Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
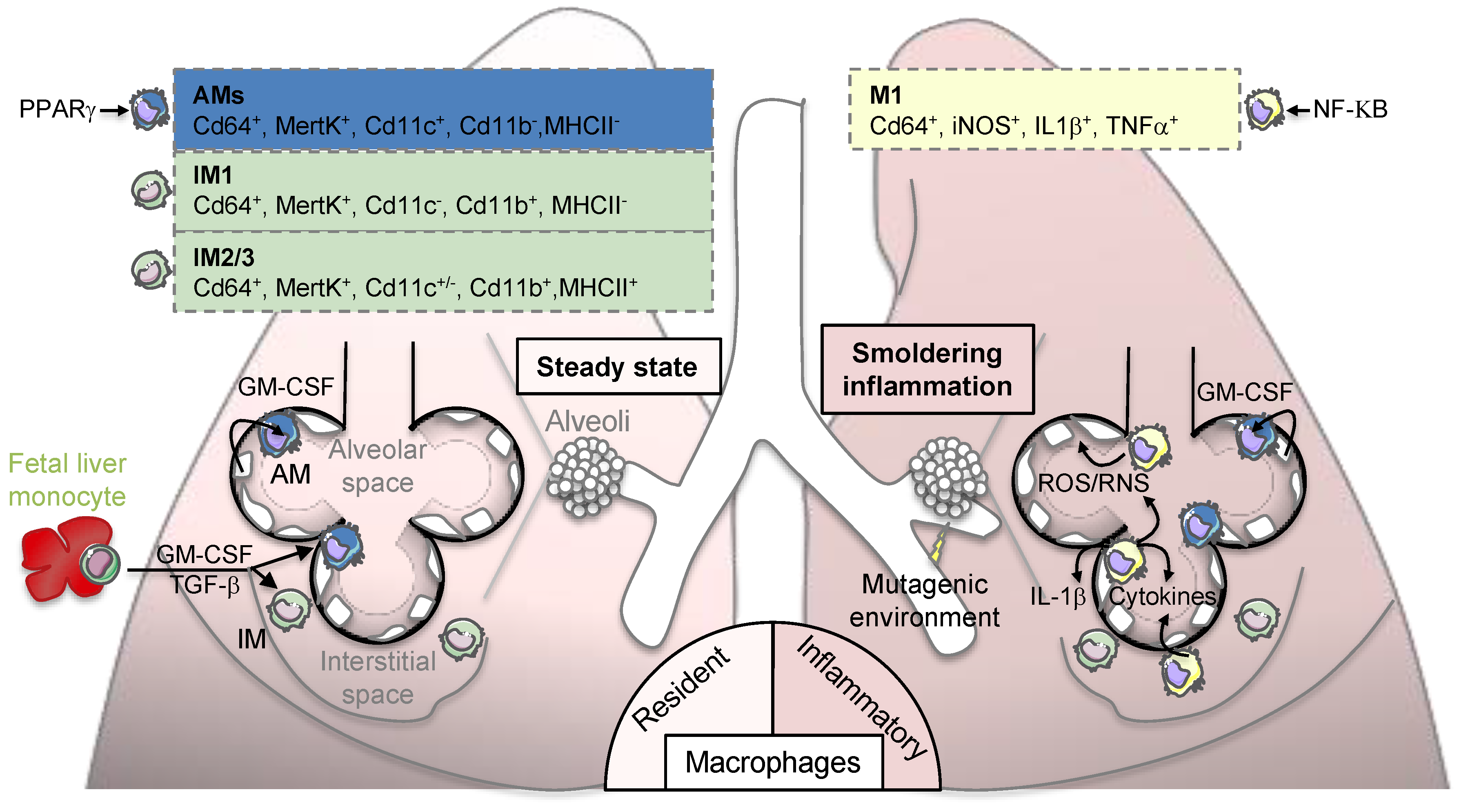
2. Environment-Dependent Maintenance of Lung Macrophages during Homeostasis
2.1. Lung Macrophage Ontogeny and Maintenance
2.2. Environment-Dependent Lung Macrophage Identity during Homeostasis
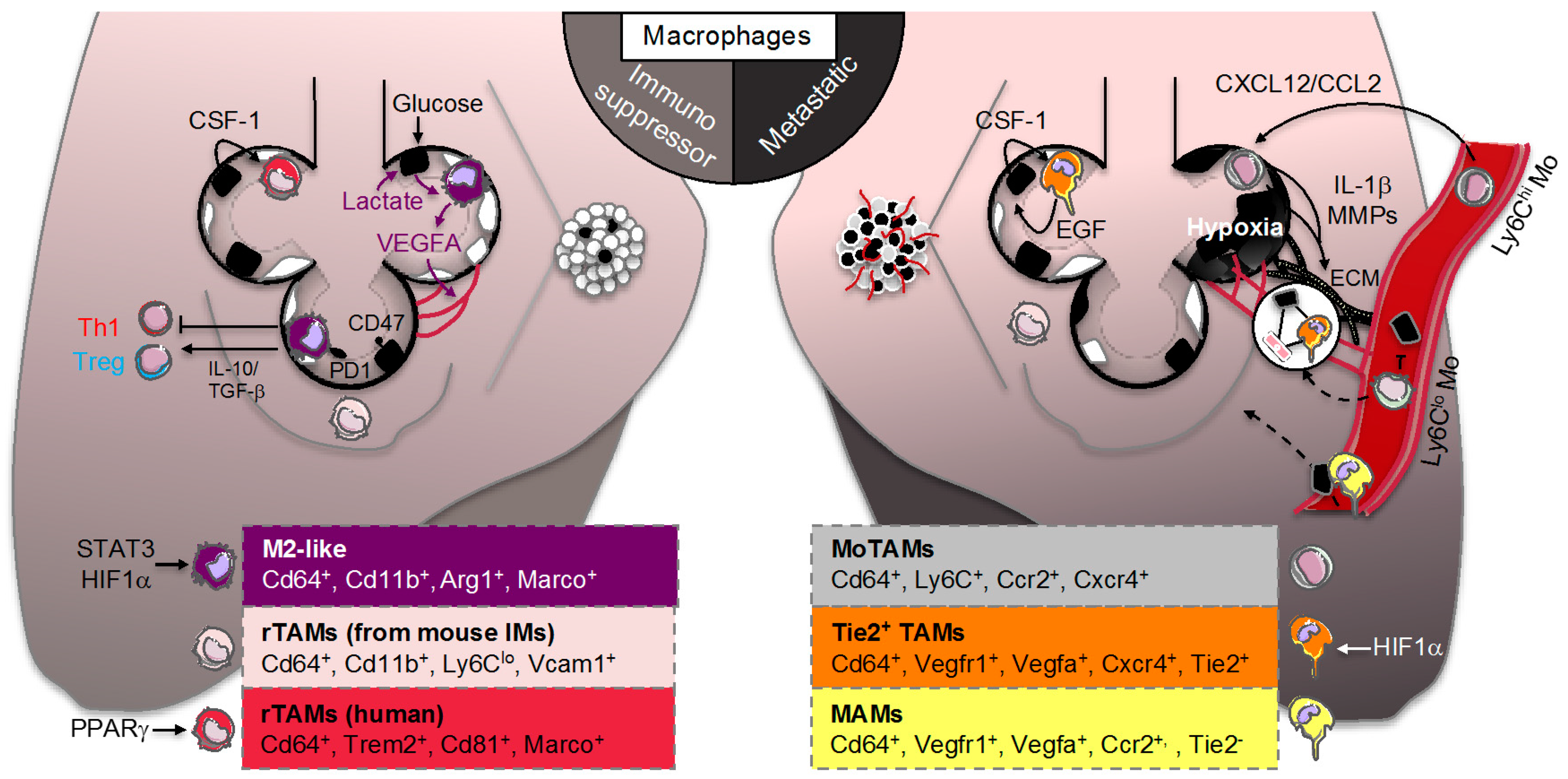
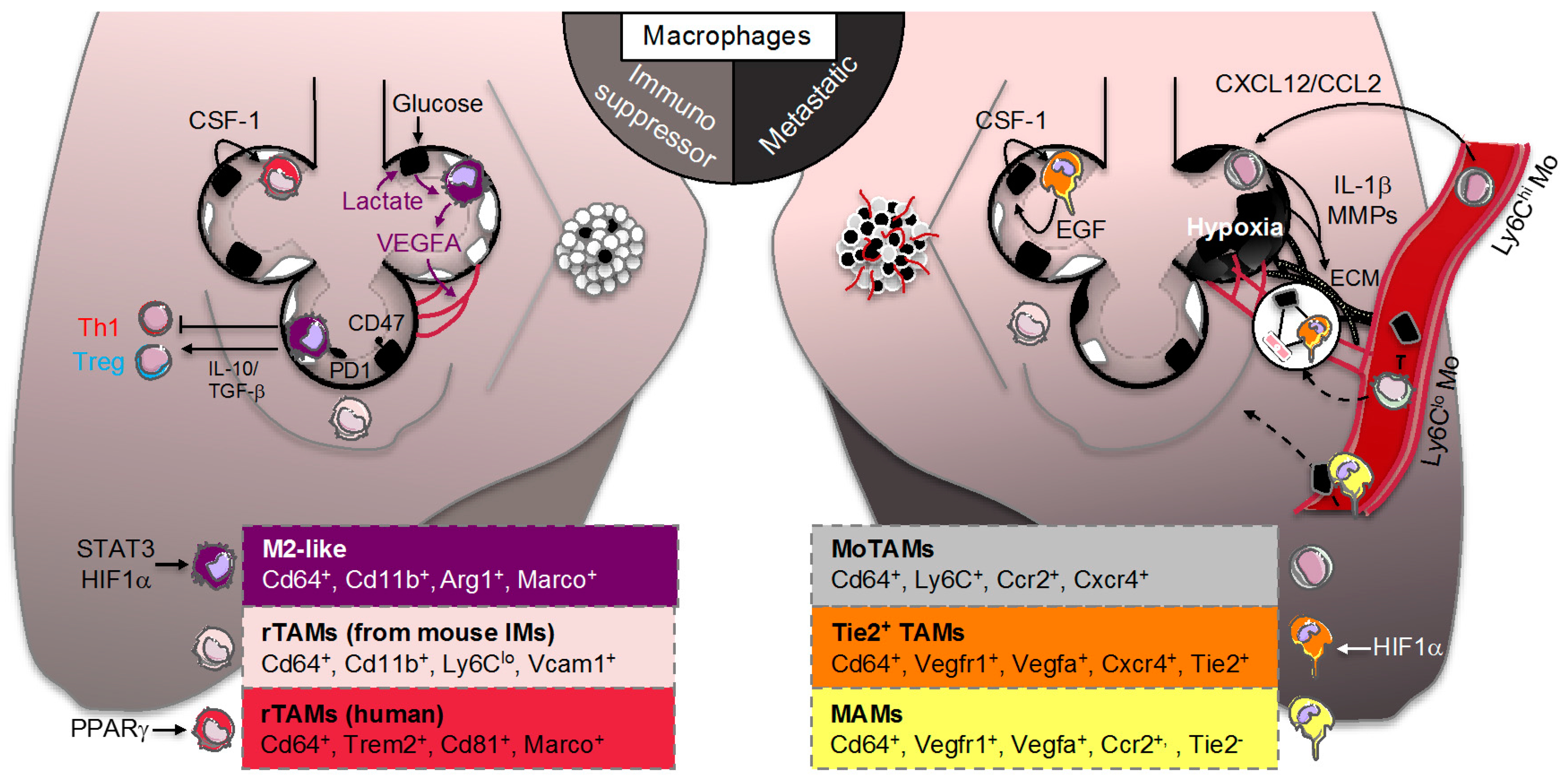
3. Lung Macrophage Origin and Diversity in Lung Adenocarcinoma
3.1. Smoldering Inflammation
3.2. Resident Tumor-Associated Macrophages (rTAMs)
3.3. Monocyte-Derived TAMs (MoTAMs)
4. Lung Macrophage Immunometabolism and Function in Lung Adenocarcinoma
4.1. TAM Immunomodulation
4.2. Adaptation of TAM to the “Warburg Effect”
4.3. TAM Immunometabolism beyond Glycolytic Activity
4.4. Tie2+ TAMs and Metastasis-Associated Macrophages (MAMs)
4.5. Bone Marrow Macrophages and Bone Metastasis
5. IL-1β Signaling and Immunometabolism: A New Role in Lung Adenocarcinoma?
5.1. CANTOS Trial and Lung Adenocarcinoma
5.2. IL-1β Signaling and Lung Adenocarcinoma
5.3. Immunometabolism: The Missing Link?
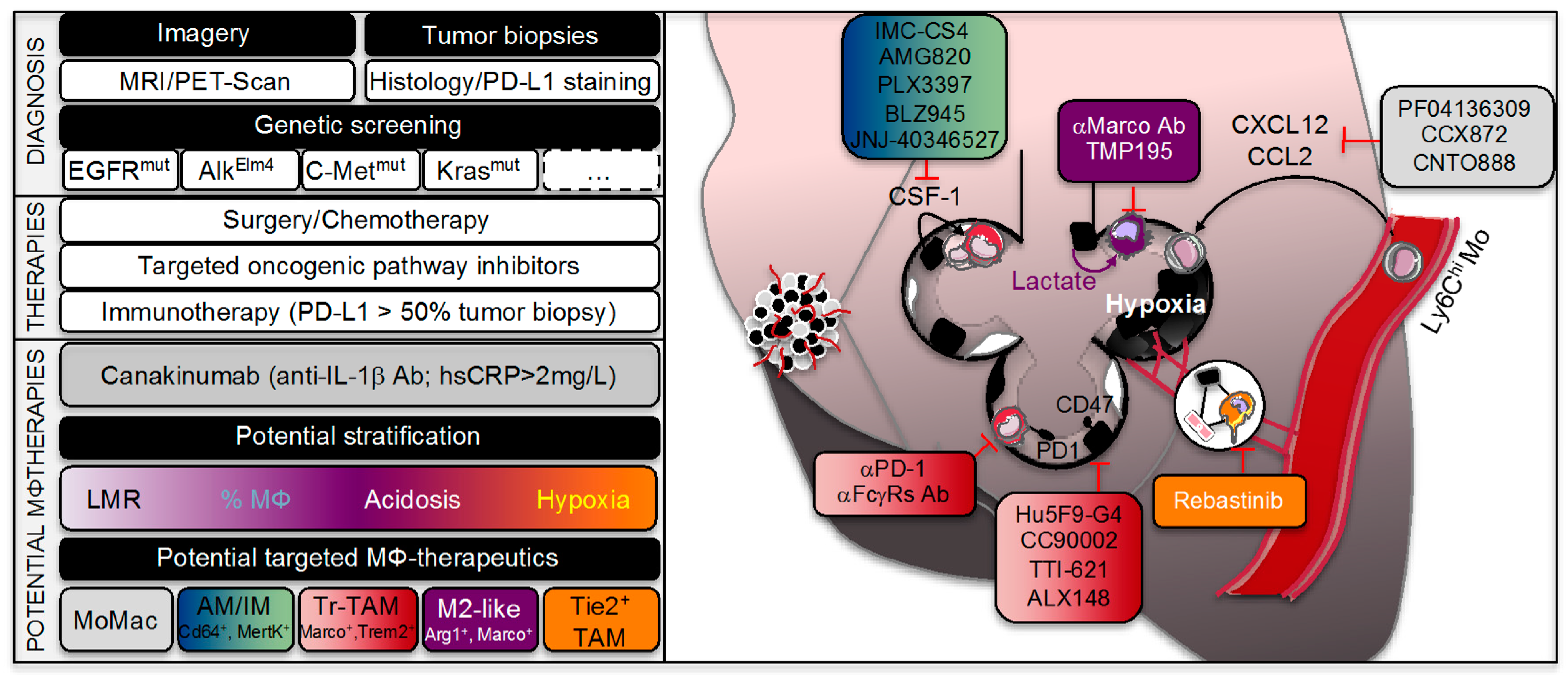
6. Lung Adenocarcinoma Treatment and Macrophage Interaction
6.1. Lung Cancer Therapy Options
6.2. Emerging Immunotherapy
6.3. TAMs and Lung Cancer Therapy Responses
7. Conclusions
Funding
Conflicts of Interest
References
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Harney, A.S.; Pollard, J.W. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell 2016, 30, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; McKay, D.; Pollard, J.W.; Lewis, C.E. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018, 78, 5492–5503. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Remark, R.; Becker, C.; Gomez, J.E.; Damotte, D.; Dieu-Nosjean, M.-C.; Sautès-Fridman, C.; Fridman, W.-H.; Powell, C.A.; Altorki, N.K.; Merad, M.; et al. The Non–Small Cell Lung Cancer Immune Contexture. A Major Determinant of Tumor Characteristics and Patient Outcome. Am. J. Respir. Crit. Care Med. 2015, 191, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Poh, A.R.; Ernst, M. Targeting Macrophages in Cancer: From Bench to Bedside. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Natoli, G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Z.; Ravichandran, K.S. Metabolic connections during apoptotic cell engulfment. Cell 2011, 147, 1442–1445. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Yvan-Charvet, L. Understanding macrophage diversity at the ontogenic and transcriptomic levels. Immunol. Rev. 2014, 262, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.Y.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2016, 193, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Ryan, R.; Pucci, F.; Sio, S.W.; Kuswanto, W.; Rauch, P.J.; Chudnovskiy, A.; Iwamoto, Y.; et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity 2013, 38, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R. The Origin and Kinetics of Mononuclear Phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Coggle, J.E.; Tarling, J.D. The Proliferation Kinetics of Pulmonary Alveolar Macrophages. J. Leukoc. Biol. 1984, 35, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, R.T. The Ontogeny of Pulmonary Alveolar Macrophages in Parabiotic Mice. J. Leukoc. Biol. 1986, 40, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and Tissue Injury: Agents of Defense or Destruction? Annu. Rev. Pharmacol. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.S.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Lambrecht, B.N.; Hammad, H. Division of labor between lung dendritic cells and macrophages in the defense against pulmonary infections. Mucosal Immunol. 2013, 6, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 2015, 16, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Buttgereit, A.; Lelios, I.; Utz, S.G.; Cansever, D.; Becher, B.; Greter, M. The Cytokine TGF-β Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity 2017, 47, 903–912.e4. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Y.S.