The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models


 , ,
, ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines and Cell Cultures
2.2. Reagents and Enzymatic Activities
2.3. Immunofluorescence Studies
2.4. Fluorescence-Activated Cell Sorter (FACS) Analyses
2.5. Growth Assays
2.6. Cell Viability and Apoptosis Assay
2.7. Western Blot
2.8. In Vivo Experiments: Xenograft Model
2.9. Immunohistochemical Analyses
2.10. Orthotopic Intra-Brain Model
2.11. Statistical Details
3. Results
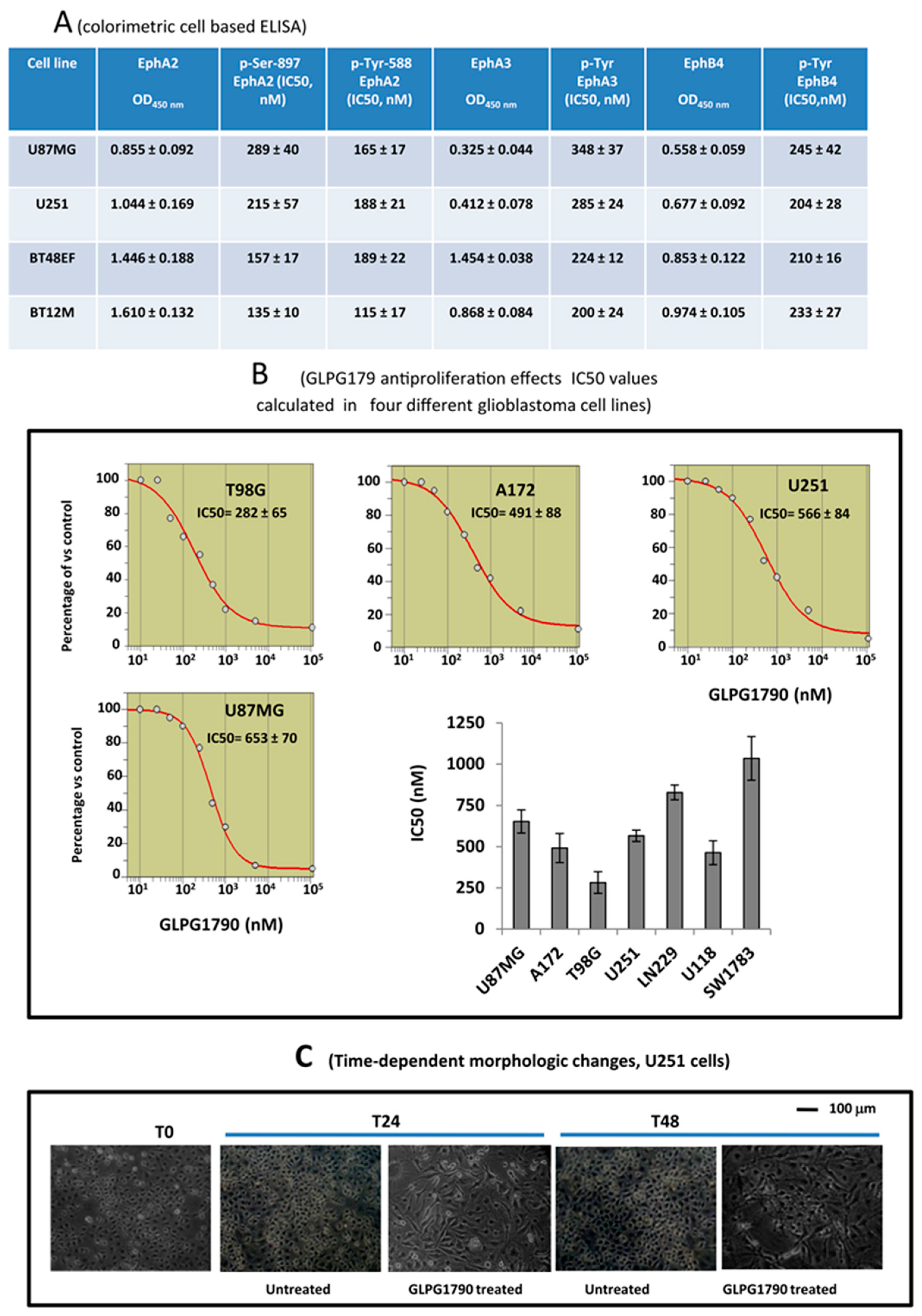
3.1. GLPG1790 Inhibits Phosphorylation of Both Tyrosine and Serine in U87MG Cells
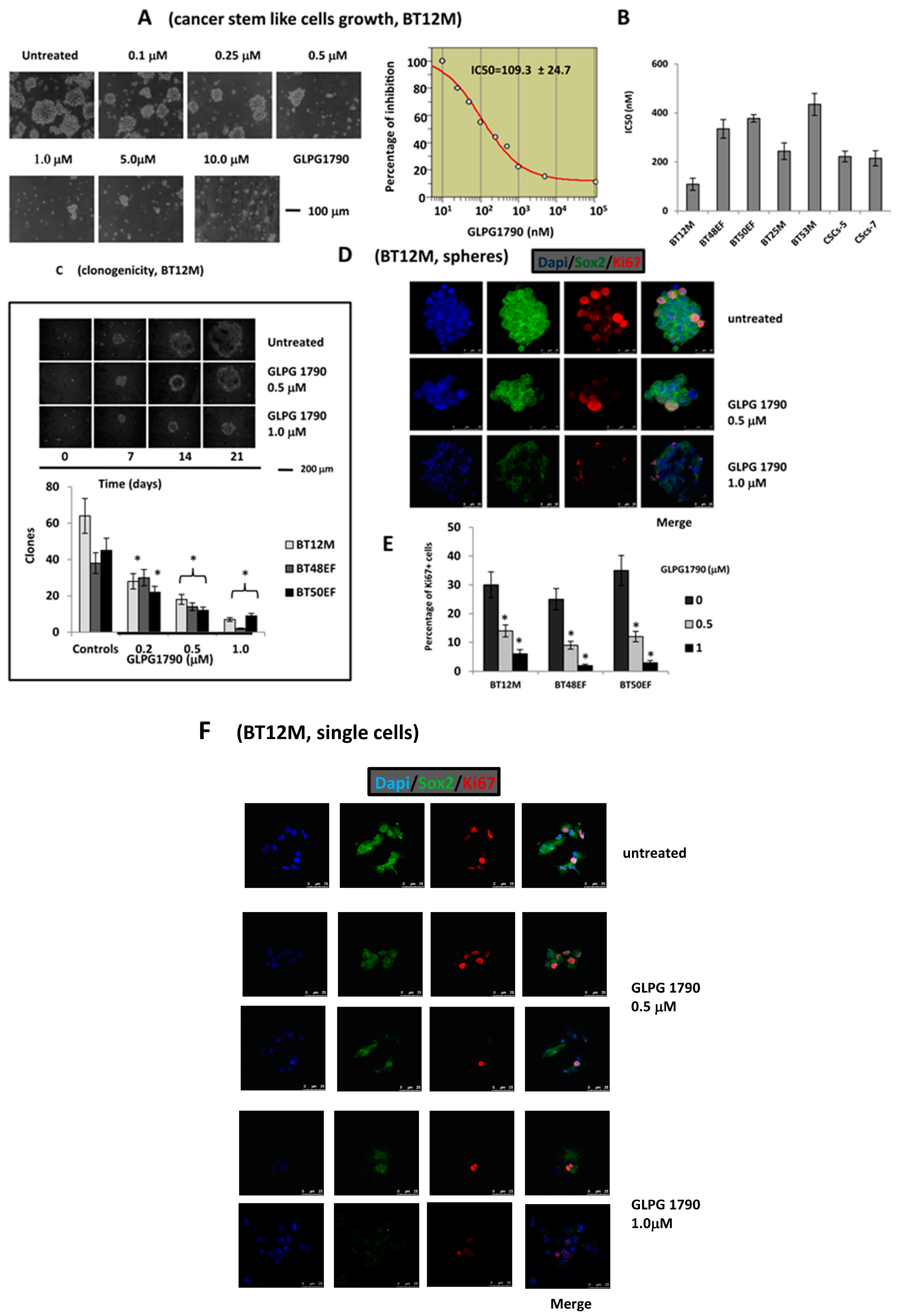
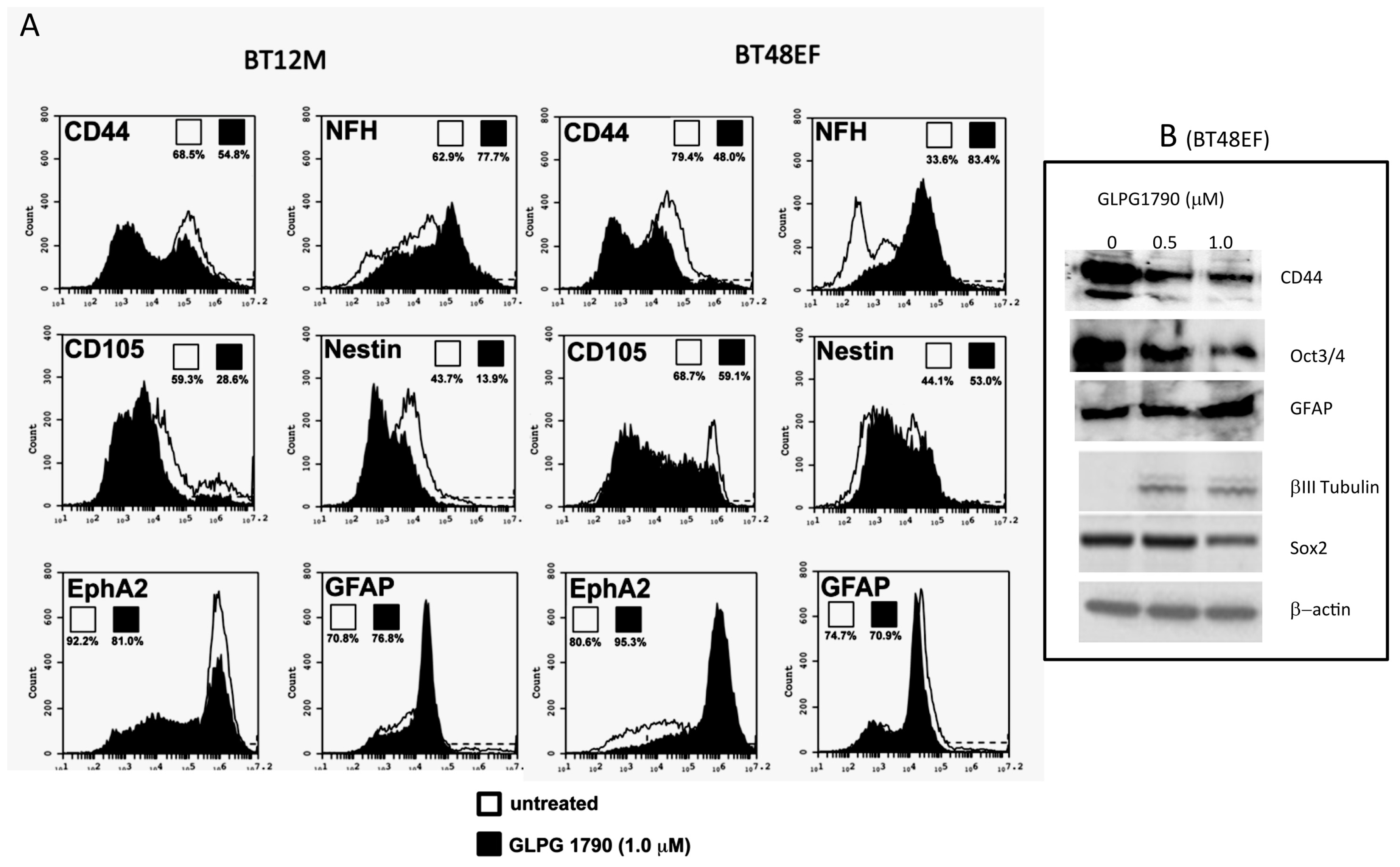
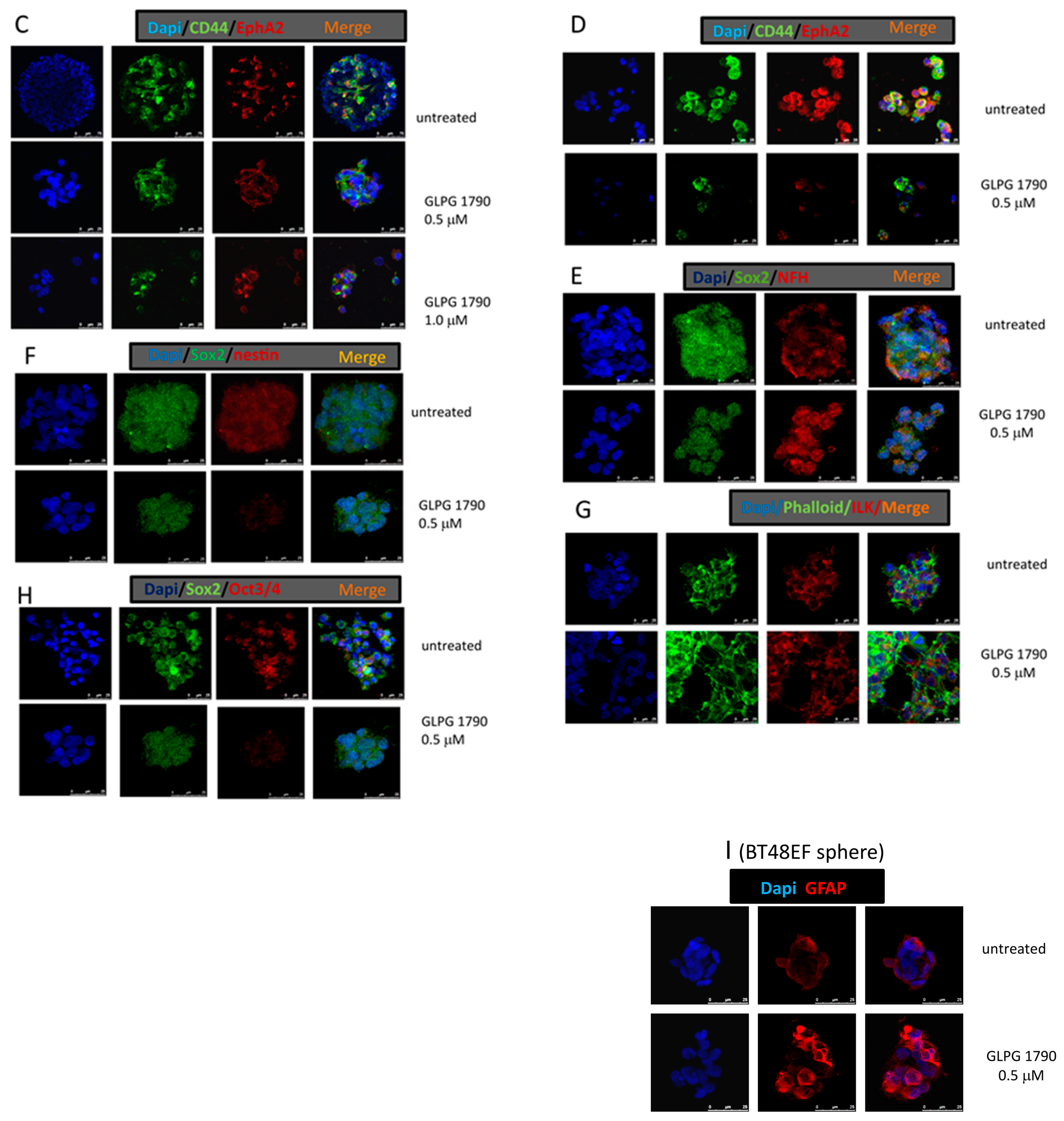
3.2. GLPG1790 Reduces Mesenchymal/Stem Cell Marker Expression in GICs
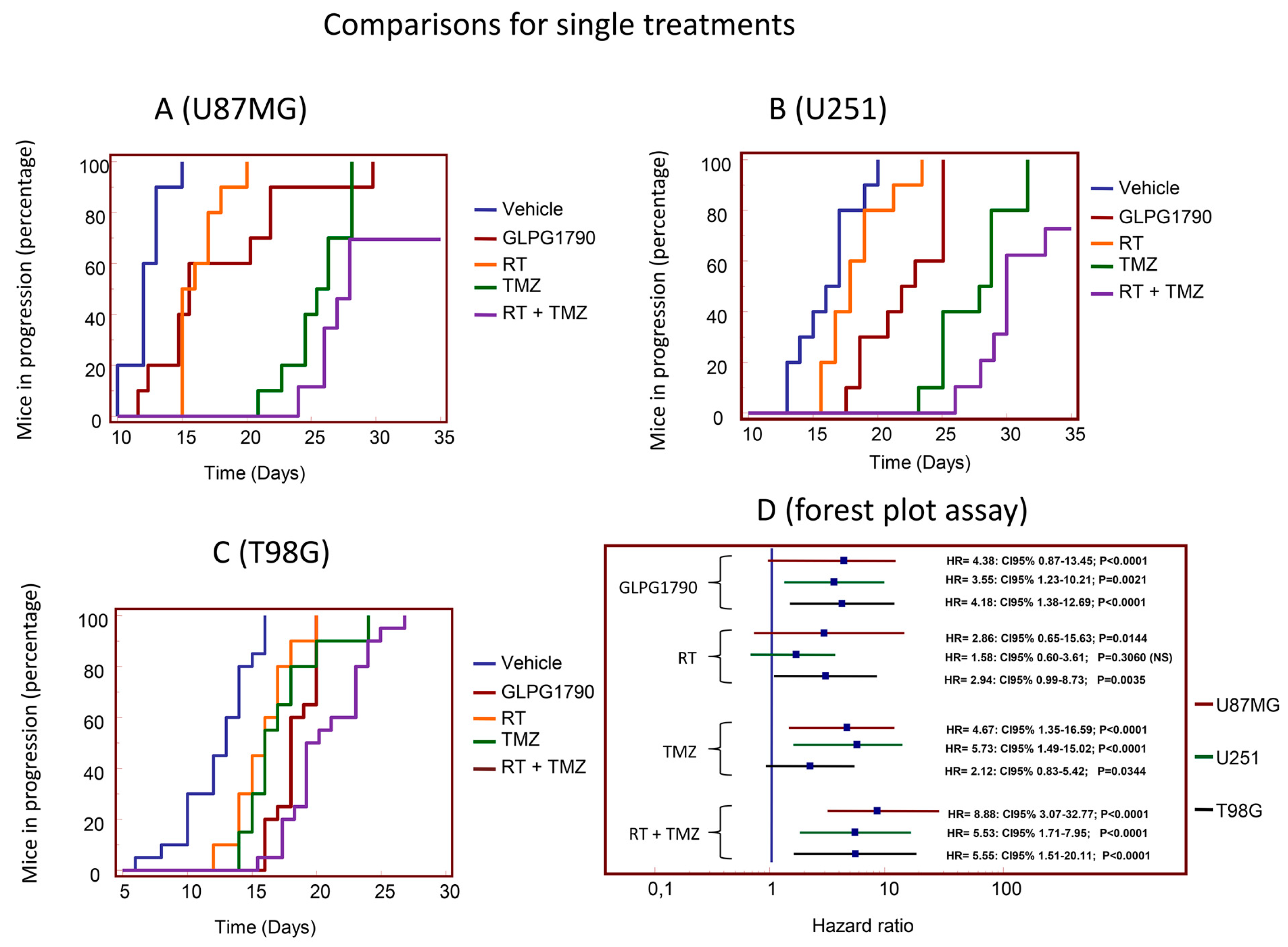
3.3. GLPG1790 Modifies Tumour Growth of GBM Cells Subcutaneously Injected in Female nu/nu Mice (Xenograft Model)
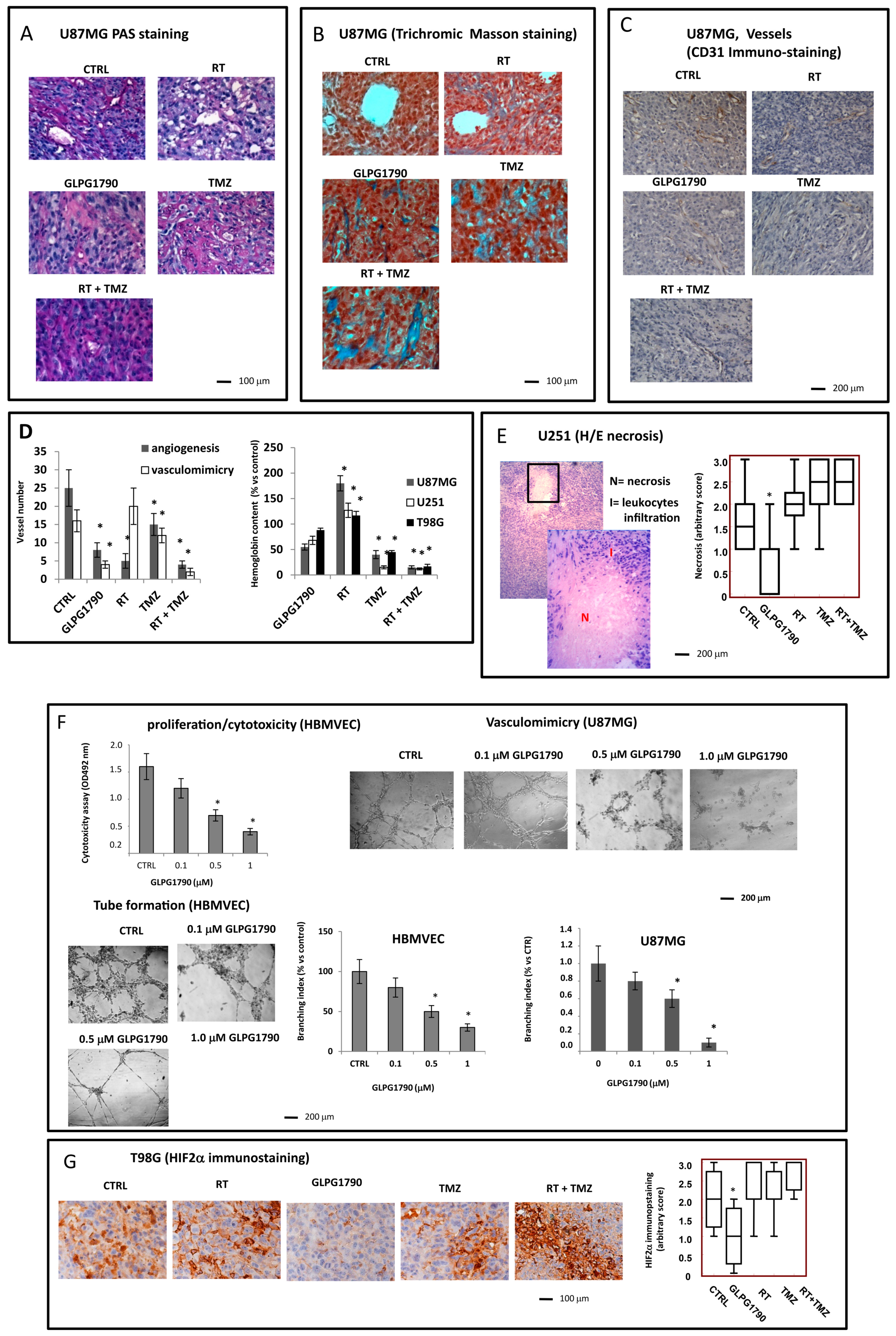
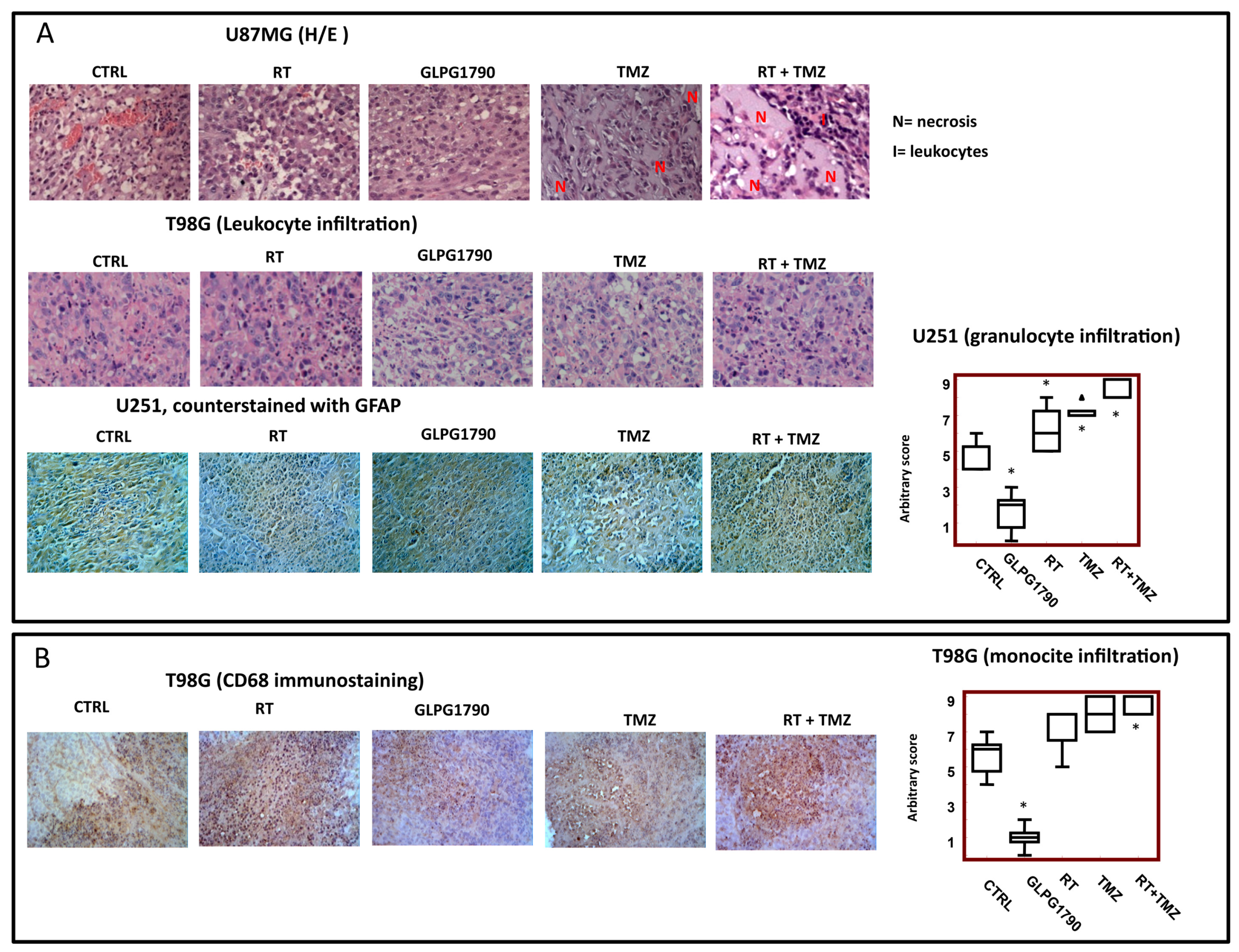
3.4. Histological Evaluation of GLPG1790 Treated Xenografts: Involvement of Tumour Microenvironment in the GLPG1790-Mediated Anti-Tumour Effects
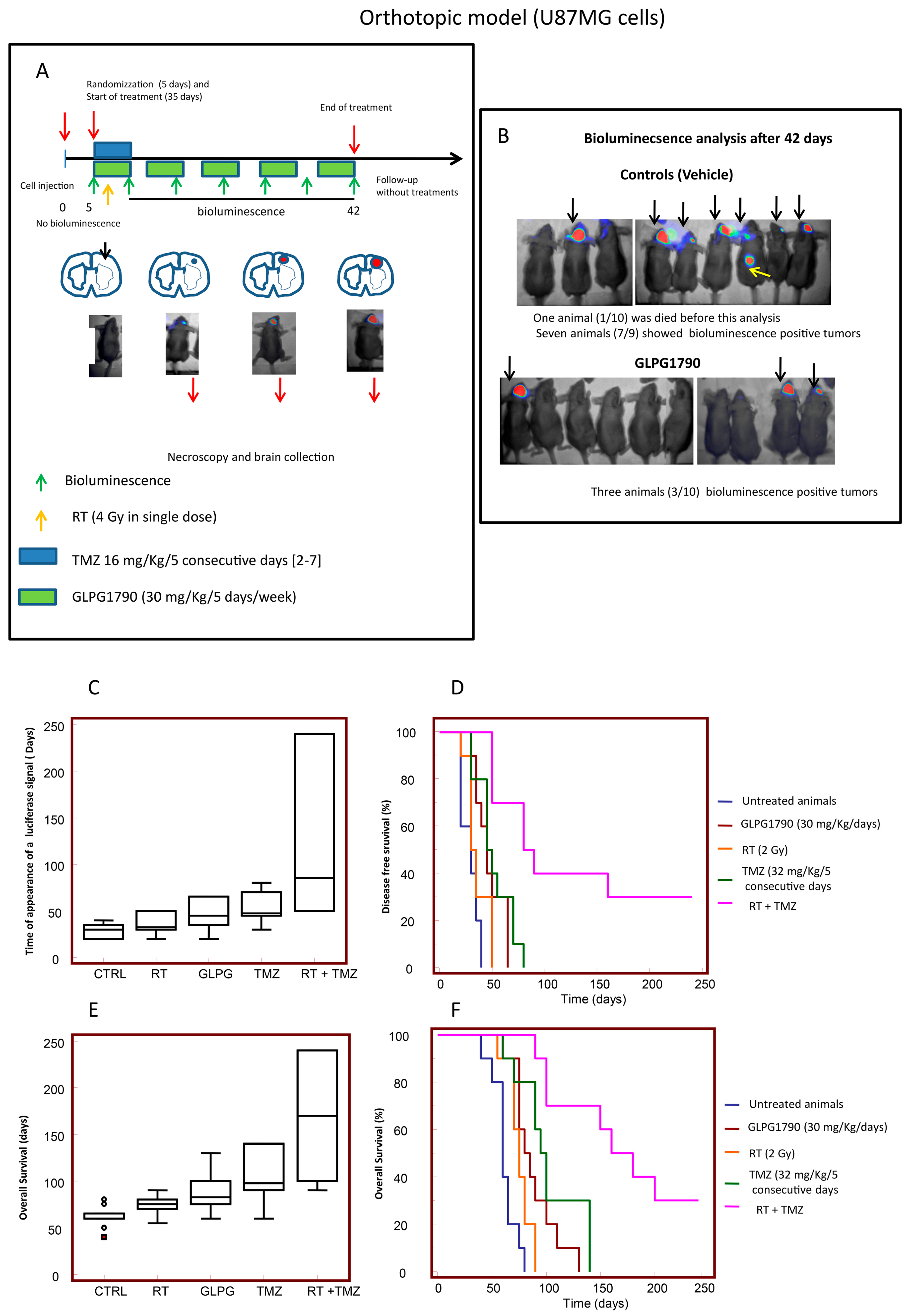
3.5. GLPG1790 Increased Disease-Free and Overall Survival in Orthotopic Intra-Brain Tumours as Determined by Using Differentiated U87MG Cells
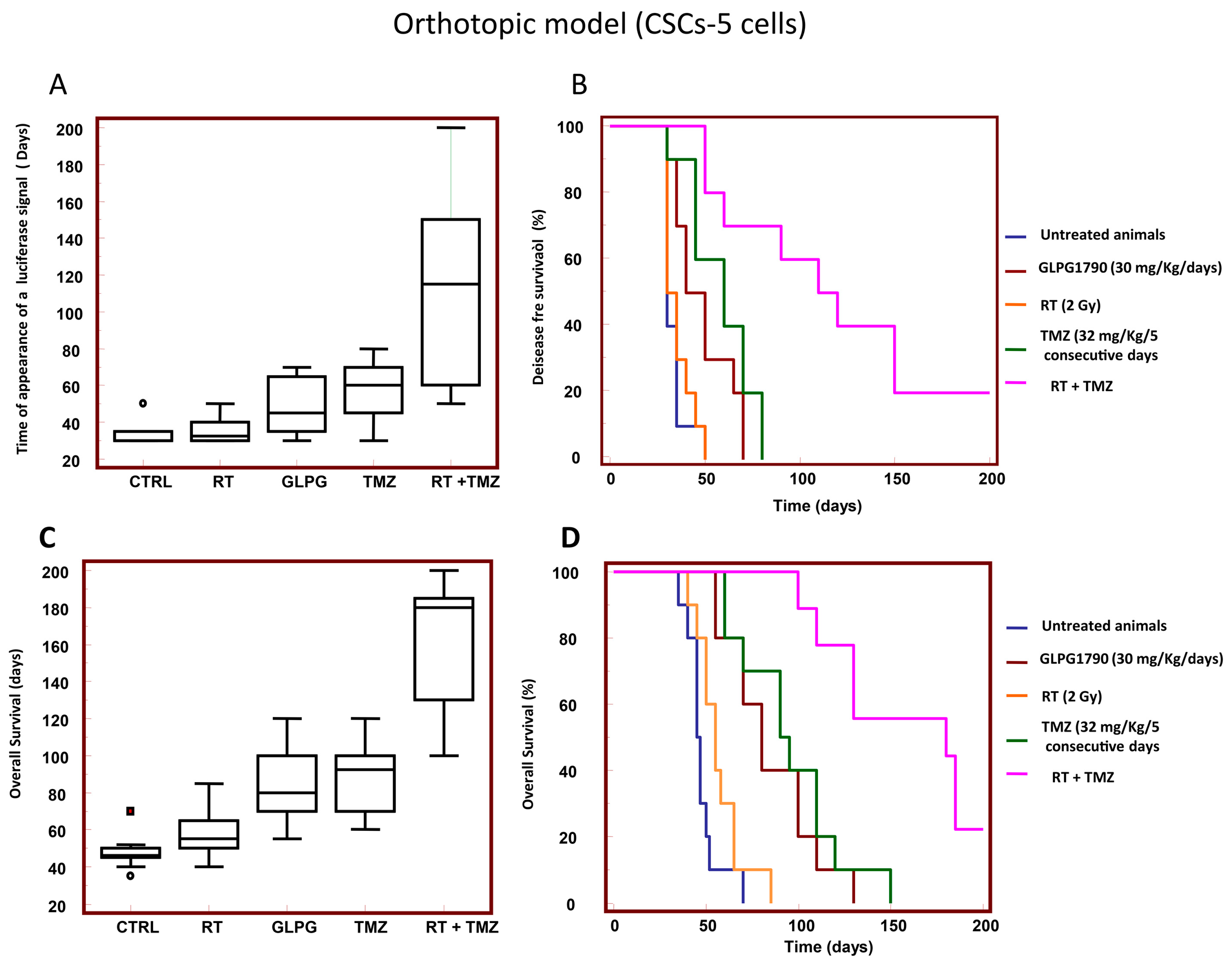
3.6. GLPG1790 Increased DFS and OS in Orthotopic Intra-Brain Tumours Generated from Patient Derived Stem-Like GSCs-5 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gittleman, H.; Boscia, A.; Ostrom, Q.T.; Truitt, G.; Fritz, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. Survivorship in Adults with Malignant Brain and other Central Nervous System Tumor from 2000–2014. Neuro Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Bougnaud, S.; Golebiewska, A.; Oudin, A.; Keunen, O.; Harter, P.N.; Mäder, L.; Azuaje, F.; Fritah, S.; Stieber, D.; Kaoma, T.; et al. Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget 2016, 7, 31955–31971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Rajappa, P.; Hu, W.; Hoffman, C.; Cisse, B.; Kim, J.H.; Gorge, E.; Yanowitch, R.; Cope, W.; Vartanian, E.; et al. A proangiogenic signaling axis in myeloid cells promotes malignant progression of glioma. J. Clin. Investig. 