A H2AX–CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cloning of Complementary DNAs
2.3. Immunoprecipitation (IP), Gst-Pulldown, Western Blot (WB), and MTT Assays
2.4. Synthesis and Purification of Peptides
2.5. DNA Damage and Apoptosis Assay
2.6. Immunofluorescence Staining and Confocal Microscopy
2.7. Kinetics of CARP-1–H2AX Interaction
2.8. Statistical Analyses
3. Results
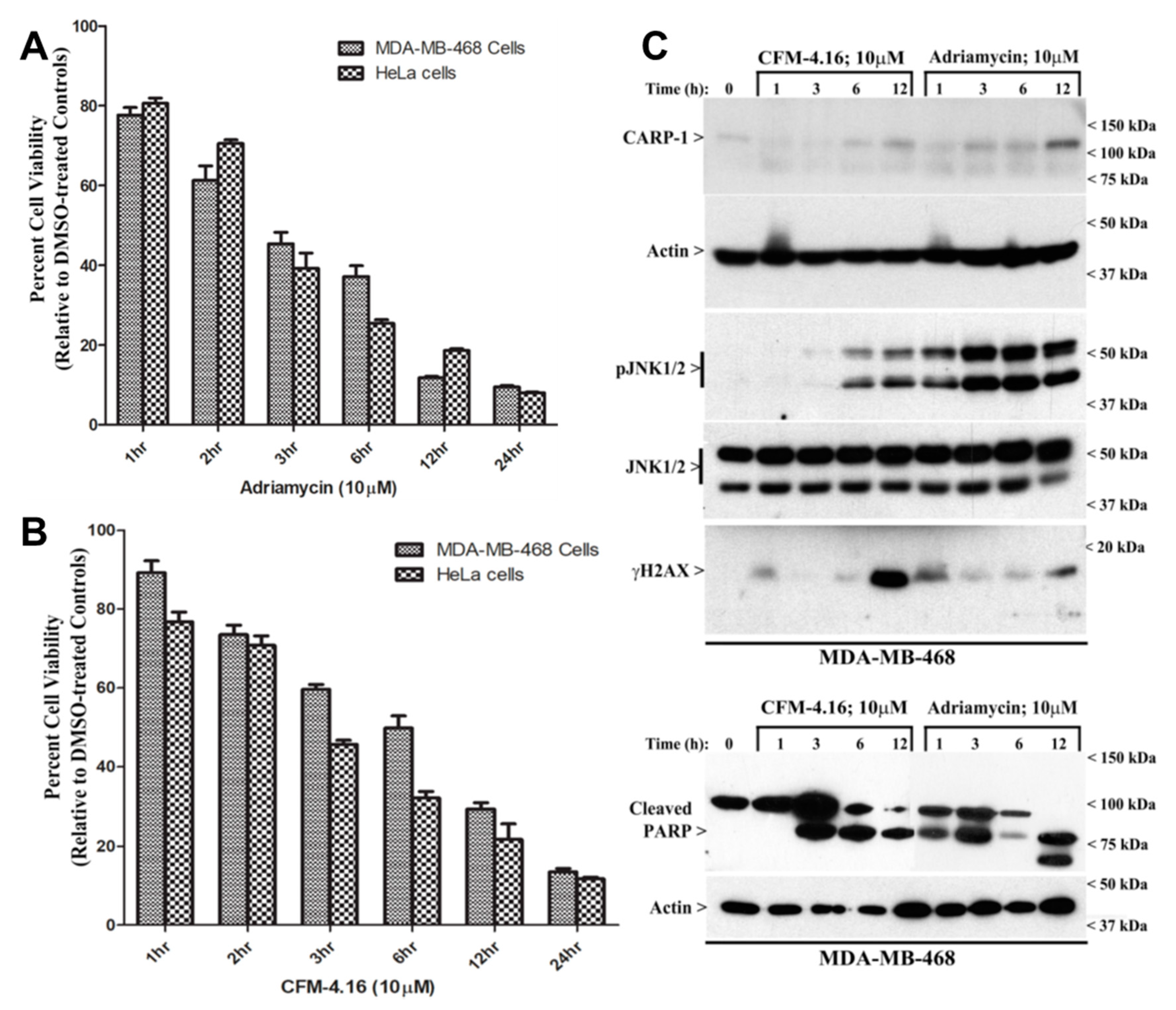
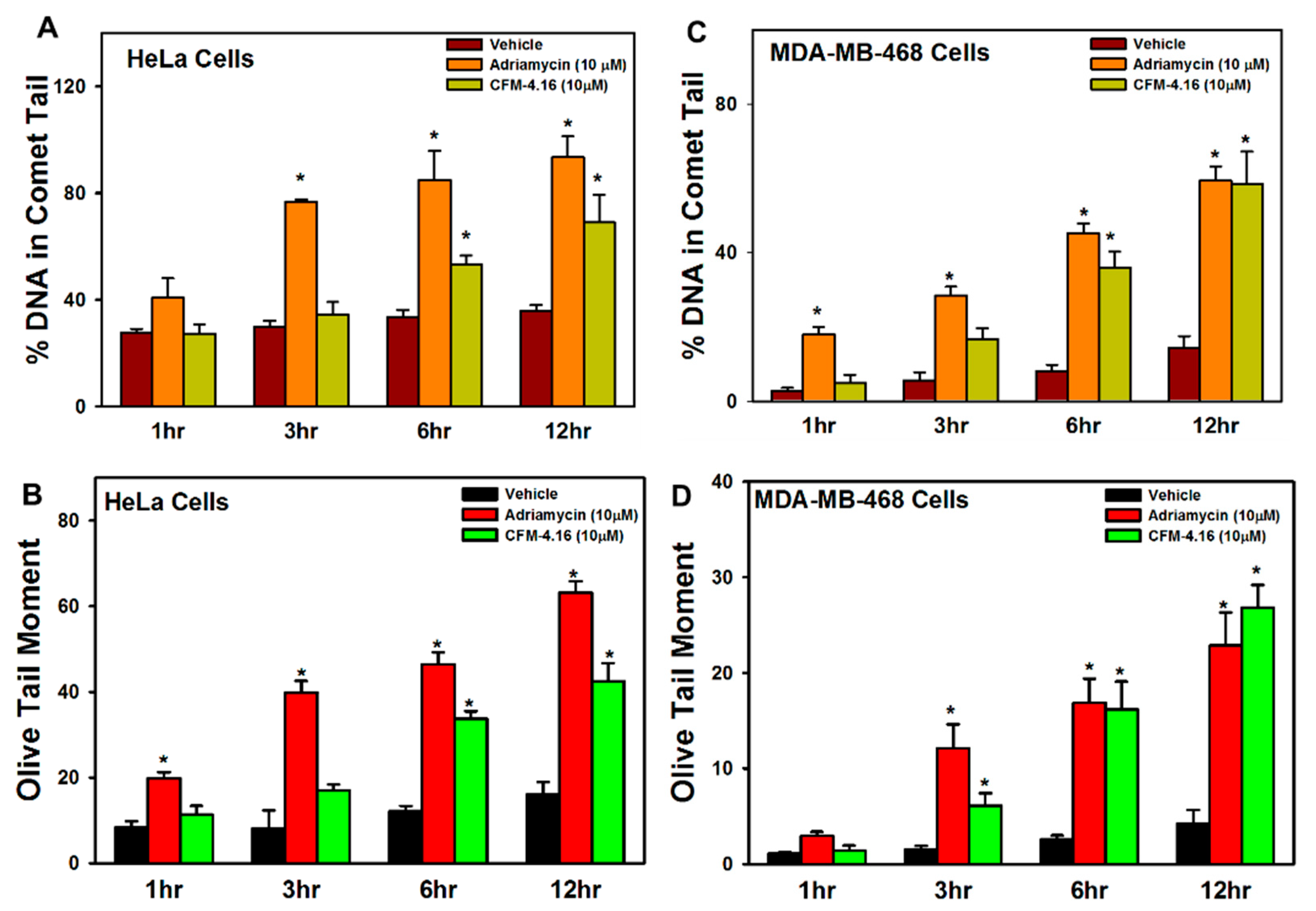
3.1. Adriamycin or CFMs Inhibit Cell Growth, in Part by Stimulating CARP-1 Expression, Phosphorylation of JNKs and H2AX, and DNA Damage
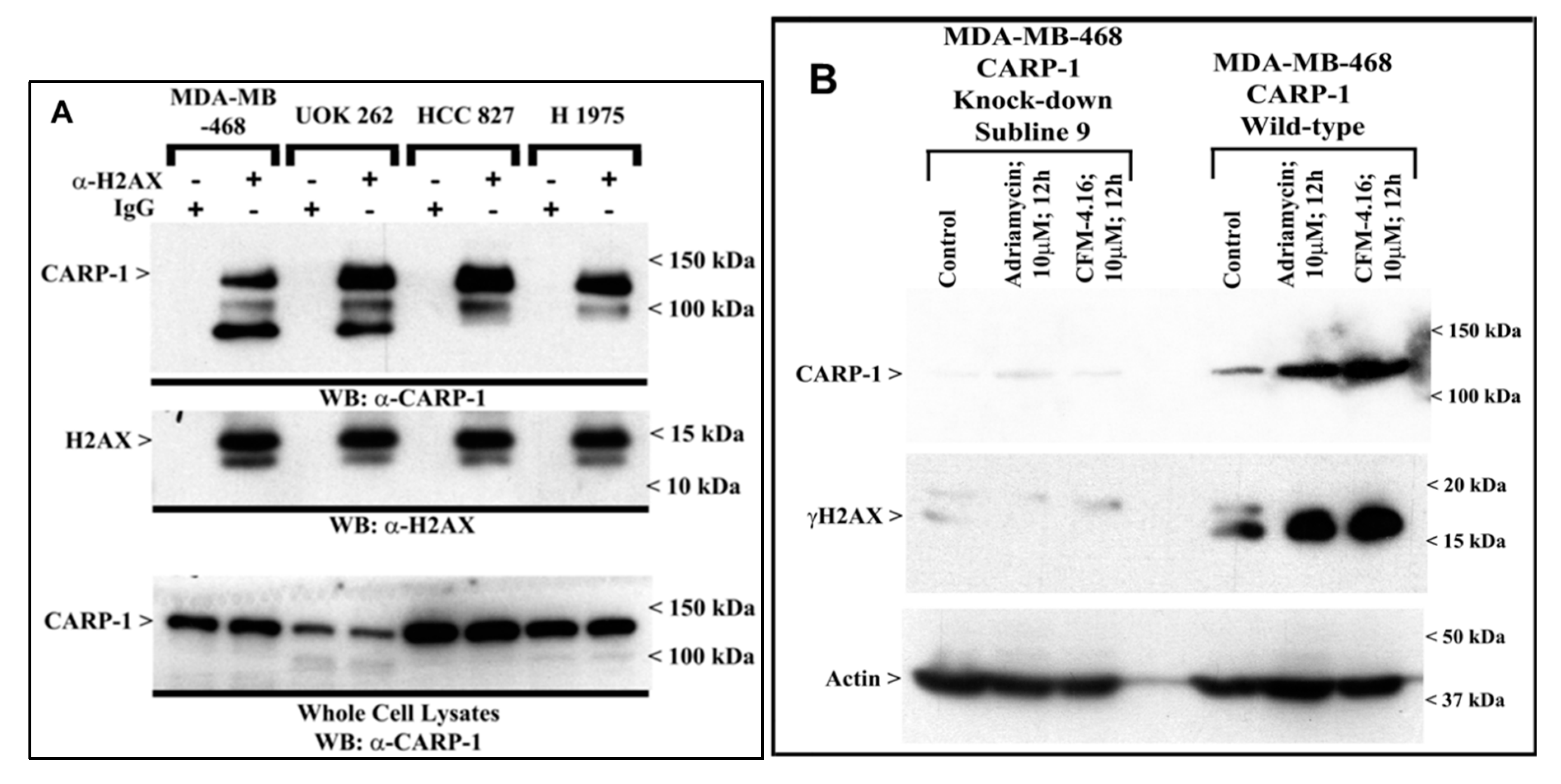
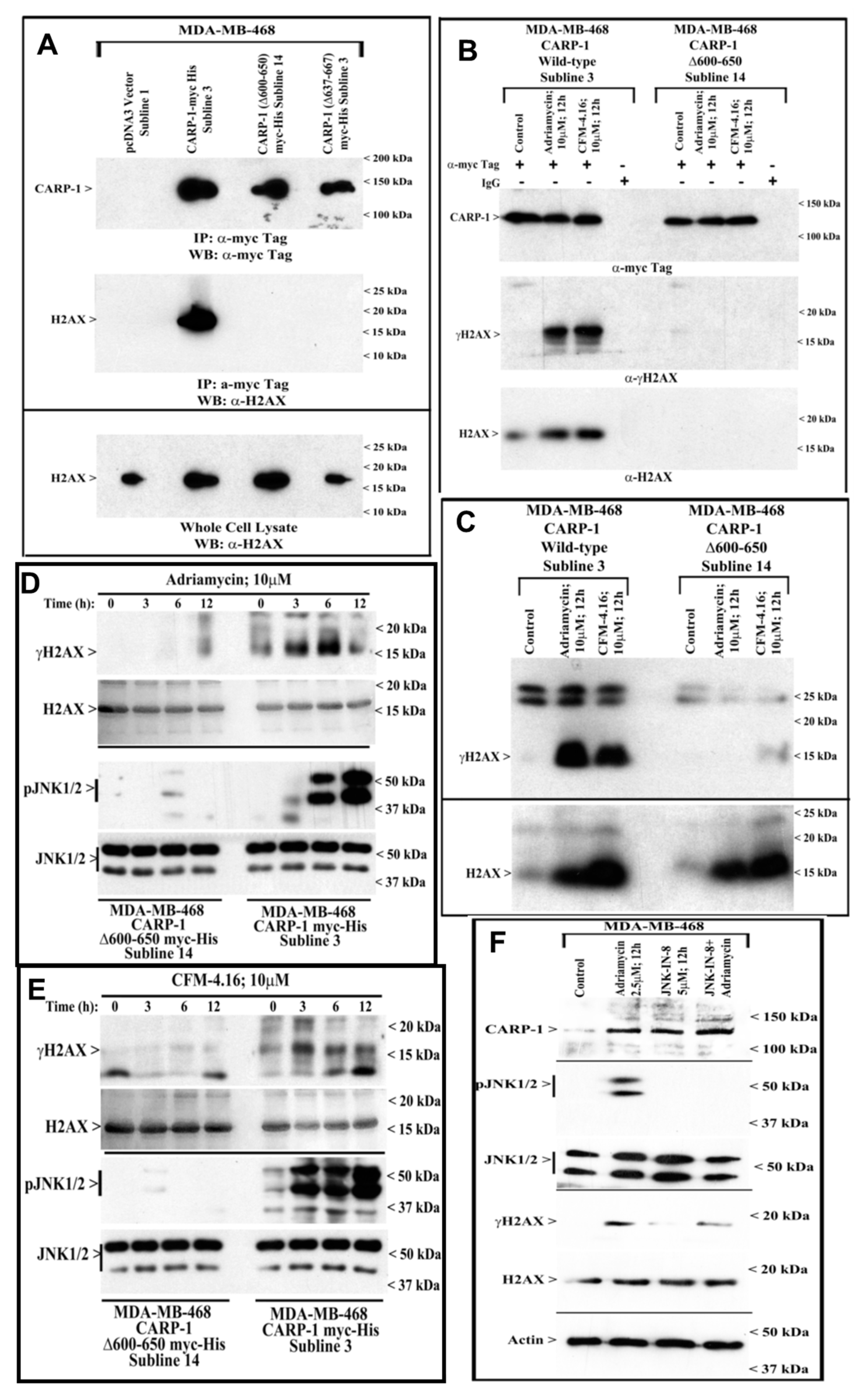
3.2. CARP-1 Interacts with H2AX, and Knock-Down of CARP-1 Leads to Reduced H2AX in Cells Treated with Adriamycin or CFM-4.16
3.3. CARP-1 (636–650) Binds with H2AX (1–35)
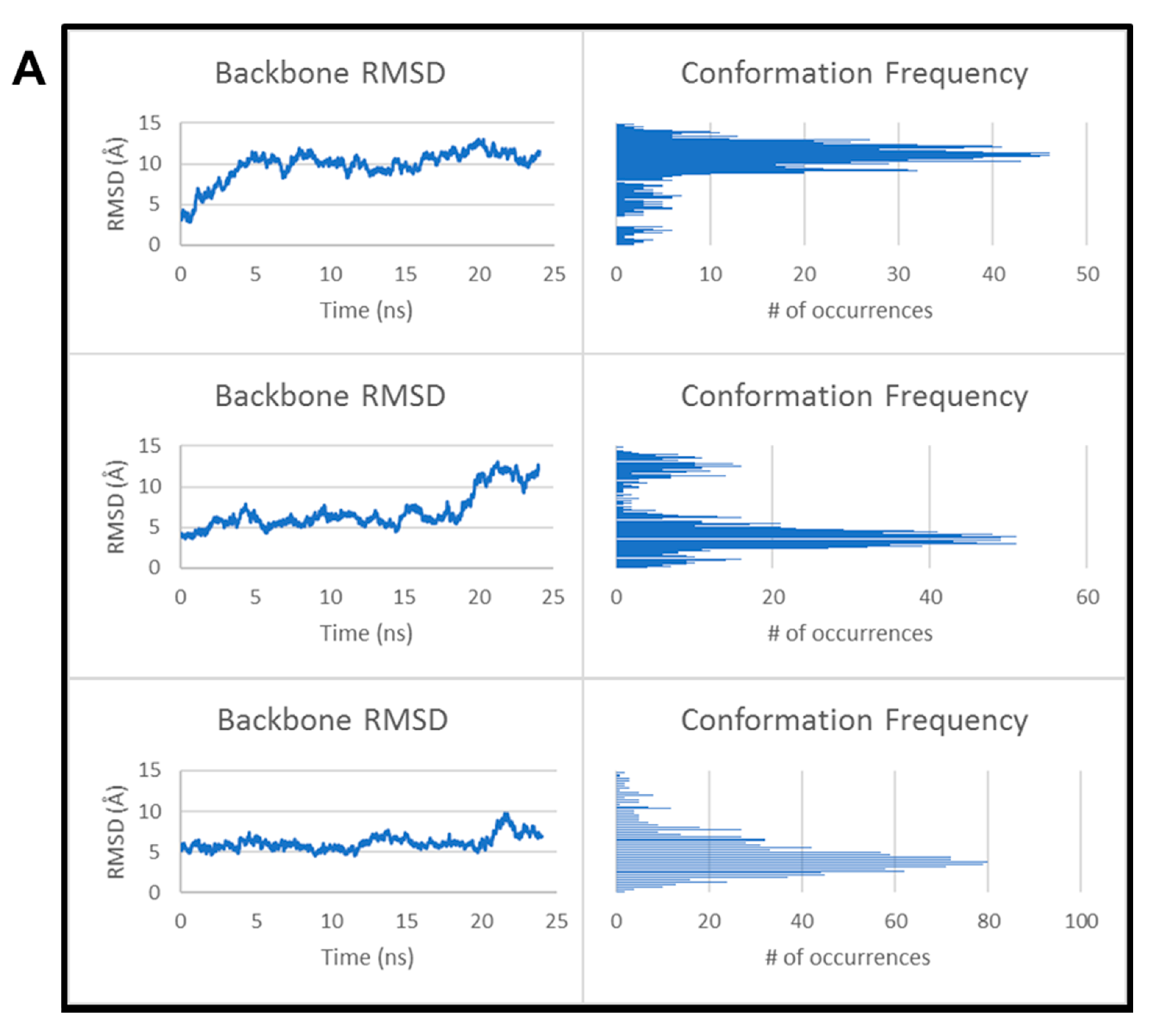
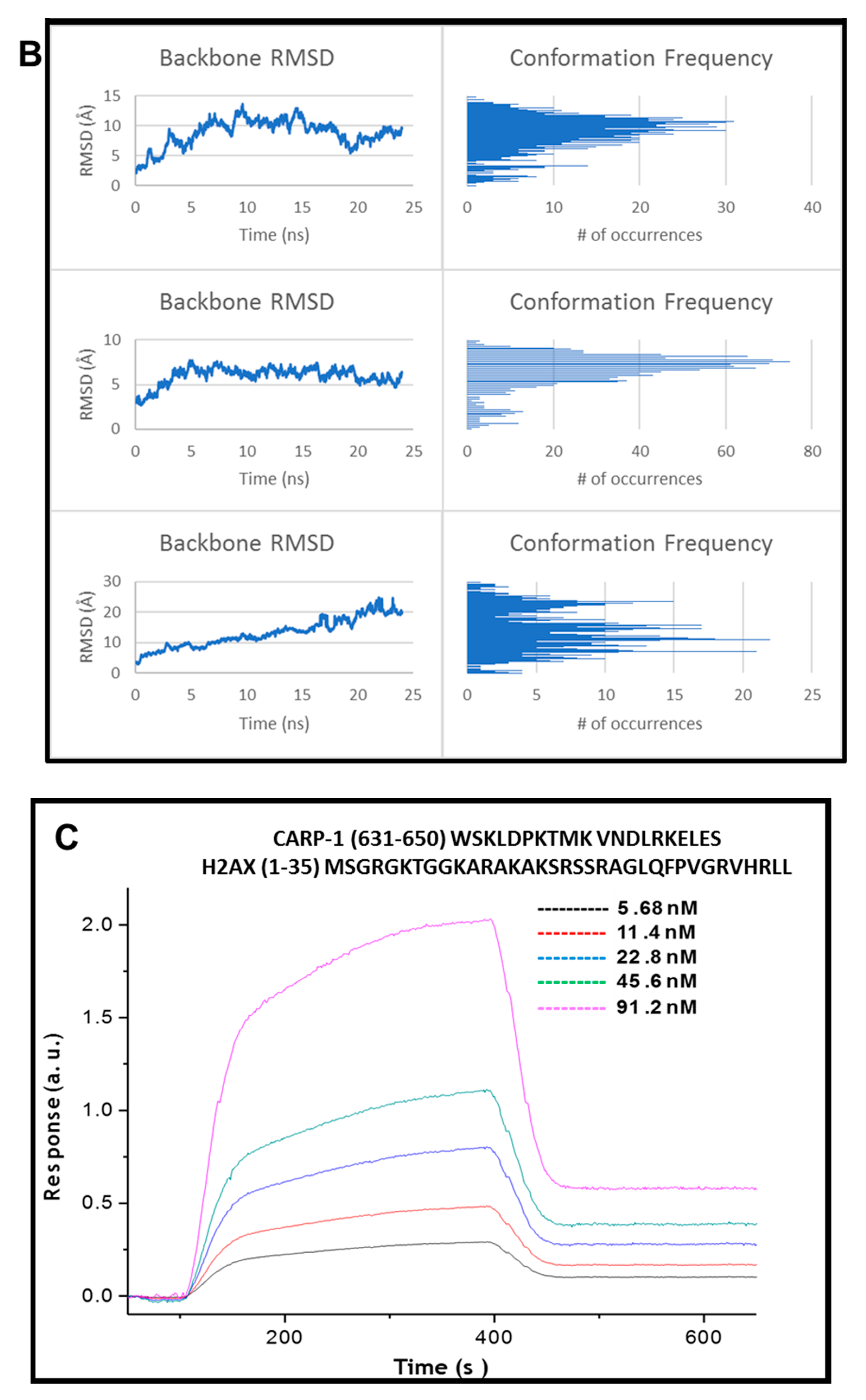
3.4. Kinetics of CARP-1 (636–650) Binding with H2AX (1–35)