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Dev. Camb. Engl. 2016, 143, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Gibbings, S.L.; Thomas, S.M.; Atif, S.M.; McCubbrey, A.L.; Desch, A.N.; Danhorn, T.; Leach, S.M.; Bratton, D.L.; Henson, P.M.; Janssen, W.J.; et al. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017, 57, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Liegeois, M.; Legrand, C.; Desmet, C.J.; Marichal, T.; Bureau, F. The interstitial macrophage: A long-neglected piece in the puzzle of lung immunity. Cell Immunol. 2018, 330, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Sabatel, C.; Radermecker, C.; Fievez, L.; Paulissen, G.; Chakarov, S.; Fernandes, C.; Olivier, S.; Toussaint, M.; Pirottin, D.; Xiao, X.; et al. Exposure to Bacterial CpG DNA Protects from Airway Allergic Inflammation by Expanding Regulatory Lung Interstitial Macrophages. Immunity 2017, 46, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Schyns, J.; Bureau, F.; Marichal, T. Lung Interstitial Macrophages: Past, Present, and Future. J. Immunol. Res. 2018, 2018, 5160794. [Google Scholar] [CrossRef] [PubMed]
- Rodero, M.P.; Poupel, L.; Loyher, P.-L.; Hamon, P.; Licata, F.; Pessel, C.; Hume, D.A.; Combadière, C.; Boissonnas, A. Immune surveillance of the lung by migrating tissue monocytes. eLife 2015. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; Allison, K.C.; Lee-Sherick, A.B.; Jakubzick, C.V.; Janssen, W.J. Promoter Specificity and Efficacy in Conditional and Inducible Transgenic Targeting of Lung Macrophages. Front. Immunol. 2017, 8, 1618. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Washko, G.R.; Dransfield, M.T.; Spira, A.; Han, M.K.; Kalhan, R. Defining Impaired Respiratory Health. A Paradigm Shift for Pulmonary Medicine. Am. J. Respir. Crit. Care Med. 2018, 198, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immunol. 2016, 28, 417–424. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Andrés, J.; Joosten, L.A.; Netea, M.G. Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 2018, 56, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, B.C.; Whitsett, J.A. Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 2002, 64, 775–802. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.D.; Malur, A.; Barna, B.P.; Ghosh, S.; Kavuru, M.S.; Malur, A.G.; Thomassen, M.J. Targeted PPAR{gamma} deficiency in alveolar macrophages disrupts surfactant catabolism. J. Lipid Res. 2010, 51, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Nobs, S.P.; Kurrer, M.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 2014, 15, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Chow, A.; Spanbroek, R.; Marcelin, G.; Greter, M.; Jakubzick, C.; Bogunovic, M.; Leboeuf, M.; van Rooijen, N.; Habenicht, A.J.; et al. Systemic analysis of PPARγ in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J. Immunol. 2012, 189, 2614–2624. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jeyanathan, M.; Haddadi, S.; Barra, N.G.; Vaseghi-Shanjani, M.; Damjanovic, D.; Lai, R.; Afkhami, S.; Chen, Y.; Dvorkin-Gheva, A.; et al. Induction of Autonomous Memory Alveolar Macrophages Requires T Cell Help and Is Critical to Trained Immunity. Cell 2018, 175, 1634–1650.e17. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e9. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, M.; Fievez, L.; Drion, P.-V.; Cataldo, D.; Bureau, F.; Lekeux, P.; Desmet, C.J. Myeloid hypoxia-inducible factor 1α prevents airway allergy in mice through macrophage-mediated immunoregulation. Mucosal Immunol. 2013, 6, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.C.; Kumar, S.; Janssen, W.J.; Graham, B.B.; Frid, M.G.; Riddle, S.R.; Kasmi, K.C.E.; Stenmark, K.R. A Time- and Compartment-Specific Activation of Lung Macrophages in Hypoxic Pulmonary Hypertension. J. Immunol. 2017, 198, 4802–4812. [Google Scholar] [CrossRef] [PubMed]
