Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma



{kind=link}
{kind=link}
Abstract
:1. Immunosurveillance and NK Cell Signaling in Melanoma
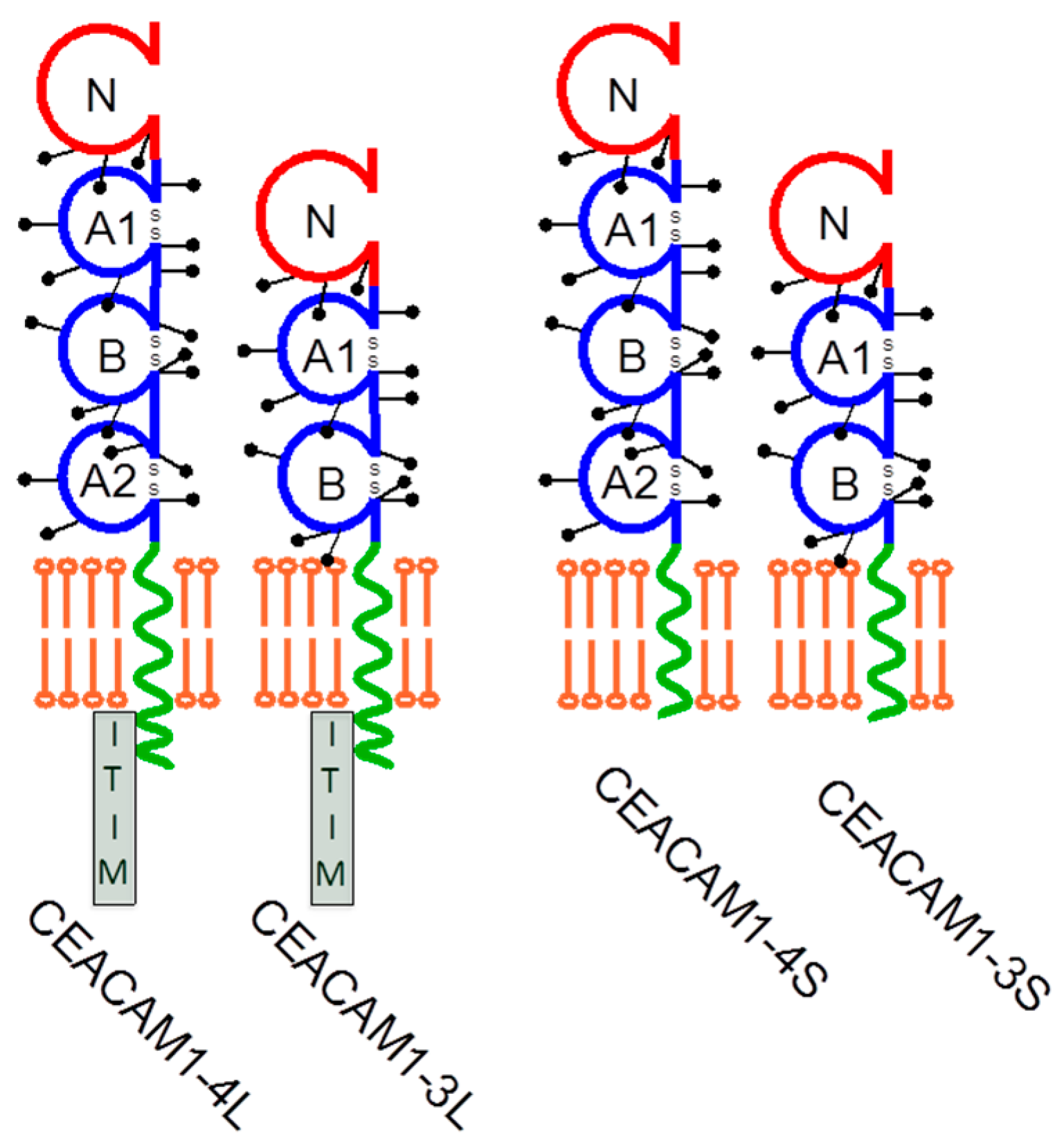
2. CEACAM1 Signaling and Its Function in Melanoma
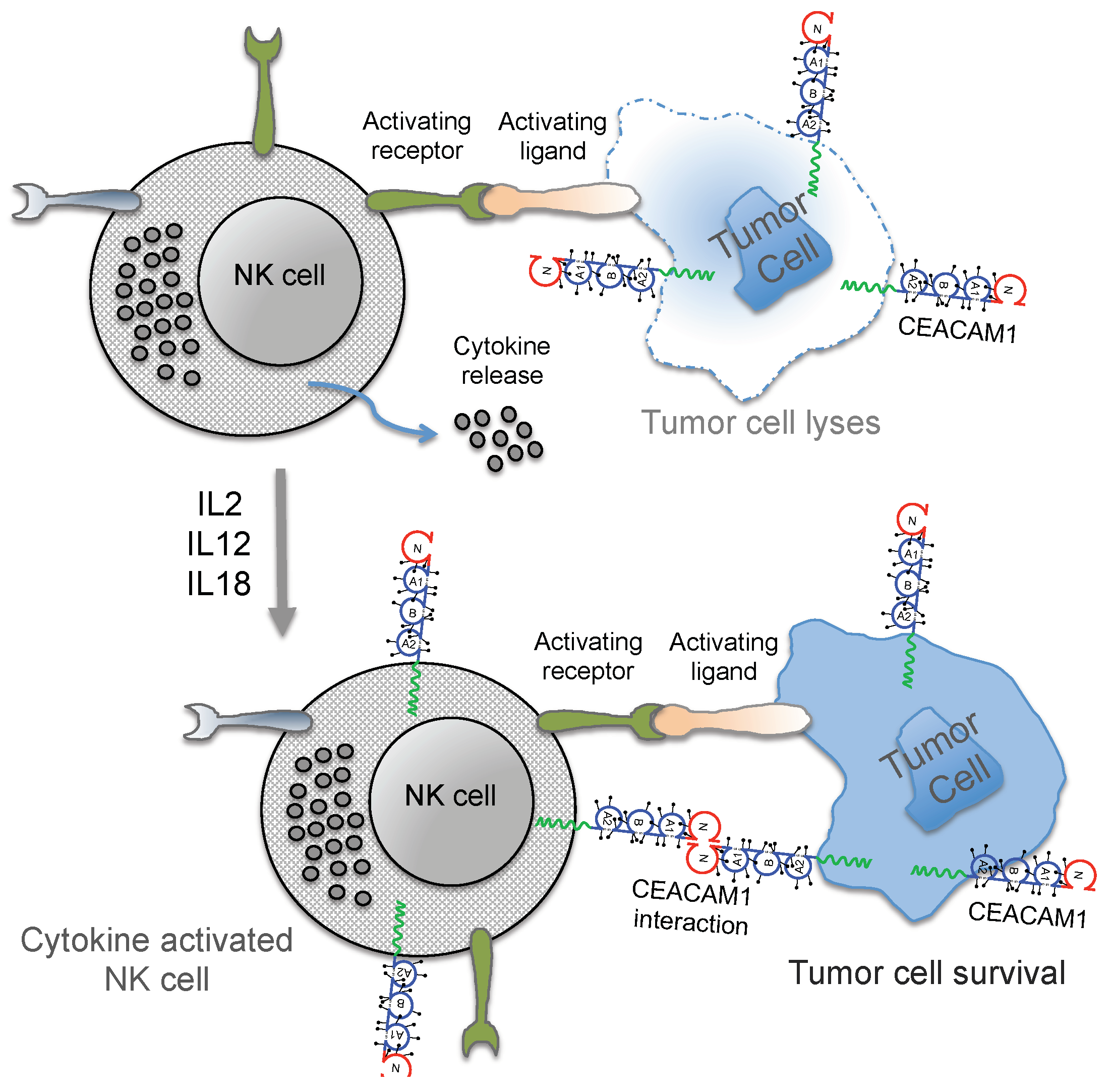
3. Regulation of NK Cell Function by CEACAM1
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hauschild, A. Melanoma in 2013: Melanoma—The run of success continues. Nat. Rev. Clin. Oncol. 2014, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.A.; Saraiya, M.; Geller, A.C.; Heneghan, M.K.; Jorgensen, C. Sun exposure and risk of melanoma. Arch. Dis. Child. 2006, 91, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.; Green, A. Melanoma and sunburn. Cancer Causes Control 1994, 5, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkwood, J.M.; Strawderman, M.H.; Ernstoff, M.S.; Smith, T.J.; Borden, E.C.; Blum, R.H. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: The Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 1996, 14, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 1994, 271, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, A.F.; Dzhandzhugazyan, K.; Zeuthen, J. The immunogenic properties of melanoma-associated antigens recognized by cytotoxic T lymphocytes. Exp. Clin. Immunogenet. 1998, 15, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageshita, T.; Wang, Z.; Calorini, L.; Yoshii, A.; Kimura, T.; Ono, T.; Gattoni-Celli, S.; Ferrone, S. Selective loss of human leukocyte class I allospecificities and staining of melanoma cells by monoclonal antibodies recognizing monomorphic determinants of class I human leukocyte antigens. Cancer Res. 1993, 53, 3349–3354. [Google Scholar]
- Pende, D.; Accame, L.; Pareti, L.; Mazzocchi, A.; Moretta, A.; Parmiani, G.; Moretta, L. The susceptibility to natural killer cell-mediated lysis of HLA class I-positive melanomas reflects the expression of insufficient amounts of different HLA class I alleles. Eur. J. Immunol. 1998, 28, 2384–2394. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar] [PubMed]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Casado, J.G.; Pawelec, G.; Morgado, S.; Sanchez-Correa, B.; Delgado, E.; Gayoso, I.; Duran, E.; Solana, R.; Tarazona, R. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol. Immunother. 2009, 58, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Investig. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschen, A.; Sucker, A.; Hill, B.; Moll, I.; Zapatka, M.; Nguyen, X.D.; Sim, G.C.; Gutmann, I.; Hassel, J.; Becker, J.C.; et al. Differential clinical significance of individual NKG2D ligands in melanoma: Soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin. Cancer Res. 2009, 15, 5208–5215. [Google Scholar] [CrossRef] [PubMed]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A.A. Role of chemokines in the biology of natural killer cells. Curr. Top. Microbiol. Immunol. 2010, 341, 37–58. [Google Scholar] [PubMed]