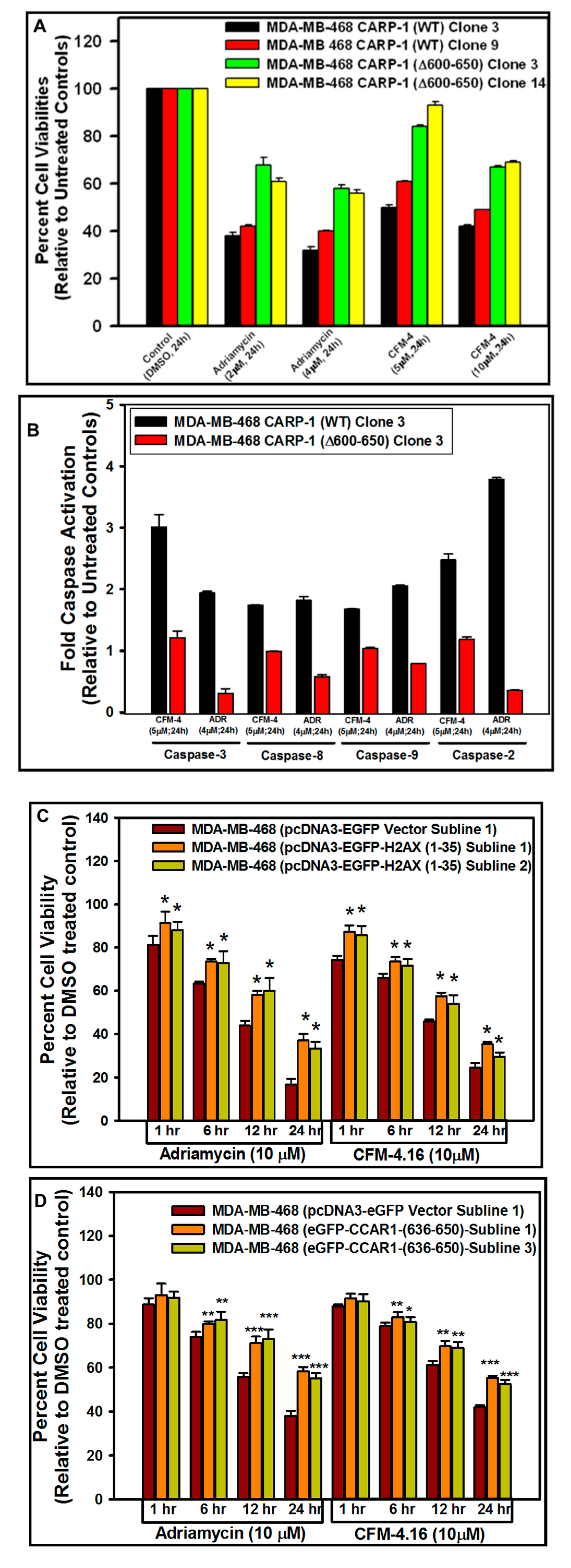
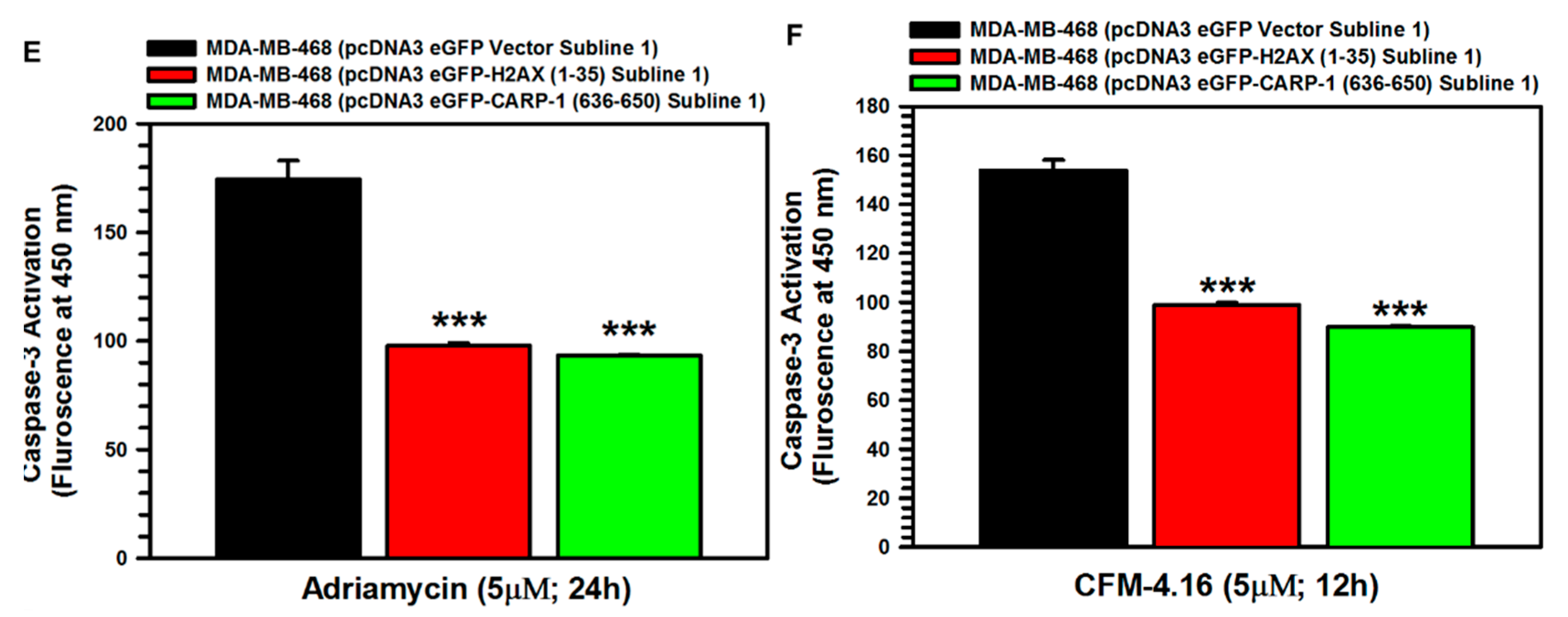
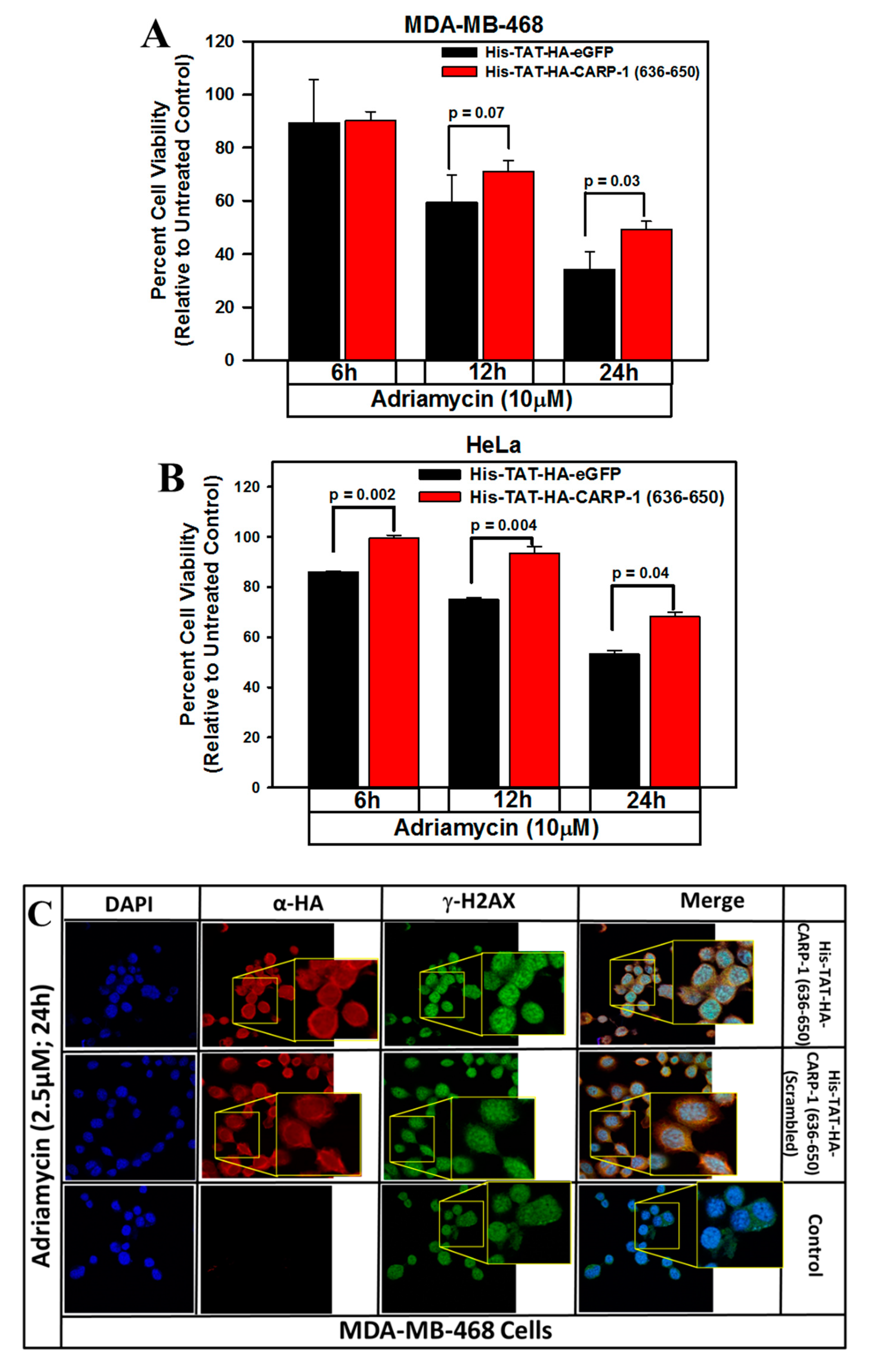
3.5. Interference with CARP-1–H2AX Binding Abrogates Cell-Growth Inhibition by Adriamycin or CFM Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartwell, L.H.; Kastan, M.B. Cell cycle control and cancer. Science 1994, 266, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Evan, G.I.; Vousden, K.I. Proliferation, cell-cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Rishi, A.K.; Zhang, L.; Boyanapalli, M.; Wali, A.; Mohammad, R.M.; Yu, Y.; Fontana, J.A.; Hatfield, J.S.; Dawson, M.I.; Majumdar, A.P.N.; et al. Identification and characterization of a Cell-Cycle and Apoptosis Regulatory Protein (CARP)-1 as a novel mediator of apoptosis signaling by Retinoid CD437. J. Biol. Chem. 2003, 278, 33422–33435. [Google Scholar] [CrossRef] [PubMed]
- Rishi, A.K.; Zhang, L.; Yu, Y.; Jiang, Y.; Nautiyal, J.; Wali, A.; Fontana, J.A.; Levi, E.; Majumdar, A.P.N. Cell cycle and apoptosis regulatory protein (CARP)-1 is involved in apoptosis signaling by epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 13188–13198. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yang, C.K.; Heo, K.; Roeder, R.G.; An, W.; Stallcup, M.R. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 2008, 31, 510–519. [Google Scholar] [CrossRef]
- Ou, C.Y.; Kim, J.H.; Yang, C.K.; Stallcup, M.R. Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and beta-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem. 2009, 284, 20629–20637. [Google Scholar] [CrossRef]
- Ou, C.Y.; Chen, T.C.; Lee, J.V.; Wang, J.C.; Stallcup, M.R. Coregulator cell cycle and apoptosis regulator 1 (CCAR1) positively regulates adipocyte differentiation through the glucocorticoid signaling pathway. J. Biol. Chem. 2014, 289, 17078–17086. [Google Scholar] [CrossRef]
- Puliyappadamba, V.T.; Wu, W.; Bevis, D.; Zhang, L.; Polin, L.; Kilkuskie, R.; Finley, R.L., Jr.; Larsen, S.D.; Levi, E.; Miller, F.R.; et al. Antagonists of anaphase promoting complex [APC]-2-cell cycle and apoptosis regulatory protein (CARP)-1 interaction are novel regulators of cell growth and apoptosis. J. Biol. Chem. 2011, 286, 38000–38017. [Google Scholar] [CrossRef]
- Muthu, M.; Cheriyan, V.T.; Rishi, A.K. CARP-1/CCAR1: A biphasic regulator of cancer cell growth and apoptosis. Oncotarget 2015, 6, 6499–6510. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.; Cheriyan, V.T.; Muthu, M.; Munie, S.; Levi, E.; Ashour, A.E.; Pass, H.I.; Wali, A.; Singh, M.; Rishi, A.K. CARP-1 functional mimetics are a novel class of small molecule inhibitors of malignant pleural mesothelioma cells. PLoS ONE 2014, 9, e89146. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.E.; Jamal, S.; Cheryan, V.T.; Muthu, M.; Zoheir, K.M.A.; Alafeefy, A.M.; Abd-allah, A.R.; Levi, E.; Tarca, A.L.; Polin, L.A.; et al. CARP-1 functional mimetics: A novel class of small molecule inhibitors of medulloblastoma cell growth. PLoS ONE 2013, 8, e66733. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.; Cheriyan, V.T.; Munie, M.; Levi, E.; Frank, J.; Ashour, A.E.; Singh, M.; Rishi, A.K. Mechanisms of neuroblastoma cell growth inhibition by CARP-1 functional mimetics. PLoS ONE 2014, 9, e102567. [Google Scholar] [CrossRef] [PubMed]
- Muthu, M.; Somagoni, J.M.; Cheriyan, V.T.; Munie, S.; Levi, E.; Ashour, A.E.; Alafeefy, A.M.; Sochacki, P.; Polin, L.A.; Reddy, K.B.; et al. Identification and testing of novel CARP-1 functional mimetic compounds as inhibitors of non-small cell lung and triple-negative breast cancers. J. Biomed. Nanotechnol. 2015, 11, 1–20. [Google Scholar] [CrossRef]
- Cheriyan, V.T.; Muthu, M.; Sekhar, S.; Patel, K.; Larsen, S.D.; Rajeswaran, W.; Polin, L.A.; Levi, E.; Singh, M.; Rishi, A.K. CARP-1 functional mimetics are novel inhibitors of drug-resistant triple-negative breast cancers. Oncotarget 2016, 7, 73370–73388. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, V.T.; Alsaab, H.; Sekhar, S.; Venkatesh, J.; Mondal, A.; Vohra, I.; Sau, S.; Muthu, M.; Polin, L.A.; Levi, E.; et al. A CARP-1 