- Maci, E.; Comito, F.; Frezza, A.M.; Tonini, G.; Pezzuto, A. Lung nodule and functional changes in smokers after smoking cessation short-term treatment. Cancer Investig. 2014, 32, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Demirjian, L.; Abboud, R.T.; Li, H.; Duronio, V. Acute effect of cigarette smoke on TNF-alpha release by macrophages mediated through the erk1/2 pathway. Biochim. Biophys. Acta 2006, 1762, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Tamaoki, J.; Nagai, A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L1392–L1401. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.; D’Onofrio, L.; Dell’Aquila, E.; Pezzuto, A. New molecular insights in tobacco-induced lung cancer. Future Oncol. 2013, 9, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226. [Google Scholar] [PubMed]
- Moghaddam, S.J.; Li, H.; Cho, S.-N.; Dishop, M.K.; Wistuba, I.I.; Ji, L.; Kurie, J.M.; Dickey, B.F.; DeMayo, F.J. Promotion of Lung Carcinogenesis by Chronic Obstructive Pulmonary Disease–Like Airway Inflammation in a K-ras–Induced Mouse Model. Am. J. Respir. Cell Mol. Biol. 2009, 40, 443–453. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 SDG Collaborators. Measuring progress from 1990 to 2017 and projecting attainment to 2030 of the health-related Sustainable Development Goals for 195 countries and territories: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 2091–2138. [Google Scholar] [CrossRef]
- Barnes, P.J.; Burney, P.G.J.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F.M. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- Wouters, E.F.M. Obesity and Metabolic Abnormalities in Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2017, 14, S389–S394. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Loyher, P.-L.; Hamon, P.; Laviron, M.; Meghraoui-Kheddar, A.; Goncalves, E.; Deng, Z.; Torstensson, S.; Bercovici, N.; Baudesson de Chanville, C.; Combadière, B.; et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med. 2018, 215, 2536–2553. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [PubMed]
- Merino Salvador, M.; Gómez de Cedrón, M.; Moreno Rubio, J.; Falagán Martínez, S.; Sánchez Martínez, R.; Casado, E.; Ramírez de Molina, A.; Sereno, M. Lipid metabolism and lung cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Politi, K.; Pitteri, S.J.; Lockwood, W.W.; Faça, V.M.; Kelly-Spratt, K.; Wong, C.-H.; Zhang, Q.; Chin, A.; Park, K.-S.; et al. Lung Cancer Signatures in Plasma Based on Proteome Profiling of Mouse Tumor Models. Cancer Cell 2011, 20, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Tammemagi, C.M.; Lam, S.; Barnett, M.J.; Duan, X.; Tam, A.; Auman, H.; Feng, Z.; Goodman, G.E.; Hanash, S.; et al. Pro-surfactant protein B as a biomarker for lung cancer prediction. J. Clin. Oncol. 2013, 31, 4536–4543. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Hanash, S.; DeFelice, B.; Miyamoto, S.; Barnett, M.; Zhao, Y.; Goodman, G.; Feng, Z.; Gandara, D.; Fiehn, O.; et al. Diacetylspermine is a Novel Prediagnostic Serum Biomarker for Non-Small-Cell Lung Cancer has Additive Performance with Pro-Surfactant Protein, B. J. Clin. Oncol. 2015, 33, 3880–3886. [Google Scholar] [CrossRef] [PubMed]
- Carus, A.; Ladekarl, M.; Hager, H.; Pilegaard, H.; Nielsen, P.S.; Donskov, F. Tumor-associated neutrophils and macrophages in non-small cell lung cancer: No immediate impact on patient outcome. Lung Cancer 2013, 81, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, D.; Zhao, L.; Huang, L.; Chen, G.; Shen, G.; Huang, J.; Chai, Y. Inverse role of distinct subsets and distribution of macrophage in lung cancer prognosis: A meta-analysis. Oncotarget 2016, 7, 40451–40460. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.J.H.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Cecchinato, V.; Uguccioni, M. Chemokine Heterocomplexes and Cancer: A Novel Chapter to Be Written in Tumor Immunity. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The Role of CXC Chemokine Receptors 1-4 on Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Ying, C.; Craig, M.; Day, L.L.; Sargent, E.; Neeley, C.; Wojno, K.; Snyder, L.A.; Yan, L.; Pienta, K.J. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007, 67, 9417–9424. [Google Scholar] [CrossRef] [PubMed]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, R.; Yamasaki, T.; Kanno, T.; Shibasaki, N.; Sakamoto, H.; Utsunomiya, N.; Sumiyoshi, T.; Shibuya, S.; Tsuruyama, T.; Nakamura, E.; et al. CCL2 as a potential therapeutic target for clear cell renal cell carcinoma. Cancer Med. 2016, 5, 2920–2933. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef]