- Messaoudene, M.; Avril, M.F.; Caignard, A. When unity makes strength: Combinatorial NK cell-based immunotherapies against melanoma. Oncoimmunology 2014, 3, e28048. [Google Scholar] [CrossRef]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Frazao, A.; Colombo, M.; Fourmentraux-Neves, E.; Messaoudene, M.; Rusakiewicz, S.; Zitvogel, L.; Vivier, E.; Vely, F.; Faure, F.; Dreno, B.; et al. Shifting the Balance of Activating and Inhibitory Natural Killer Receptor Ligands on BRAF(V600E) Melanoma Lines with Vemurafenib. Cancer Immunol. Res. 2017, 5, 582–593. [Google Scholar] [CrossRef]
- Lopez-Cobo, S.; Pieper, N.; Campos-Silva, C.; Garcia-Cuesta, E.M.; Reyburn, H.T.; Paschen, A.; Vales-Gomez, M. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology 2018, 7, e1392426. [Google Scholar] [CrossRef] [PubMed]
- Edward, M. Integrins and other adhesion molecules involved in melanocytic tumor progression. Curr. Opin. Oncol. 1995, 7, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Grunert, F.; Zimmermann, W. Carcinoembryonic antigen gene family: Molecular biology and clinical perspectives. J. Clin. Lab. Anal. 1991, 5, 344–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Simpson, J.F.; Glackin, C.; Riethorf, L.; Wagener, C.; Shively, J.E. Expression of biliary glycoprotein (CD66a) in normal and malignant breast epithelial cells. Anticancer Res. 1998, 18, 3203–3212. [Google Scholar] [PubMed]
- Huang, J.; Hardy, J.D.; Sun, Y.; Shively, J.E. Essential role of biliary glycoprotein (CD66a) in morphogenesis of the human mammary epithelial cell line MCF10F. J. Cell Sci. 1999, 112, 4193–4205. [Google Scholar]
- Muller, M.M.; Singer, B.B.; Klaile, E.; Obrink, B.; Lucka, L. Transmembrane CEACAM1 affects integrin-dependent signaling and regulates extracellular matrix protein-specific morphology and migration of endothelial cells. Blood 2005, 105, 3925–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, B.B.; Scheffrahn, I.; Heymann, R.; Sigmundsson, K.; Kammerer, R.; Obrink, B. Carcinoembryonic antigen-related cell adhesion molecule 1 expression and signaling in human, mouse, and rat leukocytes: Evidence for replacement of the short cytoplasmic domain isoform by glycosylphosphatidylinositol-linked proteins in human leukocytes. J. Immunol. 2002, 168, 5139–5146. [Google Scholar] [CrossRef] [PubMed]
- Singer, B.B.; Klaile, E.; Scheffrahn, I.; Muller, M.M.; Kammerer, R.; Reutter, W.; Obrink, B.; Lucka, L. CEACAM1 (CD66a) mediates delay of spontaneous and Fas ligand-induced apoptosis in granulocytes. Eur. J. Immunol. 2005, 35, 1949–1959. [Google Scholar] [CrossRef] [Green Version]
- Singer, B.B.; Opp, L.; Heinrich, A.; Schreiber, F.; Binding-Liermann, R.; Berrocal-Almanza, L.C.; Heyl, K.A.; Muller, M.M.; Weimann, A.; Zweigner, J.; et al. Soluble CEACAM8 interacts with CEACAM1 inhibiting TLR2-triggered immune responses. PLoS ONE 2014, 9, e94106. [Google Scholar] [CrossRef]
- Teixeira, A.M.; Fawcett, J.; Simmons, D.L.; Watt, S.M. The N-domain of the biliary glycoprotein (BGP) adhesion molecule mediates homotypic binding: Domain interactions and epitope analysis of BGPc. Blood 1994, 84, 211–219. [Google Scholar]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef] [PubMed]
