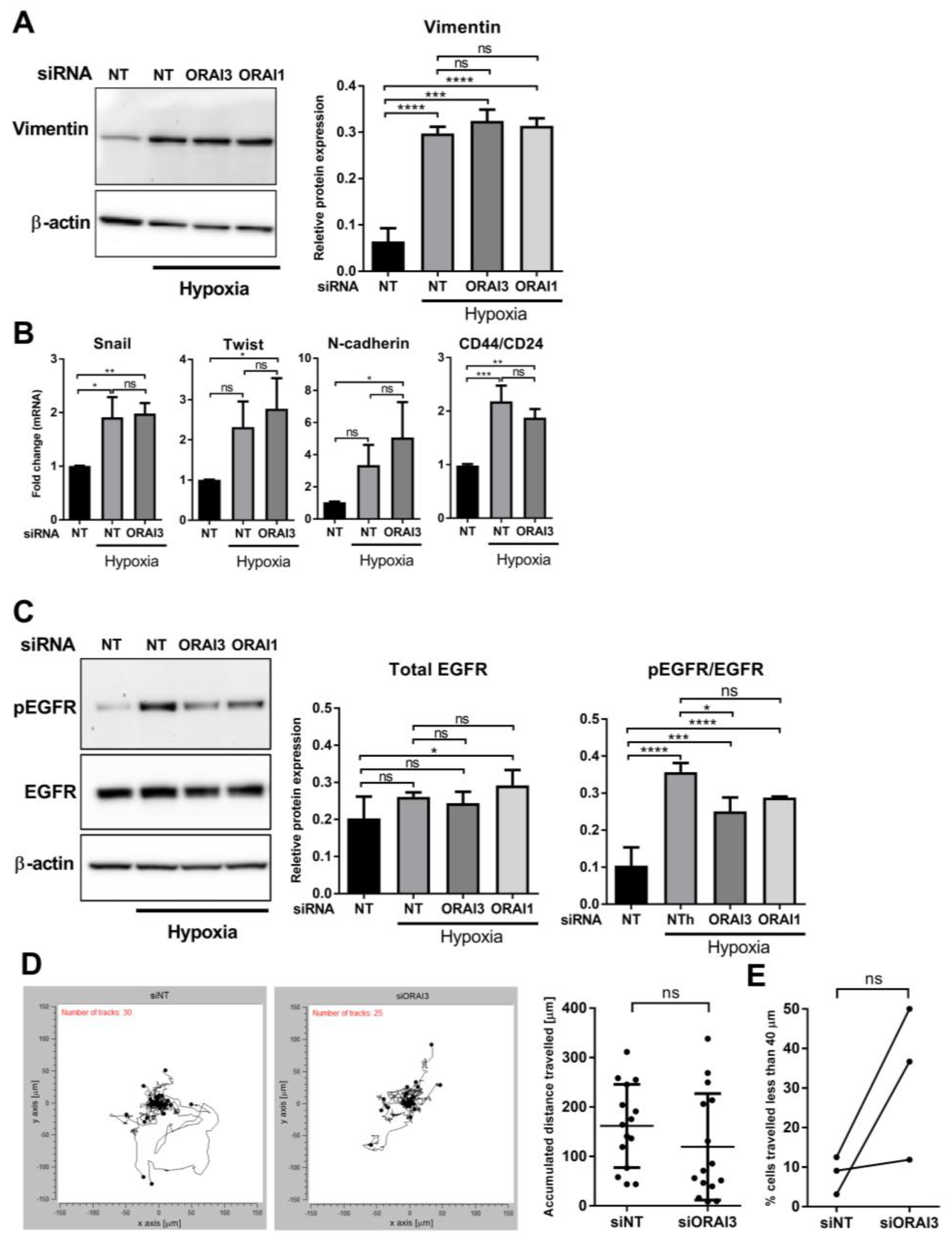
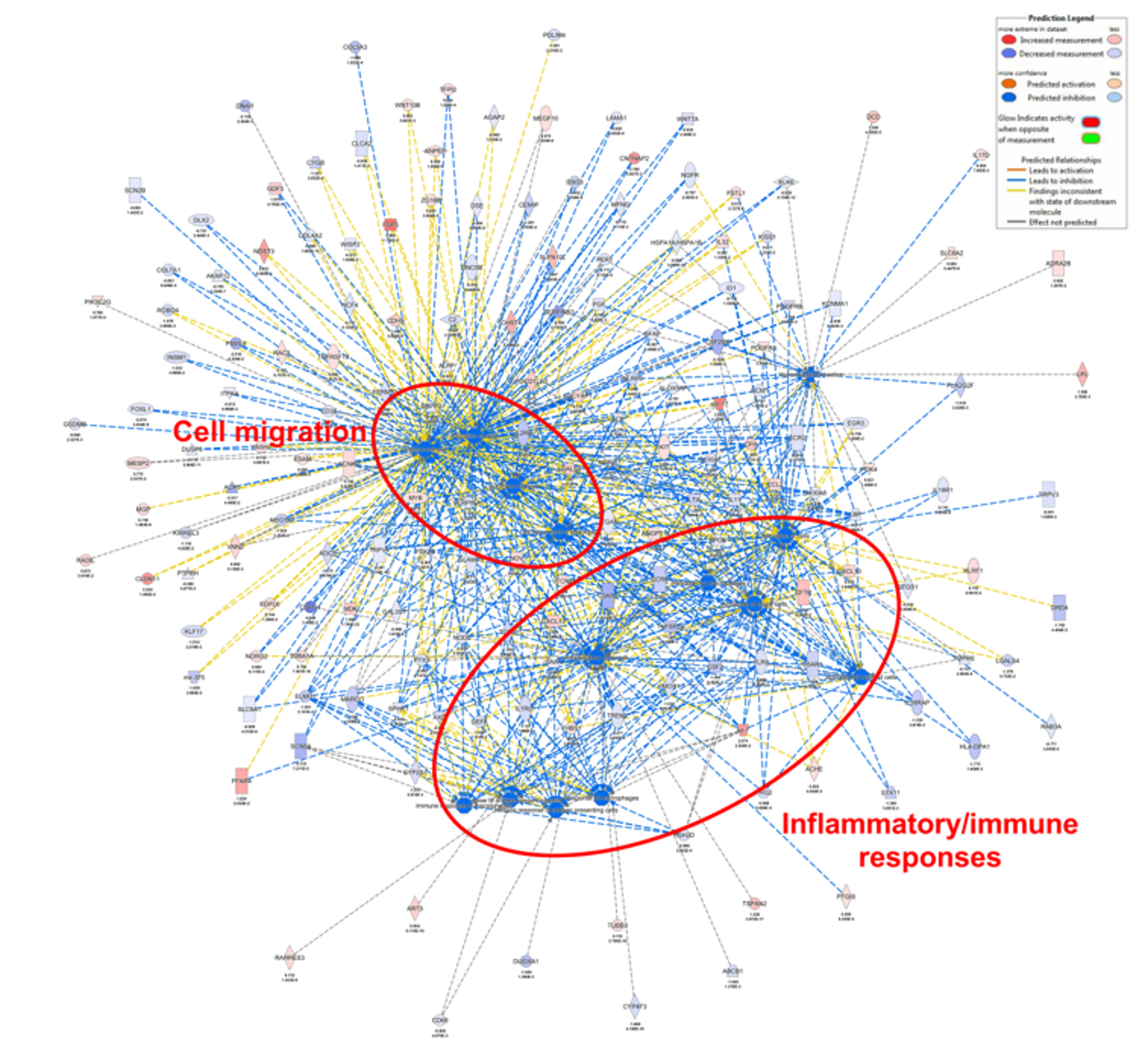
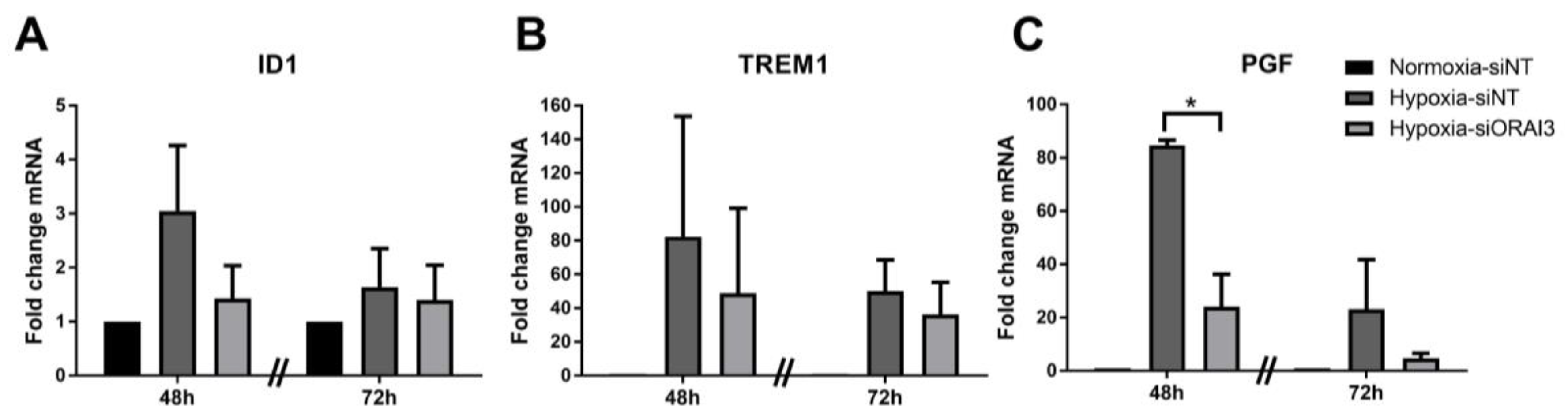
3.4. ORAI3 Is Involved in the Hypoxic Regulation of Genes Associated with Cell Migration and Inflammatory/Immune Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bittremieux, M.; Parys, J.B.; Pinton, P.; Bultynck, G. ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca2+ signaling. BBA-Mol. Cell Res. 2016, 1863, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Davis, F.M.; Roberts-Thomson, S.J.; Monteith, G.R. Ion channels and transporters in cancer. 4. Remodeling of Ca2+ signaling in tumorigenesis: Role of Ca2+ transport. Am. J. Physiol.-Cell Physiol. 2011, 301, C969–C976. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and Cellular Heterogeneity in Breast Cancer Challenges for Personalized Medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Kalimutho, M.; Parsons, K.; Mittal, D.; López, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.Y.; Zhou, Y.D.; Nwokonko, R.M.; Loktionova, N.A.; Wang, X.; Xin, P.; Trebak, M.; Wang, Y.; Gill, D.L. The Orai1 Store-operated Calcium Channel Functions as a Hexamer. J. Biol. Chem. 2016, 291, 25764–25775. [Google Scholar] [CrossRef]
- Putney, J.W.; Steinckwich-Besancon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. BBA-Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Peters, A.A.; Simpson, P.T.; Bassett, J.J.; Lee, J.M.; Da Silva, L.; Reid, L.E.; Song, S.; Parat, M.O.; Lakhani, S.R.; Kenny, P.A.; et al. Calcium channel TRPV6 as a potential therapeutic target in estrogen receptor-negative breast cancer. Mol. Cancer Ther. 2012, 11, 2158–2168. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Stanisz, H.; Vultur, A.; Herlyn, M.; Roesch, A.; Bogeski, I. The role of Orai-STIM calcium channels in melanocytes and melanoma. J. Physiol.-Lond. 2016, 594, 2825–2835. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol.-Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim. Biophys. Acta 2016, 1863, 1408–1417. [Google Scholar] [CrossRef]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef]
- Shuttleworth, T.J. Orai3—The “exceptional” Orai? J. Physiol. 2012, 590, 241–257. [Google Scholar] [CrossRef]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef] [PubMed]