- Linehan, D.; Noel, M.S.; Hezel, A.F.; Wang-Gillam, A.; Eskens, F.; Sleijfer, S.; Desar, I.M.E.; Erdkamp, F.; Wilmink, J.; Diehl, J.; et al. Overall survival in a trial of orally administered CCR2 inhibitor CCX872 in locally advanced/metastatic pancreatic cancer: Correlation with blood monocyte counts. J. Clin. Oncol. 2018, 36, 92. [Google Scholar] [CrossRef]
- Phillips, R.J.; Burdick, M.D.; Lutz, M.; Belperio, J.A.; Keane, M.P.; Strieter, R.M. The Stromal Derived Factor–1/CXCL12–CXC Chemokine Receptor 4 Biological Axis in Non–Small Cell Lung Cancer Metastases. Am. J. Respir. Crit. Care Med. 2003, 167, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Wald, O. CXCR4 Based Therapeutics for Non-Small Cell Lung Cancer (NSCLC). J. Clin. Med. 2018, 7, 303. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Cekic, C.; Sag, D.; Tacke, R.; Thomas, G.D.; Nowyhed, H.; Herrley, E.; Rasquinha, N.; McArdle, S.; Wu, R.; et al. Patrolling monocytes control tumor metastasis to the lung. Science 2015, 350, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shen, H.; Wang, G.; Zhang, P.; Liu, Q.; Du, J. Prognostic significance of systemic inflammation-based lymphocyte- monocyte ratio in patients with lung cancer: Based on a large cohort study. PLoS ONE 2014, 9, e108062. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, K.M.; Belcher, E.; Raevsky, E.; Nicholson, A.G.; Goldstraw, P.; Lim, E. Neutrophil/lymphocyte ratio and its association with survival after complete resection in non-small cell lung cancer. J. Thorac. Cardiovasc. Surg. 2009, 137, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, S.-H.; Li, H.; Li, Y.; Chen, X.-L.; Zhang, W.-Q.; Chen, H.-G.; Gu, L.-J. Preoperative lymphocyte count is a favorable prognostic factor of disease-free survival in non-small-cell lung cancer. Med. Oncol. 2013, 30, 352. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Usui, S.; Kikuchi, S.; Goto, Y.; Sakai, M.; Onizuka, M.; Sato, Y. Preoperative lymphocyte count is an independent prognostic factor in node-negative non-small cell lung cancer. Lung Cancer 2012, 75, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Barbieri, V.; Ciliberto, D.; Rossi, A.; Rocco, D.; Addeo, R.; Staropoli, N.; Pastina, P.; Marvaso, G.; Martellucci, I.; et al. Systemic inflammatory status at baseline predicts bevacizumab benefit in advanced non-small cell lung cancer patients. Cancer Biol. Ther. 2013, 14, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yao, H.; Song, G.; Liao, X.; Xian, Y.; Li, W. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am. J. Transl. Res. 2015, 7, 1699–1711. [Google Scholar] [PubMed]
- Song, W.; Mazzieri, R.; Yang, T.; Gobe, G.C. Translational Significance for Tumor Metastasis of Tumor-Associated Macrophages and Epithelial-Mesenchymal Transition. Front. Immunol. 2017, 8, 1106. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Chen, B.; Jiang, R.; Li, J.; Wang, B. Resveratrol inhibits lung cancer growth by suppressing M2-like polarization of tumor associated macrophages. Cell Immunol. 2017, 311, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.-W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Pello, O.M.; Andrés, V. Role of c-MYC in tumor-associated macrophages and cancer progression. Oncoimmunology 2013, 2, e22984. [Google Scholar] [CrossRef] [PubMed]
- Georgoudaki, A.-M.; Prokopec, K.E.; Boura, V.F.; Hellqvist, E.; Sohn, S.; Östling, J.; Dahan, R.; Harris, R.A.; Rantalainen, M.; Klevebring, D.; et al. Reprogramming Tumor-Associated Macrophages by Antibody Targeting Inhibits Cancer Progression and Metastasis. Cell Rep. 2016, 15, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Weissman, I. How One Thing Led to Another. Annu. Rev. Immunol. 2016, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; McCarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Kong, X.; Li, E.; Liu, Y.; Du, X.; Kang, Z.; Tang, Y.; Kuang, Y.; Yang, Z.; et al. CD47 Promotes Tumor Invasion and Metastasis in Non-small Cell Lung Cancer. Sci. Rep. 2016, 6, 29719. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.M.; Li, S.; Petti, A.A.; Soysal, S.D.; Li, L.; McLellan, M.D.; Hoog, J.; et al. Breast Cancer Neoantigens Can Induce CD8+ T-Cell Responses and Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody as a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Kim, J.-W. Convergence of Cancer Metabolism and Immunity: An Overview. Biomol. Ther. 2018, 26, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Smit, J.W.A.; Netea, M.G. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018, 413, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Maki, Y.; Soh, J.; Ichimura, K.; Shien, K.; Furukawa, M.; Muraoka, T.; Tanaka, N.; Ueno, T.; Yamamoto, H.; Asano, H.; et al. Impact of GLUT1 and Ki-67 expression on early-stage lung adenocarcinoma diagnosed according to a new international multidisciplinary classification. Oncol. Rep. 2013, 29, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Osugi, J.; Yamaura, T.; Muto, S.; Okabe, N.; Matsumura, Y.; Hoshino, M.; Higuchi, M.; Suzuki, H.; Gotoh, M. Prognostic impact of the combination of glucose transporter 1 and ATP citrate lyase in node-negative patients with non-small lung cancer. Lung Cancer 2015, 88, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase A in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Okada, M.; Takeda, H.; Kuramoto, K.; Sanomachi, T.; Togashi, K.; Seino, S.; Yamamoto, M.; Yoshioka, T.; Kitanaka, C. Involvement of GLUT1-mediated glucose transport and metabolism in gefitinib resistance of non-small-cell lung cancer cells. Oncotarget 2018, 9, 32667–32679. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Hanai, J.-I.; Ren, J.-G.; Kats, L.; Burgess, K.; Bhargava, P.; Signoretti, S.; Billiard, J.; Duffy, K.J.; Grant, A.; et al. Targeting lactate dehydrogenase—A inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 2014, 19, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. ChemMedChem 2016, 11, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Shriwas, P.; Qian, Y.; Wang, X.; Roberts, D.; Bergmeier, S.; Chen, X. Abstract 2799: New-generation glucose transporter inhibitors targeting non-small cell lung cancer and triple-negative breast cancer. Cancer Res. 2018, 78, 2799. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. When will small molecule lactate dehydrogenase inhibitors realize their potential in the cancer clinic? Future Med. Chem. 2017, 9, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Obre, E.; Rossignol, R. Emerging concepts in bioenergetics and cancer research: Metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 2015, 59, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Bucci, V.; Akkari, L.; Deforet, M.; Joyce, J.A.; Xavier, J.B. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Xie, H.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Papagiannakopoulos, T.; Olenchock, B.A.; Heyman, J.E.; Keibler, M.A.; Luengo, A.; Bauer, M.R.; Jha, A.K.; O’Brien, J.P.; Pierce, K.A.; et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016, 23, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Patiño, C.; Bossowski, J.P.; Donatis, G.M.D.; Mondragón, L.; Villa, E.; Aira, L.E.; Chiche, J.; Mhaidly, R.; Lebeaupin, C.; Marchetti, S.; et al. Low-Protein Diet Induces IRE1α-Dependent Anticancer Immunosurveillance. Cell Metab. 2018, 27, 828–842.e7. [Google Scholar] [CrossRef] [PubMed]
- Orillion, A.; Damayanti, N.P.; Shen, L.; Adelaiye-Ogala, R.; Affronti, H.; Elbanna, M.; Chintala, S.; Ciesielski, M.; Fontana, L.; Kao, C.; et al. Dietary Protein Restriction Reprograms Tumor-Associated Macrophages and Enhances Immunotherapy. Clin. Cancer Res. 2018, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Poczobutt, J.M.; Gijon, M.; Amin, J.; Hanson, D.; Li, H.; Walker, D.; Weiser-Evans, M.; Lu, X.; Murphy, R.C.; Nemenoff, R.A. Eicosanoid Profiling in an Orthotopic Model of Lung Cancer Progression by Mass Spectrometry Demonstrates Selective Production of Leukotrienes by Inflammatory Cells of the Microenvironment. PLoS ONE 2013, 8, e79633. [Google Scholar] [CrossRef] [PubMed]