- Obrink, B. On the role of CEACAM1 in cancer. Lung Cancer 2008, 60, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Obrink, B. CEA adhesion molecules: Multifunctional proteins with signal-regulatory properties. Curr. Opin. Cell Biol. 1997, 9, 616–626. [Google Scholar] [CrossRef]
- Gray-Owen, S.D.; Blumberg, R.S. CEACAM1: Contact-dependent control of immunity. Nat. Rev. Immunol. 2006, 6, 433–446. [Google Scholar] [CrossRef]
- Singer, B.B.; Scheffrahn, I.; Kammerer, R.; Suttorp, N.; Ergun, S.; Slevogt, H. Deregulation of the CEACAM expression pattern causes undifferentiated cell growth in human lung adenocarcinoma cells. PLoS ONE 2010, 5, e8747. [Google Scholar] [CrossRef] [PubMed]
- Soni, P.; Lakkis, M.; Poy, M.N.; Fernstrom, M.A.; Najjar, S.M. The differential effects of pp120 (Ceacam 1) on the mitogenic action of insulin and insulin-like growth factor 1 are regulated by the nonconserved tyrosine 1316 in the insulin receptor. Mol. Cell. Biol. 2000, 20, 3896–3905. [Google Scholar] [CrossRef]
- Hunter, I.; Lindh, M.; Obrink, B. Differential regulation of C-CAM isoforms in epithelial cells. J. Cell Sci. 1994, 107, 1205–1216. [Google Scholar]
- Klaile, E.; Vorontsova, O.; Sigmundsson, K.; Muller, M.M.; Singer, B.B.; Ofverstedt, L.G.; Svensson, S.; Skoglund, U.; Obrink, B. The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J. Cell Biol. 2009, 187, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Fournes, B.; Sadekova, S.; Turbide, C.; Letourneau, S.; Beauchemin, N. The CEACAM1-L Ser503 residue is crucial for inhibition of colon cancer cell tumorigenicity. Oncogene 2001, 20, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimnejad, A.; Flayeh, R.; Unteregger, G.; Wagener, C.; Brummer, J. Cell adhesion molecule CEACAM1 associates with paxillin in granulocytes and epithelial and endothelial cells. Exp. Cell Res. 2000, 260, 365–373. [Google Scholar] [CrossRef]
- Jin, L.; Li, Y.; Chen, C.J.; Sherman, M.A.; Le, K.; Shively, J.E. Direct interaction of tumor suppressor CEACAM1 with beta catenin: Identification of key residues in the long cytoplasmic domain. Exp. Biol. Med. (Maywood) 2008, 233, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Edlund, M.; Blikstad, I.; Obrink, B. Calmodulin binds to specific sequences in the cytoplasmic domain of C-CAM and down-regulates C-CAM self-association. J. Biol. Chem. 1996, 271, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.M.; Klaile, E.; Vorontsova, O.; Singer, B.B.; Obrink, B. Homophilic adhesion and CEACAM1-S regulate dimerization of CEACAM1-L and recruitment of SHP-2 and c-Src. J. Cell Biol. 2009, 187, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadekova, S.; Lamarche-Vane, N.; Li, X.; Beauchemin, N. The CEACAM1-L glycoprotein associates with the actin cytoskeleton and localizes to cell-cell contact through activation of Rho-like GTPases. Mol. Biol. Cell 2000, 11, 65–77. [Google Scholar] [CrossRef]