2017, 127, 1826–1838. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Gonçalves, R.M.; Klafke, K.; de Souza, P.O.; Dillenburg, F.C.; Carro, L.; Gelain, D.P.; Moreira, J.C. Inflammatory landscape of human brain tumors reveals an NFκB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017, 390, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Song, X.; Zhu, J.; Fu, A.; Li, J.; Chen, T. The interventional effect of new drugs combined with the Stupp protocol on glioblastoma: A network meta-analysis. Clin. Neurol. Neurosurg. 2017, 159, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xu, S.; Gao, X.; Wang, J.; Xue, H.; Chen, Z.; Zhang, J.; Guo, X.; Qian, M.; Qiu, W.; et al. Macrophage migration inhibitory factor promotes vasculogenic mimicry formation induced by hypoxia via CXCR4/AKT/EMT pathway in human glioblastoma cells. Oncotarget 2017, 8, 80358–80372. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Chen, W.W.; Zhang, X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches. Oncol. Lett. 2016, 12, 2283–2288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegué, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2018, 13, 206–220. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer. 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Lim, E.J.; Suh, Y.; Yoo, K.C.; Lee, J.H.; Kim, I.G.; Kim, M.J.; Chang, J.H.; Kang, S.G.; Lee, S.J. Tumor-associated mesenchymal stem-like cells provide extracellular signaling cue for invasiveness of glioblastoma cells. Oncotarget 2017, 8, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattiraju, A.; Sai, K.K.S.; Mintz, A. Glioblastoma Stem Cells and Their Microenvironment. Adv. Exp. Med. Biol. 2017, 1041, 119–140. [Google Scholar] [PubMed]
- Wang, P.; Lan, C.; Xiong, S.; Zhao, X.; Shan, Y.; Hu, R.; Wan, W.; Yu, S.; Liao, B.; Li, G.; et al. HIF1α regulates single differentiated glioma cell dedifferentiation to stem-like cell phenotypes with high tumorigenic potential under hypoxia. Oncotarget 2017, 8, 28074–28092. [Google Scholar] [PubMed] [Green Version]