functional mimetic compound is synergistic with BRAF targeting in non-small cell lung cancers. Oncotarget 2018, 9, 29680–29697. [Google Scholar] [CrossRef] [PubMed]
- Cheriyan, V.T.; Alsaab, H.O.; Sekhar, S.; Steiber, C.; Keshwani, P.; Sau, S.; Muthu, M.; Polin, L.A.; Levi, E.; Iyer, A.K.; et al. A CARP-1 functional mimetic loaded vitamin E-TPGS micellar nano-formulation for inhibition of renal cell carcinoma. Oncotarget 2017, 8, 104928–104945. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA Damage response and cancer therapy. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Picco, V.; Pagès, G. Linking JNK Activity to the DNA Damage Response. Genes Cancer 2013, 4, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Zhu, F.; Cho, Y.-Y.; Tang, F.; Zykova, T.; Ma, W.-Y.; Bode, A.M.; Dong, Z. Cell Apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Levi, E.; Majumder, P.; Yu, Y.; Aboukameel, A.; Du, J.; Xu, H.; Mohammad, R.M.; Hatfield, J.S.; Wali, A.; et al. TAT-tagged Cell Cycle and Apoptosis Regulatory Protein (CARP)-1 peptides suppress growth of human breast cancer cells in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer Ta, P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Edayathumangalam, R.S.; Weyermann, P.; Gottesfeld, J.M.; Dervan, P.B.; Luger, K. Molecular recognition of the nucleosomal “supergroove”. Proc. Natl. Acad. Sci. USA 2004, 101, 6864–6869. [Google Scholar] [CrossRef] [PubMed]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Ceruti, D.S.; Cheatham Te, I.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular-Dynamics Simulations at Constant Pressure and-or Temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Ebeit, N.P.; Kaoud, T.S.; Edupuganti, R.; Ravenstein, S.V.; Dalby, K.N.; VanDenBerg, C.L. A c-Jun N-terminal kinase inhibitor, JNK-IN-8, sensitizes triple negative breast cancer cells to lapatinib. Oncotarget 2017, 8, 104894–104912. [Google Scholar]
- Gauld, J.W.; (University of Windsor, Windsor, ON N9B 3P4, Canada). Personal communication, 2019.