- Schumann, D.; Chen, C.J.; Kaplan, B.; Shively, J.E. Carcinoembryonic antigen cell adhesion molecule 1 directly associates with cytoskeleton proteins actin and tropomyosin. J. Biol. Chem. 2001, 276, 47421–47433. [Google Scholar] [CrossRef] [PubMed]
- Khairnar, V.; Duhan, V.; Patil, A.M.; Zhou, F.; Bhat, H.; Thoens, C.; Sharma, P.; Adomati, T.; Friendrich, S.K.; Bezgovsek, J.; et al. CEACAM1 promotes CD8(+) T cell responses and improves control of a chronic viral infection. Nat. Commun. 2018, 9, 2561. [Google Scholar] [CrossRef] [PubMed]
- Klaile, E.; Muller, M.M.; Kannicht, C.; Singer, B.B.; Lucka, L. CEACAM1 functionally interacts with filamin A and exerts a dual role in the regulation of cell migration. J. Cell Sci. 2005, 118, 5513–5524. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2016, 536, 359. [Google Scholar] [CrossRef] [PubMed]
- Singer, B.B.; Scheffrahn, I.; Obrink, B. The tumor growth-inhibiting cell adhesion molecule CEACAM1 (C-CAM) is differently expressed in proliferating and quiescent epithelial cells and regulates cell proliferation. Cancer Res. 2000, 60, 1236–1244. [Google Scholar]
- Ebrahimnejad, A.; Streichert, T.; Nollau, P.; Horst, A.K.; Wagener, C.; Bamberger, A.M.; Brummer, J. CEACAM1 enhances invasion and migration of melanocytic and melanoma cells. Am. J. Pathol. 2004, 165, 1781–1787. [Google Scholar] [CrossRef]
- Fiori, V.; Magnani, M.; Cianfriglia, M. The expression and modulation of CEACAM1 and tumor cell transformation. Ann. Istituto Super. Sanita 2012, 48, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Thies, A.; Moll, I.; Berger, J.; Wagener, C.; Brummer, J.; Schulze, H.J.; Brunner, G.; Schumacher, U. CEACAM1 expression in cutaneous malignant melanoma predicts the development of metastatic disease. J. Clin. Oncol. 2002, 20, 2530–2536. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Tapolsky, M.; Earley, K.; Wood, C.G.; Wilson, D.R.; Logothetis, C.J.; Lin, S.H. Tumor-suppressive activity of CD66a in prostate cancer. Cancer Gene Ther. 1999, 6, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumaier, M.; Paululat, S.; Chan, A.; Matthaes, P.; Wagener, C. Biliary glycoprotein, a potential human cell adhesion molecule, is down-regulated in colorectal carcinomas. Proc. Natl. Acad. Sci. USA 1993, 90, 10744–10748. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, L.; Lisboa, B.W.; Henkel, U.; Naumann, M.; Wagener, C.; Loning, T. Differential expression of CD66a (BGP), a cell adhesion molecule of the carcinoembryonic antigen family, in benign, premalignant, and malignant lesions of the human mammary gland. J. Histochem. Cytochem. 1997, 45, 957–963. [Google Scholar] [CrossRef]
- Brummer, J.; Ebrahimnejad, A.; Flayeh, R.; Schumacher, U.; Loning, T.; Bamberger, A.M.; Wagener, C. cis Interaction of the cell adhesion molecule CEACAM1 with integrin beta(3). Am. J. Pathol. 2001, 159, 537–546. [Google Scholar] [CrossRef]
- Markel, G.; Ortenberg, R.; Seidman, R.; Sapoznik, S.; Koren-Morag, N.; Besser, M.J.; Bar, J.; Shapira, R.; Kubi, A.; Nardini, G.; et al. Systemic dysregulation of CEACAM1 in melanoma patients. Cancer Immunol. Immunother. 2010, 59, 215–230. [Google Scholar] [CrossRef]