- Saul, S.; Gibhardt, C.S.; Schmidt, B.; Lis, A.; Pasieka, B.; Conrad, D.; Jung, P.; Gaupp, R.; Wonnenberg, B.; Diler, E.; et al. A calcium-redox feedback loop controls human monocyte immune responses: The role of ORAI Ca2+ channels. Sci. Signal. 2016, 9, ra26. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-Induced Reactive Oxygen Species Cause Chromosomal Abnormalities in Endothelial Cells in the Tumor Microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, M.; Wouters, B.G. The Roles of Reactive Oxygen Species and Autophagy in Mediating the Tolerance of Tumor Cells to Cycling Hypoxia. Semin. Radiat. Oncol. 2013, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden, A.F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. BBA-Mol. Cell Res. 2013, 1833, 752–760. [Google Scholar] [CrossRef]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 63–75. [Google Scholar]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Ackland, M.L.; Michalczyk, A.; Whitehead, R.H. PMC42, a novel model for the differentiated human breast. Exp. Cell Res. 2001, 263, 14–22. [Google Scholar] [CrossRef]
- Ackland, M.L.; Newgreen, D.F.; Fridman, M.; Waltham, M.C.; Arvanitis, A.; Minichiello, J.; Price, J.T.; Thompson, E.W. Epidermal growth factor-induced epithelio-mesenchymal transition in human breast carcinoma cells. Lab. Investig. 2003, 83, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Flanagan, J.U.; Stevenson, R.J.; Inserra, M.; Vetter, I.; Monteith, G.R.; Denny, W.A. Evaluation of known and novel inhibitors of Orai1-mediated store operated Ca2+ entry in MDA-MB-231 breast cancer cells using a Fluorescence Imaging Plate Reader assay. Bioorg. Med. Chem. 2017, 25, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: Consequences on calcium signalling, cell migration and proliferation. Cell. Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef]
- Bassett, J.J.; Bong, A.H.L.; Janke, E.K.; Robitaille, M.; Roberts-Thomson, S.J.; Peters, A.A.; Monteith, G.R. Assessment of cytosolic free calcium changes during ceramide-induced cell death in MDA-MB-231 breast cancer cells expressing the calcium sensor GCaMP6m. Cell Calcium 2018, 72, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Chen, X.; Takada, M.; Fan, C.; Zheng, X.; Wen, H.; Liu, Y.; Wang, C.; Pestell, R.G.; et al. EglN2 associates with the NRF1-PGC1alpha complex and controls mitochondrial function in breast cancer. EMBO J. 2015, 34, 2953–2970. [Google Scholar] [CrossRef] [PubMed]