- Gauden, A.J.; Hunn, A.; Erasmus, A.; Waites, P.; Dubey, A.; Gauden, S.J. Combined modality treatment of newly diagnosed glioblastoma multiforme in a regional neurosurgical centre. J. Clin. Neurosci. 2009, 16, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Day, B.W.; Stringer, B.W.; Boyd, A.W. Eph receptors as therapeutic targets in glioblastoma. Br. J. Cancer 2014, 111, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yamashita, Y.; Noda, H.; Okafuji, T.; Go, M.J.; Tanaka, H. EphA receptor tyrosine kinases interact with co-expressed ephrin—A ligands in cis. Neurosci. Res. 2004, 48, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Saha, N.; Robev, D.; Mason, E.O.; Himanen, J.P.; Nikolov, D.B. Therapeutic potential of targeting the Eph/ephrin signaling complex. Int. J. Biochem. Cell Biol. 2018, 105, 123–133. [Google Scholar] [CrossRef]
- Tognolini, M.; Incerti, M.; Pala, D.; Russo, S.; Castelli, R.; Hassan-Mohamed, I.; Giorgio, C.; Lodola, A. Target hopping as a useful tool for the identification of novel EphA2 protein-protein antagonists. ChemMedChem 2014, 9, 67–72. [Google Scholar] [CrossRef]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef]
- Tu, Y.; He, S.; Fu, J.; Li, G.; Xu, R.; Lu, H.; Deng, J. Expression of EphrinB2 and EphB4 in glioma tissues correlated to the progression of glioma and the prognosis of glioblastoma patients. Clin. Transl. Oncol. 2012, 14, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, W.; Amato, K. Eph receptor tyrosine kinases in cancer stem cells. Cytokine Growth Factor Rev. 2015, 26, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lodola, A.; Giorgio, C.; Incerti, M.; Zanotti, I.; Tognolini, M. Targeting Eph/ephrin system in cancer therapy. Eur. J. Med. Chem. 2017, 142, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Gale, N.W.; Guo, H.; Qian, J.; Petty, A.; Kaspar, J.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.; Hambardzumyan, D.; et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene 2015, 34, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Ferluga, S.; Tomé, C.M.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 is a competitive small molecule Eph-ephrin antagonist blocking in vitro angiogenesis at low micromolar concentrations. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujuguet, P.; Beirinckx, F.; Delachaume, C.; Shenton, D.D.; Huck, J.; van der Aar, E.; Brys, R.; van Rompaey, L.; Wigerinck, P.; Saniere, L. Abstract 1753: GLPG1790: The first ephrin (EPH) receptor tyrosine kinase inhibitor for the treatment of triple negative breast cancer. In Proceedings of the Annual Meeting Cancer Research, San Diego, CA, USA, 5–9 April 2014. [Google Scholar]