- Sivan, S.; Suzan, F.; Rona, O.; Tamar, H.; Vivian, B.; Tamar, P.; Jacob, S.; Gal, M.; Michal, L. Serum CEACAM1 Correlates with Disease Progression and Survival in Malignant Melanoma Patients. Clin. Dev. Immunol. 2012, 2012, 290536. [Google Scholar] [CrossRef]
- Thies, A.; Berlin, A.; Brunner, G.; Schulze, H.J.; Moll, I.; Pfuller, U.; Wagener, C.; Schachner, M.; Altevogt, P.; Schumacher, U. Glycoconjugate profiling of primary melanoma and its sentinel node and distant metastases: Implications for diagnosis and pathophysiology of metastases. Cancer Lett. 2007, 248, 68–80. [Google Scholar] [CrossRef]
- Liu, W.; Wei, W.; Winer, D.; Bamberger, A.M.; Bamberger, C.; Wagener, C.; Ezzat, S.; Asa, S.L. CEACAM1 impedes thyroid cancer growth but promotes invasiveness: A putative mechanism for early metastases. Oncogene 2007, 26, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.; Heinemann, A.; Nilewski, E.; Scheffrahn, I.; Klode, J.; Scherag, A.; Schadendorf, D.; Singer, B.B.; Helfrich, I. CEACAM1-3S Drives Melanoma Cells into NK Cell-Mediated Cytolysis and Enhances Patient Survival. Cancer Res. 2015, 75, 1897–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Di, G.; Wu, C.T.; Hu, X.; Duan, H. CEACAM1 inhibits cell-matrix adhesion and promotes cell migration through regulating the expression of N-cadherin. Biochem. Biophys. Res. Commun. 2013, 430, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Ortenberg, R.; Sapir, Y.; Raz, L.; Hershkovitz, L.; Ben, A.A.; Sapoznik, S.; Barshack, I.; Avivi, C.; Berkun, Y.; Besser, M.J.; et al. Novel immunotherapy for malignant melanoma with a monoclonal antibody that blocks CEACAM1 homophilic interactions. Mol. Cancer Ther. 2012, 11, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Loffek, S.; Franzke, C.W.; Helfrich, I. Tension in Cancer. Int. J. Mol. Sci. 2016, 17, 1910. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Wegwitz, F.; Lenfert, E.; Gerstel, D.; von, E.L.; Einhoff, J.; Schmidt, G.; Logsdon, M.; Brandner, J.; Tiegs, G.; Beauchemin, N.; et al. CEACAM1 controls the EMT switch in murine mammary carcinoma in vitro and in vivo. Oncotarget 2016, 7, 63730–63746. [Google Scholar] [PubMed] [Green Version]
- Loffek, S.; Ullrich, N.; Gorgens, A.; Murke, F.; Eilebrecht, M.; Menne, C.; Giebel, B.; Schadendorf, D.; Singer, B.B.; Helfrich, I. CEACAM1-4L Promotes Anchorage-Independent Growth in Melanoma. Front. Oncol. 2015, 5, 234. [Google Scholar] [CrossRef]
- Hoek, K.S.; Schlegel, N.C.; Eichhoff, O.M.; Widmer, D.S.; Praetorius, C.; Einarsson, S.O.; Valgeirsdottir, S.; Bergsteinsdottir, K.; Schepsky, A.; Dummer, R.; et al. Novel MITF targets identified using a two-step DNA microarray strategy. Pigment Cell Melanoma Res. 2008, 21, 665–676. [Google Scholar] [CrossRef]
- Ullrich, N.; Loffek, S.; Horn, S.; Ennen, M.; Sanchez-Del-Campo, L.; Zhao, F.; Breitenbuecher, F.; Davidson, I.; Singer, B.B.; Schadendorf, D.; et al. MITF is a critical regulator of the carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) in malignant melanoma. Pigment Cell Melanoma Res. 2015, 28, 736–740. [Google Scholar] [CrossRef]