- Poczobutt, J.M.; De, S.; Yadav, V.K.; Nguyen, T.T.; Li, H.; Sippel, T.R.; Weiser-Evans, M.C.M.; Nemenoff, R.A. Expression Profiling of Macrophages Reveals Multiple Populations with Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J. Immunol. 2016, 196, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-F.; Chao, T.-T.; Su, Y.-F.; Hsu, C.-C.; Chien, C.-Y.; Chiu, K.-C.; Shiah, S.-G.; Lee, C.-H.; Liu, S.-Y.; Shieh, Y.-S. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-κB signaling. Oncotarget 2017, 8, 20706–20718. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Murdoch, C.; Venneri, M.A.; Naldini, L.; Lewis, C.E. Tie2-expressing monocytes: Regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007, 28, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Venneri, M.A.; Biziato, D.; Nonis, A.; Moi, D.; Sica, A.; Di Serio, C.; Naldini, L.; De Palma, M. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 2009, 114, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Franklin, R.A.; Adler, M.; Jacox, J.B.; Bailis, W.; Shyer, J.A.; Flavell, R.A.; Mayo, A.; Alon, U.; Medzhitov, R. Circuit Design Features of a Stable Two-Cell System. Cell 2018, 172, 744–757.e17. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, M.; Schröder, C.P.; Oosting, S.F.; Dafni, U.; Kleibeuker, J.E.; de Vries, E.G.E. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: From bench to bedside. Oncotarget 2016, 7, 21247–21258. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.; Soong, D.Y.H.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, X.H.-F.; Massagué, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.J.; Wedin, R. Surgery for skeletal metastases in lung cancer. Acta Orthop. 2011, 82, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Rove, K.O.; Crawford, E.D. Metastatic cancer in solid tumors and clinical outcome: Skeletal-related events. Oncology 2009, 23, 21–27. [Google Scholar] [PubMed]
- Luis-Ravelo, D.; Antón, I.; Zandueta, C.; Valencia, K.; Ormazábal, C.; Martínez-Canarias, S.; Guruceaga, E.; Perurena, N.; Vicent, S.; De Las Rivas, J.; et al. A gene signature of bone metastatic colonization sensitizes for tumor-induced osteolysis and predicts survival in lung cancer. Oncogene 2014, 33, 5090–5099. [Google Scholar] [CrossRef] [PubMed]
- Marx, J. Cell biology. Podosomes and invadopodia help mobile cells step lively. Science 2006, 312, 1868–1869. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, T.R.; Romero-Moreno, R.; Mason, D.E.; Nystrom, L.; Boerckel, J.D.; Niebur, G.; Littlepage, L.E. Bone: A Fertile Soil for Cancer Metastasis. Curr. Drug Targets 2017, 18, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.M.; Takayanagi, H. The immune system, bone and RANKL. Arch. Biochem. Biophys. 2014, 561, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Shih, L.-Y.; Shih, H.-N.; Chen, T.-H. Bone resorption activity of osteolytic metastatic lung and breast cancers. J. Orthop. Res. 2004, 22, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Hernández, I.; Moreno, J.L.; Zandueta, C.; Montuenga, L.; Lecanda, F. Novel alternatively spliced ADAM8 isoforms contribute to the aggressive bone metastatic phenotype of lung cancer. Oncogene 2010, 29, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.-L.; Liao, S.-H.; Hung, J.-Y.; Huang, M.-S.; Hsu, Y.-L. MicroRNA-33a functions as a bone metastasis suppressor in lung cancer by targeting parathyroid hormone related protein. Biochim. Biophys. Acta 2013, 1830, 3756–3766. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.C.; He, Y.; Broomfield, A.; Paatan, N.J.; Harrington, B.S.; Tseng, H.-W.; Beaven, E.A.; Kiernan, D.M.; Swindle, P.; Clubb, A.B.; et al. CD169(+) macrophages mediate pathological formation of woven bone in skeletal lesions of prostate cancer. J. Pathol. 2016, 239, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Zhang, L.; Qin, F.; Li, X.; Guo, X.; Li, Y.; Qiu, C.; Zhao, Y. Targeting macrophages for cancer therapy disrupts bone homeostasis and impairs bone marrow erythropoiesis in mice bearing Lewis lung carcinoma tumors. Cell Immunol. 2018, 331, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, M.C.; Shipley, R.T.; Corcoran, H.L.; Dickson, B.A. CT demonstration of calcification in carcinoma of the lung. AJR Am. J. Roentgenol. 