- Megiorni, F.; Gravina, G.L.; Camero, S.; Ceccarelli, S.; Del Fattore, A.; Desiderio, V.; Papaccio, F.; McDowell, H.P.; Shukla, R.; Pizzuti, A.; et al. Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells. J. Hematol. Oncol. 2017, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Krusche, B.; Ottone, C.; Clements, M.P.; Johnstone, E.R.; Goetsch, K.; Lieven, H.; Mota, S.G.; Singh, P.; Khadayate, S.; Ashraf, A.; et al. EphrinB2 drives perivascular invasion and proliferation of glioblastoma stem-like cells. eLife 2016, 5, e14845. [Google Scholar] [CrossRef]
- Luchman, H.A.; Stechishin, O.D.; Dang, N.H.; Blough, M.D.; Chesnelong, C.; Kelly, J.J.; Nguyen, S.A.; Chan, J.A.; Weljie, A.M.; Cairncross, J.G.; et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol. 2012, 14, 184–191. [Google Scholar] [CrossRef]
- Mendiburu-Eliçabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of rapamycin against glioblastoma cancer stem cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Bruckheimer, E.M.; Fazenbaker, C.A.; Gallagher, S.; Mulgrew, K.; Fuhrmann, S.; Coffman, K.T.; Walsh, W.; Ready, S.; Cook, K.; Damschroder, M.; et al. Antibody-dependent cell-mediated cytotoxicity effector-enhanced EphA2 agonist monoclonal antibody demonstrates potent activity against human tumors. Neoplasia 2009, 11, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Masuko, K.; Okazaki, S.; Satoh, M.; Tanaka, G.; Ikeda, T.; Torii, R.; Ueda, E.; Nakano, T.; Danbayashi, M.; Tsuruoka, T.; et al. Anti-tumor effect against human cancer xenografts by a fully human monoclonal antibody to a variant 8-epitope of CD44R1 expressed on cancer stem cells. PLoS ONE 2012, 7, e29728. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Marampon, F.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Richardson, P.J.; Patient, L.; Burbidge, S.; et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J. Hematol. Oncol. 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Giorgio, C.; Mancini, A.; Pellegrini, C.; Colapietro, A.; Delle Monache, S.; Maturo, M.G.; Sferra, R.; Chiodelli, P.; et al. UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget 2018, 9, 24347–24363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Mancini, A.; Mattei, C.; Vitale, F.; Marampon, F.; Colapietro, A.; Rossi, G.; Ventura, L.; Vetuschi, A.; Di Cesare, E.; et al. Enhancement of radiosensitivity by the novel anticancer quinolone derivative vosaroxin in preclinical glioblastoma models. Oncotarget 2017, 8, 29865–29886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Tortoreto, M.; Mancini, A.; Addis, A.; Di Cesare, E.; Lenzi, A.