- Bentley, N.J.; Eisen, T.; Goding, C.R. Melanocyte-specific expression of the human tyrosinase promoter: Activation by the microphthalmia gene product and role of the initiator. Mol. Cell. Biol. 1994, 14, 7996–8006. [Google Scholar] [CrossRef] [PubMed]
- Berson, J.F.; Harper, D.C.; Tenza, D.; Raposo, G.; Marks, M.S. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 2001, 12, 3451–3464. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Tenza, D.; Murphy, D.M.; Berson, J.F.; Marks, M.S. Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 2001, 152, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Cheli, Y.; Ohanna, M.; Ballotti, R.; Bertolotto, C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010, 23, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Laurette, P.; Strub, T.; Koludrovic, D.; Keime, C.; Le, G.S.; Seberg, H.; Van, O.E.; Imrichova, H.; Siddaway, R.; Aerts, S.; et al. Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. Elife 2015, 4, e06857. [Google Scholar] [CrossRef] [PubMed]
- Abou-Rjaily, G.A.; Lee, S.J.; May, D.; Al-Share, Q.Y.; Deangelis, A.M.; Ruch, R.J.; Neumaier, M.; Kalthoff, H.; Lin, S.H.; Najjar, S.M. CEACAM1 modulates epidermal growth factor receptor—Mediated cell proliferation. J. Clin. Investig. 2004, 114, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, R.; Hahn, S.; Singer, B.B.; Luo, J.S.; von Kleist, S. Biliary glycoprotein (CD66a), a cell adhesion molecule of the immunoglobulin superfamily, on human lymphocytes: Structure, expression and involvement in T cell activation. Eur. J. Immunol. 1998, 28, 3664–3674. [Google Scholar] [CrossRef]
- Khairnar, V.; Duhan, V.; Maney, S.K.; Honke, N.; Shaabani, N.; Pandyra, A.A.; Seifert, M.; Pozdeev, V.; Xu, H.C.; Sharma, P.; et al. CEACAM1 induces B-cell survival and is essential for protective antiviral antibody production. Nat. Commun. 2015, 6, 6217. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Pan, H.; Shively, J.E. CEACAM1 negatively regulates IL-1beta production in LPS activated neutrophils by recruiting SHP-1 to a SYK-TLR4-CEACAM1 complex. PLoS Pathog. 2012, 8, e1002597. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Shively, J.E. Carcinoembryonic antigen-related cell adhesion molecule-1 regulates granulopoiesis by inhibition of granulocyte colony-stimulating factor receptor. Immunity 2010, 33, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Rueckschloss, U.; Kuerten, S.; Ergun, S. The role of CEA-related cell adhesion molecule-1 (CEACAM1) in vascular homeostasis. Histochem. Cell Biol. 2016, 146, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Slevogt, H.; Zabel, S.; Opitz, B.; Hocke, A.; Eitel, J.; N’Guessan, P.D.; Lucka, L.; Riesbeck, K.; Zimmermann, W.; Zweigner, J.; et al. CEACAM1 inhibits Toll-like receptor 2-triggered antibacterial responses of human pulmonary epithelial cells. Nat. Immunol. 