- Schodel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 117, E207–E217. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Getz, G.; Nghiemphu, L.; Barretina, J.; Hsueh, T.; Linhart, D.; Vivanco, I.; Lee, J.C.; Huang, J.H.; Alexander, S.; et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc. Natl. Acad. Sci. USA 2007, 104, 20007–20012. [Google Scholar] [CrossRef] [PubMed]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Davis, F.M.; Kenny, P.A.; Soo, E.T.; van Denderen, B.J.; Thompson, E.W.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Remodeling of purinergic receptor-mediated Ca2+ signaling as a consequence of EGF-induced epithelial-mesenchymal transition in breast cancer cells. PLoS ONE 2011, 6, e23464. [Google Scholar] [CrossRef]
- Stewart, T.A.; Azimi, I.; Brooks, A.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Janus kinases and Src family kinases in the regulation of EGF-induced vimentin expression in MDA-MB-468 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 76, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Beilby, H.; Davis, F.M.; Marcial, D.L.; Kenny, P.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Altered purinergic receptor-Ca signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol. Oncol. 2015, 10, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Starmans, M.H.W.; Chu, K.C.; Haider, S.; Nguyen, F.; Seigneuric, R.; Magagnin, M.G.; Koritzinsky, M.; Kasprzyk, A.; Boutros, P.C.; Wouters, B.G.; et al. The prognostic value of temporal in vitro and in vivo derived hypoxia gene-expression signatures in breast cancer. Radiother. Oncol. 2012, 102, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed]
- Holsken, A.; Gebhardt, M.; Buchfelder, M.; Fahlbusch, R.; Blümcke, I.; Buslei, R. EGFR Signaling Regulates Tumor Cell Migration in Craniopharyngiomas. Clin. Cancer Res. 2011, 17, 4367–4377. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Evers, A.; Zhou, W.H.; Swendeman, S.L.; Wong, P.M.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 229. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef]
- Sobradillo, D.; Hernandez-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef]