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J. Hematol. Oncol. 2014, 7, 46. [Google Scholar] [CrossRef]
- Yuan, J.P.; Wang, L.W.; Qu, A.P.; Chen, J.M.; Xiang, Q.M.; Chen, C.; Sun, S.; Pang, D.; Liu, J.; Li, J. Quantum Dots-Based Quantitative and In Situ Multiple Imaging on Ki67 and Cytokeratin to Improve Ki67 Assessment in Breast Cancer. PLoS ONE 2015, 10, e0122734. [Google Scholar] [CrossRef] [PubMed]
- Sysel, A.M.; Valli, V.E.; Bauer, J.A. Immunohistochemical quantification of the cobalamin transport protein, cell surface receptor and Ki-67 in naturally occurring canine and feline malignant tumors and in adjacent normal tissues. Oncotarget 2015, 6, 2331–2348. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neurooncol. 2017, 132, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Carvalho, B.; Vaz, R.; Linhares, P. Influence of neutrophil-lymphocyte ratio in prognosis of glioblastoma multiforme. J. Neurooncol. 2018, 136, 173–180. [Google Scholar] [CrossRef]
- Saito, T.; Sugiyama, K.; Hama, S.; Yamasaki, F.; Takayasu, T.; Nosaka, R.; Muragaki, Y.; Kawamata, T.; Kurisu, K. Prognostic importance of temozolomide-induced neutropenia in glioblastoma, IDH-wildtype patients. Neurosurg. Rev. 2018, 41, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Curran, C.S.; Bertics, P.J. Eosinophils in glioblastoma biology. J. Neuroinflamm. 2012, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaoka, Y.; Negishi, M.; Katoh, H. Tyrosine kinase activity of EphA2 promotes its S897 phosphorylation and glioblastoma cell proliferation. Biochem. Biophys. Res. Commun. 2018, 499, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef]
- El-Khoueiry, A.; Gitlitz, B.; Cole, S.; Tsao-Wei, D.; Goldkorn, A.; Quinn, D.; Lenz, H.J.; Nieva, J.; Dorff, T.; Oswald, M.; et al. A first-in-human phase I study of sEphB4-HSA in patients with advanced solid tumors with expansion at the maximum tolerated dose (MTD) or recommended phase II dose (RP2D). Eur. J. Cancer 2016, 69, S11. [Google Scholar] [CrossRef]