2008, 9, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Greicius, G.; Severinson, E.; Beauchemin, N.; Obrink, B.; Singer, B.B. CEACAM1 is a potent regulator of B cell receptor complex-induced activation. J. Leukoc. Biol. 2003, 74, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Chow, E.M.; Wong, H.; Gu, J.; Mandelboim, O.; Gray-Owen, S.D.; Ostrowski, M.A. CEACAM1 (CD66a) promotes human monocyte survival via a phosphatidylinositol 3-kinase- and AKT-dependent pathway. J. Biol. Chem. 2006, 281, 39179–39193. [Google Scholar] [CrossRef] [PubMed]
- Skubitz, K.M.; Skubitz, A.P. Interdependency of CEACAM-1, -3, -6, and -8 induced human neutrophil adhesion to endothelial cells. J. Transl. Med. 2008, 6, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, A.K.; Wegscheid, C.; Schaefers, C.; Schiller, B.; Neumann, K.; Lunemann, S.; Langeneckert, A.E.; Oldhafer, K.J.; Weiler-Normann, C.; Lang, K.S.; et al. Carcinoembryonic antigen-related cell adhesion molecule 1 controls IL-2-dependent regulatory T-cell induction in immune-mediated hepatitis in mice. Hepatology 2018, 68, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.J.; Edwards, A.M.; Rowe, H.A.; Virji, M. Carcinoembryonic antigen-related cell adhesion molecule (CEACAM)-binding recombinant polypeptide confers protection against infection by respiratory and urogenital pathogens. Mol. Microbiol. 2005, 55, 1515–1527. [Google Scholar] [CrossRef] [Green Version]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejias-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virji, M.; Evans, D.; Hadfield, A.; Grunert, F.; Teixeira, A.M.; Watt, S.M. Critical determinants of host receptor targeting by Neisseria meningitidis and Neisseria gonorrhoeae: Identification of Opa adhesiotopes on the N-domain of CD66 molecules. Mol. Microbiol. 1999, 34, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, M.; Hoe, C.J.; Makepeace, K.; van der Ley, P.; Pollard, A.J. Phase variation of Opa proteins of Neisseria meningitidis and the effects of bacterial transformation. J. Biosci. 2016, 41, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Markel, G.; Lieberman, N.; Katz, G.; Arnon, T.I.; Lotem, M.; Drize, O.; Blumberg, R.S.; Bar-Haim, E.; Mader, R.; Eisenbach, L.; et al. CD66a interactions between human melanoma and NK cells: A novel class I MHC-independent inhibitory mechanism of cytotoxicity. J. Immunol. 2002, 168, 2803–2810. [Google Scholar] [CrossRef] [PubMed]
- Markel, G.; Wolf, D.; Hanna, J.; Gazit, R.; Goldman-Wohl, D.; Lavy, Y.; Yagel, S.; Mandelboim, O. Pivotal role of CEACAM1 protein in the inhibition of activated decidual lymphocyte functions. J. Clin. Investig. 2002, 110, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markel, G.; Seidman, R.; Stern, N.; Cohen-Sinai, T.; Izhaki, O.; Katz, G.; Besser, M.; Treves, A.J.; Blumberg, R.S.; Loewenthal, R.; et al. Inhibition of human tumor-infiltrating lymphocyte effector functions by the homophilic carcinoembryonic cell adhesion molecule 1 interactions. J. Immunol. 2006, 177, 6062–6071. [Google Scholar] [CrossRef] [PubMed]