- Ay, A.S.; Benzerdjerb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 Constitutes a Native Store-Operated Calcium Entry That Regulates Non Small Cell Lung Adenocarcinoma Cell Proliferation. PLoS ONE 2013, 8, e0124201. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- Azimi, I.; Milevskiy, M.J.G.; Kaemmerer, E.; Turner, D.; Yapa, K.T.D.S.; Brown, M.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. TRPC1 is a differential regulator of hypoxia-mediated events and Akt signalling in PTEN-deficient breast cancer cells. J. Cell Sci. 2017, 130, 2292–2305. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I. The interplay between HIF-1 and calcium signalling in cancer. Int. J. Biochem. Cell Biol. 2018, 97, 73–77. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef]
- Incio, J.; Tam, J.; Rahbari, N.N.; Suboj, P.; McManus, D.T.; Chin, S.M.; Vardam, T.D.; Batista, A.; Babykutty, S.; Jung, K.; et al. PIGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin. Cancer Res. 2016, 22, 2993–3004. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Gilkes, D.M.; Wong, C.C.L.; Kshitiz Luo, W.; Zhang, H.; Wei, H.; Takano, N.; Schito, L.; Levchenko, A. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J. Clin. Investig. 2013, 123, 189–205. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azimi, I.; Milevskiy, M.J.G.; Chalmers, S.B.; Yapa, K.T.D.S.; Robitaille, M.; Henry, C.; Baillie, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers 2019, 11, 208. https://doi.org/10.3390/cancers11020208
Azimi I, Milevskiy MJG, Chalmers SB, Yapa KTDS, Robitaille M, Henry C, Baillie GJ, Thompson EW, Roberts-Thomson SJ, Monteith GR. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers. 2019; 11(2):208. https://doi.org/10.3390/cancers11020208
Chicago/Turabian StyleAzimi, Iman, Michael J.G. Milevskiy, Silke B. Chalmers, Kunsala T.D.S. Yapa, Mélanie Robitaille, Christopher Henry, Gregory J. Baillie, Erik W. Thompson, Sarah J. Roberts-Thomson, and Gregory R. Monteith. 2019. "ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses" Cancers 11, no. 2: 208. https://doi.org/10.3390/cancers11020208
APA StyleAzimi, I., Milevskiy, M. J. G., Chalmers, S. B., Yapa, K. T. D. S., Robitaille, M., Henry, C., Baillie, G. J., Thompson, E. W., Roberts-Thomson, S. J., & Monteith, G. R. (2019). ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers, 11(2), 208. https://doi.org/10.3390/cancers11020208