- Swords, R.T.; Greenberg, P.L.; Wei, A.H.; Durrant, S.; Advani, A.S.; Hertzberg, M.S.; Lewis, I.D.; Rivera, G.; Gratzinger, D.; Fan, A.C.; et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: Results from a phase 1 study. Leuk Res. 2016, 50, 123–131. [Google Scholar] [CrossRef]
- Incerti, M.; Tognolini, M.; Russo, S.; Pala, D.; Giorgio, C.; Hassan-Mohamed, I.; Noberini, R.; Pasquale, E.B.; Vicini, P.; Piersanti, S.; et al. Amino Acid Conjugates of Lithocholic Acid As Antagonists of the EphA2 Receptor. J. Med. Chem. 2013, 56, 2936–2947. [Google Scholar] [CrossRef]
- Castelli, R.; Tognolini, M.; Vacondio, F.; Incerti, M.; Pala, D.; Callegari, D.; Bertoni, S.; Giorgio, C.; Hassan-Mohamed, I.; Zanotti, I.; et al. Δ(5)-Cholenoyl-amino acids as selective and orally available antagonists of the Eph-ephrin system. Eur. J. Med. Chem. 2015, 103, 312–324. [Google Scholar] [CrossRef]
- Lamour, V.; Henry, A.; Kroonen, J.; Nokin, M.J.; von Marschall, Z.; Fisher, L.W.; Chau, T.L.; Chariot, A.; Sanson, M.; Delattre, J.Y.; et al. Targeting osteopontin suppresses glioblastoma stem-like cell character and tumorigenicity in vivo. Int. J. Cancer 2015, 137, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Nakada, M.; Yamada, D.; Nakano, I.; Todo, T.; Ino, Y.; Hoshii, T.; Tadokoro, Y.; Ohta, K.; Ali, M.A.; et al. Strong therapeutic potential of γ-secretase inhibitor MRK003 for CD44-high and CD133-low glioblastoma initiating cells. J. Neurooncol. 2015, 121, 239–250. [Google Scholar] [CrossRef]
- Shankar, A.; Kumar, S.; Iskander, A.S.; Varma, N.R.; Janic, B.; deCarvalho, A.; Mikkelsen, T.; Frank, J.A.; Ali, M.M.; Knight, R.A.; et al. Subcurative radiation significantly increases cell proliferation, invasion, and migration of primary glioblastoma multiforme in vivo. Chin. J. Cancer 2014, 33, 148–158. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. 2012, 11, M111.010744. [Google Scholar] [CrossRef] [PubMed]
- Song, W.S.; Yang, Y.P.; Huang, C.S.; Lu, K.H.; Liu, W.H.; Wu, W.W.; Lee, Y.Y.; Lo, W.L.; Lee, S.D.; Chen, Y.W.; et al. Sox2, a stemness gene, regulates tumor-initiating and drug-resistant properties in CD133-positive glioblastoma stem cells. J. Chin. Med. Assoc. 2016, 79, 538–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagat, M.; Palanichamy, J.K.; Ramalingam, P.; Mudassir, M.; Irshad, K.; Chosdol, K.; Sarkar, C.; Seth, P.; Goswami, S.; Sinha, S.; et al. HIF-2α mediates a marked increase in migration and stemness characteristics in a subset of glioma cells under hypoxia by activating an Oct-4/Sox-2-Mena (INV) axis. Int. J. Biochem. Cell Biol. 2016, 74, 60–71. [Google Scholar] [CrossRef]