1990, 154, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, J.; Takashima, T.; Miyata, S.; Kitagawa, M. CT demonstration of calcification in an adenoid cystic carcinoma of the lung. AJR Am. J. Roentgenol. 1990, 154, 419. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Al-Jahdali, H.H.; Allen, C.M.; Irion, K.L.; Al Ghanem, S.; Koteyar, S.S. The calcified lung nodule: What does it mean? Ann. Thorac. Med. 2010, 5, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Chabner, B.A.; Nabel, C.S. Canakinumab and Lung Cancer: Intriguing, but Is It Real? Oncologist 2018, 23, 637–638. [Google Scholar] [CrossRef] [PubMed]
- Crossman, D.; Rothman, A.M.K. Interleukin-1 β inhibition with canakinumab and reducing lung cancer—Subset analysis of the canakinumab anti-inflammatory thrombosis outcome study trial (CANTOS). J. Thorac. Dis. 2018, 10, S3084–3087. [Google Scholar] [CrossRef] [PubMed]
- Haux, J. Infection and cancer. Lancet 2001, 358, 155–156. [Google Scholar] [CrossRef]
- Vernon, L.F. William Bradley Coley, MD, and the phenomenon of spontaneous regression. Immunol. Targets Ther. 2018, 7, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Interleukin-1 β as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010, 29, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Kanneganti, T.-D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.S.; Yvan-Charvet, L.; Masters, S.L.; Murphy, A.J. The modern interleukin-1 superfamily: Divergent roles in obesity. Semin. Immunol. 2016, 28, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, J.W.; Kim, R.Y.; Robertson, A.A.B.; Hirota, J.A.; Wood, L.G.; Knight, D.A.; Cooper, M.A.; O’Neill, L.A.J.; Horvat, J.C.; Hansbro, P.M. Inflammasomes in the lung. Mol. Immunol. 2017, 86, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Howrylak, J.A.; Nakahira, K. Inflammasomes: Key Mediators of Lung Immunity. Annu. Rev. Physiol. 2017, 79, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.; Garofalo, A.; Padura, I.M.; Mantovani, A.; Giavazzi, R. Interleukin 1 receptor antagonist inhibits the augmentation of metastasis induced by interleukin 1 or lipopolysaccharide in a human melanoma/nude mouse system. Cancer Res. 1993, 53, 5051–5054. [Google Scholar] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Kuwano, T.; Tsutsumi-Miyahara, C.; Ueda, S.; Kimura, Y.N.; Hamano, S.; Sonoda, K.; Saijo, Y.; Nukiwa, T.; Strieter, R.M.; et al. Infiltration of COX-2–expressing macrophages is a prerequisite for IL-1β–induced neovascularization and tumor growth. J. Clin. Investig. 2005, 115, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, J.; Han, X.; Li, H.; Xie, M.; Sun, Y.; Liu, W.; Ba, X.; Zeng, X. Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1β-mediated increase in E-selectin expression. Int. J. Cancer 2017, 140, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory cytokine IL-1 β promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Próchnicki, T.; Latz, E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.P.Y.; Duncan, J.A.; Lei, Y. How the noninflammasome NLRs function in the innate immune system. Science 2010, 327, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef] [PubMed]
- Lasithiotaki, I.; Tsitoura, E.; Samara, K.D.; Trachalaki, A.; Charalambous, I.; Tzanakis, N.; Antoniou, K.M. NLRP3/Caspase-1 inflammasome activation is decreased in alveolar macrophages in patients with lung cancer. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Pouniotis, D.S.; Plebanski, M.; Apostolopoulos, V.; McDonald, C.F. Alveolar macrophage function is altered in patients with lung cancer. Clin. Exp. Immunol. 2006, 143, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and cancer progression: The emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Wang, Y.; Zeng, X.; Wang, Z.; Wang, H.; Xie, W. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 7501–7513. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; O’Neill, L.A.J. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Edye, M.E.; Lopez-Castejon, G.; Allan, S.M.; Brough, D. Acidosis drives DAMP-induced interleukin-1 secretion via a caspase-1-independent pathway. J. Biol. Chem. 2013. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. A broken krebs cycle in macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, W. Succinate in the cancer-immune cycle. Cancer Lett. 2017, 390, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Davies, L.C.; Karwan, M.; Ileva, L.; Ozaki, M.K.; Cheng, R.Y.S.; Ridnour, L.A.; Annunziata, C.M.; Wink, D.A.; McVicar, D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 2018, 128. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.-Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; McCormack, R.; Webster, A.; Milenkova, T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: A phase-IV, open-label, single-arm study. Br. J. Cancer 2014, 110, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Millett, R.L.; Elkon, J.M.; Tabbara, I.A. Directed Therapies in Anaplastic Lymphoma Kinase-rearranged Non-small Cell Lung Cancer. Anticancer Res. 2018, 38, 4969–4975. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, G.; Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.-L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Montero, J.; Letai, A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018, 25, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Yeo, A.; Faber, A.C.; Lifshits, E.; Chen, Z.; Cheng, K.A.; Walton, Z.; Sarosiek, K.A.; Letai, A.; Heist, R.S.; et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res. 2014, 74, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Jung, J.Y.; Kim, A.; Chang, Y.S.; Kim, S.K. ABT-737 Synergizes with Cisplatin Bypassing Aberration of Apoptotic Pathway in Non-small Cell Lung Cancer. Neoplasia 2017, 19, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.C.P. Small nanobody drugs win big backing from pharma. Nat. Med. 2013, 19, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Frink, R.E.; Peyton, M.; Schiller, J.H.; Gazdar, A.F.; Shay, J.W.; Minna, J.D. Telomerase inhibitor imetelstat has preclinical activity across the spectrum of non-small cell lung cancer oncogenotypes in a telomere length dependent manner. Oncotarget 2016, 7, 31639–31651. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yin, Y.; Wang, J.; Shi, B.; Zhang, L.; Qian, D.; Li, C.; Zhang, H.; Wang, S.; Zhu, J.; et al. Kras mutations increase telomerase activity targeting telomerase is a promising therapeutic strategy for Kras-mutant, NSCLC. Oncotarget 2017, 8, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Ho, P.-C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, H.-R.; Leng, L.; Kang, I.; Jorgensen, W.L.; Cho, C.-S.; Bucala, R.; Kim, W.-U. Role of macrophage migration inhibitory factor in the regulatory T cell response of tumor-bearing mice. J. Immunol. 2012, 189, 3905–3913. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kuang, D.-M.; Wu, Y.; Xiao, X.; Li, X.-F.; Li, T.-J.; Zheng, L. Activated CD69+ T Cells Foster Immune Privilege by Regulating IDO Expression in Tumor-Associated Macrophages. J. Immunol. 2012, 188, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Kim, H.; Quail, D.F.; Foley, E.A.; Joyce, J.A. Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell Rep. 2017, 19, 101–113. [Google Scholar] [CrossRef] [PubMed]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.L.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-Small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guilbaud, E.; Gautier, E.L.; Yvan-Charvet, L. Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer. Cancers 2019, 11, 298. https://doi.org/10.3390/cancers11030298
Guilbaud E, Gautier EL, Yvan-Charvet L. Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer. Cancers. 2019; 11(3):298. https://doi.org/10.3390/cancers11030298
Chicago/Turabian StyleGuilbaud, Emma, Emmanuel L. Gautier, and Laurent Yvan-Charvet. 2019. "Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer" Cancers 11, no. 3: 298. https://doi.org/10.3390/cancers11030298
APA StyleGuilbaud, E., Gautier, E. L., & Yvan-Charvet, L. (2019). Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer. Cancers, 11(3), 298. https://doi.org/10.3390/cancers11030298