- Stern, N.; Markel, G.; Arnon, T.I.; Gruda, R.; Wong, H.; Gray-Owen, S.D.; Mandelboim, O. Carcinoembryonic antigen (CEA) inhibits NK killing via interaction with CEA-related cell adhesion molecule 1. J. Immunol. 2005, 174, 6692–6701. [Google Scholar] [CrossRef] [PubMed]
- Moller, H.; Bohrsch, V.; Lucka, L.; Hackenberger, C.P.; Hinderlich, S. Efficient metabolic oligosaccharide engineering of glycoproteins by UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) knock-down. Mol. Biosyst. 2011, 7, 2245–2251. [Google Scholar] [CrossRef]
- Moller, M.J.; Kammerer, R.; Grunert, F.; von Kleist, S. Biliary glycoprotein (BGP) expression on T cells and on a natural-killer-cell sub-population. Int. J. Cancer 1996, 65, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Agaugue, S.; Marcenaro, E.; Ferranti, B.; Moretta, L.; Moretta, A. Human natural killer cells exposed to IL-2, IL-12, IL-18, or IL-4 differently modulate priming of naive T cells by monocyte-derived dendritic cells. Blood 2008, 112, 1776–1783. [Google Scholar] [CrossRef] [Green Version]
- Thirion, G.; Feliu, A.A.; Coutelier, J.P. CD66a (CEACAM1) expression by mouse natural killer cells. Immunology 2008, 125, 535–540. [Google Scholar] [CrossRef] [Green Version]
- Sapoznik, S.; Ortenberg, R.; Schachter, J.; Markel, G. CEACAM1 in malignant melanoma: A diagnostic and therapeutic target. Curr. Top. Med. Chem. 2012, 12, 3–10. [Google Scholar] [CrossRef]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosomi, S.; Chen, Z.; Baker, K.; Chen, L.; Huang, Y.H.; Olszak, T.; Zeissig, S.; Wang, J.H.; Mandelboim, O.; Beauchemin, N.; et al. CEACAM1 on activated NK cells inhibits NKG2D-mediated cytolytic function and signaling. Eur. J. Immunol. 2013, 43, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Chen, L.; Baker, K.; Olszak, T.; Zeissig, S.; Huang, Y.H.; Kuo, T.T.; Mandelboim, O.; Beauchemin, N.; Lanier, L.L.; et al. CEACAM1 dampens antitumor immunity by down-regulating NKG2D ligand expression on tumor cells. J. Exp. Med. 2011, 208, 2633–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Markel, G.; Achdout, H.; Katz, G.; Ling, K.L.; Salio, M.; Gruda, R.; Gazit, R.; Mizrahi, S.; Hanna, J.; Gonen-Gross, T.; et al. Biological function of the soluble CEACAM1 protein and implications in TAP2-deficient patients. Eur. J. Immunol. 2004, 34, 2138–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helfrich, I.; Singer, B.B. Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma. Cancers 2019, 11, 356. https://doi.org/10.3390/cancers11030356
Helfrich I, Singer BB. Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma. Cancers. 2019; 11(3):356. https://doi.org/10.3390/cancers11030356
Chicago/Turabian StyleHelfrich, Iris, and Bernhard B. Singer. 2019. "Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma" Cancers 11, no. 3: 356. https://doi.org/10.3390/cancers11030356
APA StyleHelfrich, I., & Singer, B. B. (2019). Size Matters: The Functional Role of the CEACAM1 Isoform Signature and Its Impact for NK Cell-Mediated Killing in Melanoma. Cancers, 11(3), 356. https://doi.org/10.3390/cancers11030356