- Karmakar, S.; Seshacharyulu, P.; Lakshmanan, I.; Vaz, A.P.; Chugh, S.; Sheinin, Y.M.; Mahapatra, S.; Batra, S.K.; Ponnusamy, M.P. hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget 2017, 8, 14806–14820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC71. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Tilly, H.; Ward, J.H.; Macarthur, D.C.; Lowe, J.; Coyle, B.; Grundy, R.G. CD105 (Endoglin) exerts prognostic effects via its role in the microvascular niche of paediatric high grade glioma. Acta Neuropathol. 2012, 124, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamura, Y.; Takouda, J.; Yoshimoto, K.; Nakashima, K. New aspects of glioblastoma multiforme revealed by similarities between neural and glioblastoma stem cells. Cell Biol. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gállego Pérez-Larraya, J.; Paris, S.; Idbaih, A.; Dehais, C.; Laigle-Donadey, F.; Navarro, S.; Capelle, L.; Mokhtari, K.; Marie, Y.; Sanson, M.; et al. Diagnostic and prognostic value of preoperative combined GFAP, IGFBP-2, and YKL-40 plasma levels in patients with glioblastoma. Cancer 2014, 120, 3972–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Jin, C.; Ma, H.; Huang, M.; Shi, G.P.; Wang, J.; Xiang, M. EphrinB2/EphB4 pathway in postnatal angiogenesis: A potential therapeutic target for ischemic cardiovascular disease. Angiogenesis 2016, 19, 297–309. [Google Scholar] [CrossRef]
- Guo, J.Q.; Zheng, Q.H.; Chen, H.; Chen, L.; Xu, J.B.; Chen, M.Y.; Lu, D.; Wang, Z.H.; Tong, H.F.; Lin, S. Ginsenoside Rg3 inhibition of vasculogenic mimicry in pancreatic cancer through downregulation of VE-cadherin/EphA2/MMP9/MMP2 expression. Int. J. Oncol. 2014, 45, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W.; Zhang, W.Z.; Ge, C.Y.; Zhang, J.T.; Liu, Z.Y.; Fan, Y.Z. Inhibition of tumor vasculogenic mimicry and prolongation of host survival in highly aggressive gallbladder cancers by norcantharidin via blocking the ephrin type a receptor 2/focal adhesion kinase/paxillin signaling pathway. PLoS ONE 2014, 9, e96982. [Google Scholar] [CrossRef] [PubMed]