- Ion, B.F.; Aboelnga, M.M.; Gauld, J.W. Insights from molecular dynamics on substrate binding and effects of active site mutations in Delta(1)-pyrroline-5-carboxylate dehydrogenase. Can. J. Chem. 2016, 94, 1151–1162. [Google Scholar] [CrossRef]
- Wei, W.L.; Monard, G.; Gauld, J.W. Computational insights into substrate binding and catalytic mechanism of the glutaminase domain of glucosamine-6-phosphate synthase (GlmS). RSC Adv. 2017, 7, 29626–29638. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, M.K.; Kauffman, M.E.; Zhu, H.; Jia, Z.; Li, Y.R. Fluorescence-Based Assays for Measuring Doxorubicin in Biological Systems. React. Oxyg. Species 2016, 2, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H. Anthracyclines and anthracenediones. In Cancer and Chemotherapy and Biotherapy: Principles and Practice, 3rd ed.; Chabner, B.A., Longo, D.L., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 500–537. [Google Scholar]
- Garland, J.M.; Rudin, C. Cytochrome c induces caspase-dependent apoptosis in intact hematopoietic cells and overrides apoptosis suppression mediated by bcl-2, growth factor signaling, MAP-kinase-kinase, and malignant change. Blood 1998, 92, 1235–1246. [Google Scholar]
- Froesch, B.A.; Aime-Sempe, C.; Leber, B.; Andrews, D.; Reed, J.C. Inhibition of p53 transcriptional activity by Bcl-2 requires its membrane-anchoring domain. J. Biol. Chem. 1999, 274, 6469–6475. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Yu, J.Y.; Yin, D.; Patwardhan, G.A.; Gupta, V.; Hirabayashi, Y.; Holleran, W.M.; Giuliano, A.E.; Jazwinski, S.M.; Gouaze-Andersson, V.; et al. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008, 22, 2541–2551. [Google Scholar] [CrossRef]
- Brunquell, J.; Yuan, J.; Erwin, A.; Westerheide, S.D.; Xue, B. DBC1/CCAR2 and CCAR1 are largely disordered proteins that have evolved from one common ancestor. Biomed. Res. Int. 2014, 2014, 418458. [Google Scholar] [CrossRef]
- Zannini, L.; Buscemi, G.; Kim, J.-E.; Fontanella, E.; Delia, D. DBC1 phosphorylation by ATM/ATR inhibits SIRT1 deacetylase in response to DNA damage. J. Mol. Cell Biol. 2012, 4, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, B.; Minter-Dykhouse, K.; Yu, J.; Zhang, J.; Liu, T.; Zhang, H.; Lee, S.; Kim, J.; Wang, L.; Lou, Z. DBC1 functions as a tumor suppressor by regulating p53 stability. Cell Rep. 2015, 10, 1324–1334. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approaches | CCAR1/H2AX Model A | CCAR1/H2AX Model B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | ||||||
| B.E. | Std. Dev. | B.E. | Std. Dev. | B.E. | Std. Dev. | B.E. | Std. Dev. | B.E. | Std. Dev. | |
| MM-PBSA | −31.4 | 5.39 | −21.8 | 3.97 | −37.8 | 6.80 | −24.5 | 6.00 | −44.2 | 5.46 |
| MM-GBSA | −27.0 | 6.44 | −17.6 | 3.47 | −25.5 | 8.56 | −20.5 | 5.07 | −37.6 | 6.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekhar, S.C.; Venkatesh, J.; Cheriyan, V.T.; Muthu, M.; Levi, E.; Assad, H.; Meister, P.; Undyala, V.V.; Gauld, J.W.; Rishi, A.K. A H2AX–CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage. Cancers 2019, 11, 221. https://doi.org/10.3390/cancers11020221
Sekhar SC, Venkatesh J, Cheriyan VT, Muthu M, Levi E, Assad H, Meister P, Undyala VV, Gauld JW, Rishi AK. A H2AX–CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage. Cancers. 2019; 11(2):221. https://doi.org/10.3390/cancers11020221
Chicago/Turabian StyleSekhar, Sreeja C., Jaganathan Venkatesh, Vino T. Cheriyan, Magesh Muthu, Edi Levi, Hadeel Assad, Paul Meister, Vishnu V. Undyala, James W. Gauld, and Arun K. Rishi. 2019. "A H2AX–CARP-1 Interaction Regulates Apoptosis Signaling Following DNA Damage" Cancers 11, no. 2: 221. https://doi.org/10.3390/cancers11020221