- Shiuan, E.; Chen, J. Eph Receptor Tyrosine Kinases in Tumor Immunity. Cancer Res. 2016, 76, 6452–6457. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Arthur, A.; Hayball, J.D.; Gronthos, S. EphB and Ephrin-B interactions mediate human mesenchymal stem cell suppression of activated T-cells. Stem Cells Dev. 2013, 22, 2751–2764. [Google Scholar] [CrossRef] [PubMed]
- Ende, G.; Poitz, D.M.; Strasser, R.H.; Jellinghaus, S. The role of the Eph/ephrin-system in atherosclerotic plaque development: A complex puzzle. Cardiovasc. Pathol. 2014, 23, 251. [Google Scholar] [CrossRef] [PubMed]
- Konda, N.; Saeki, N.; Nishino, S.; Ogawa, K. Truncated EphA2 likely potentiates cell adhesion via integrins as well as infiltration and/or lodgment of a monocyte/macrophage cell line in the red pulp and marginal zone of the mouse spleen, where ephrin-A1 is prominently expressed in the vasculature. Histochem. Cell Biol. 2017, 147, 317–339. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gravina, G.L.; Mancini, A.; Colapietro, A.; Delle Monache, S.; Sferra, R.; Vitale, F.; Cristiano, L.; Martellucci, S.; Marampon, F.; Mattei, V.; et al. The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers 2019, 11, 359. https://doi.org/10.3390/cancers11030359
Gravina GL, Mancini A, Colapietro A, Delle Monache S, Sferra R, Vitale F, Cristiano L, Martellucci S, Marampon F, Mattei V, et al. The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers. 2019; 11(3):359. https://doi.org/10.3390/cancers11030359
Chicago/Turabian StyleGravina, Giovanni Luca, Andrea Mancini, Alessandro Colapietro, Simona Delle Monache, Roberta Sferra, Flora Vitale, Loredana Cristiano, Stefano Martellucci, Francesco Marampon, Vincenzo Mattei, and et al. 2019. "The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models" Cancers 11, no. 3: 359. https://doi.org/10.3390/cancers11030359
APA StyleGravina, G. L., Mancini, A., Colapietro, A., Delle Monache, S., Sferra, R., Vitale, F., Cristiano, L., Martellucci, S., Marampon, F., Mattei, V., Beirinckx, F., Pujuguet, P., Saniere, L., Lorenzon, G., van der Aar, E., & Festuccia, C. (2019). The Small Molecule Ephrin Receptor Inhibitor, GLPG1790, Reduces Renewal Capabilities of Cancer Stem Cells, Showing Anti-Tumour Efficacy on Preclinical Glioblastoma Models. Cancers, 11(3), 359. https://doi.org/10.3390/cancers11030359