Integrative Analysis Reveals Subtype-Specific Regulatory Determinants in Triple Negative Breast Cancer


Abstract
:1. Introduction
2. Results
2.1. Selection of Yin and Yang Genes
2.2. Curated Data Sets
2.3. Construction of The Tumor-Specific Lasso Regression Models
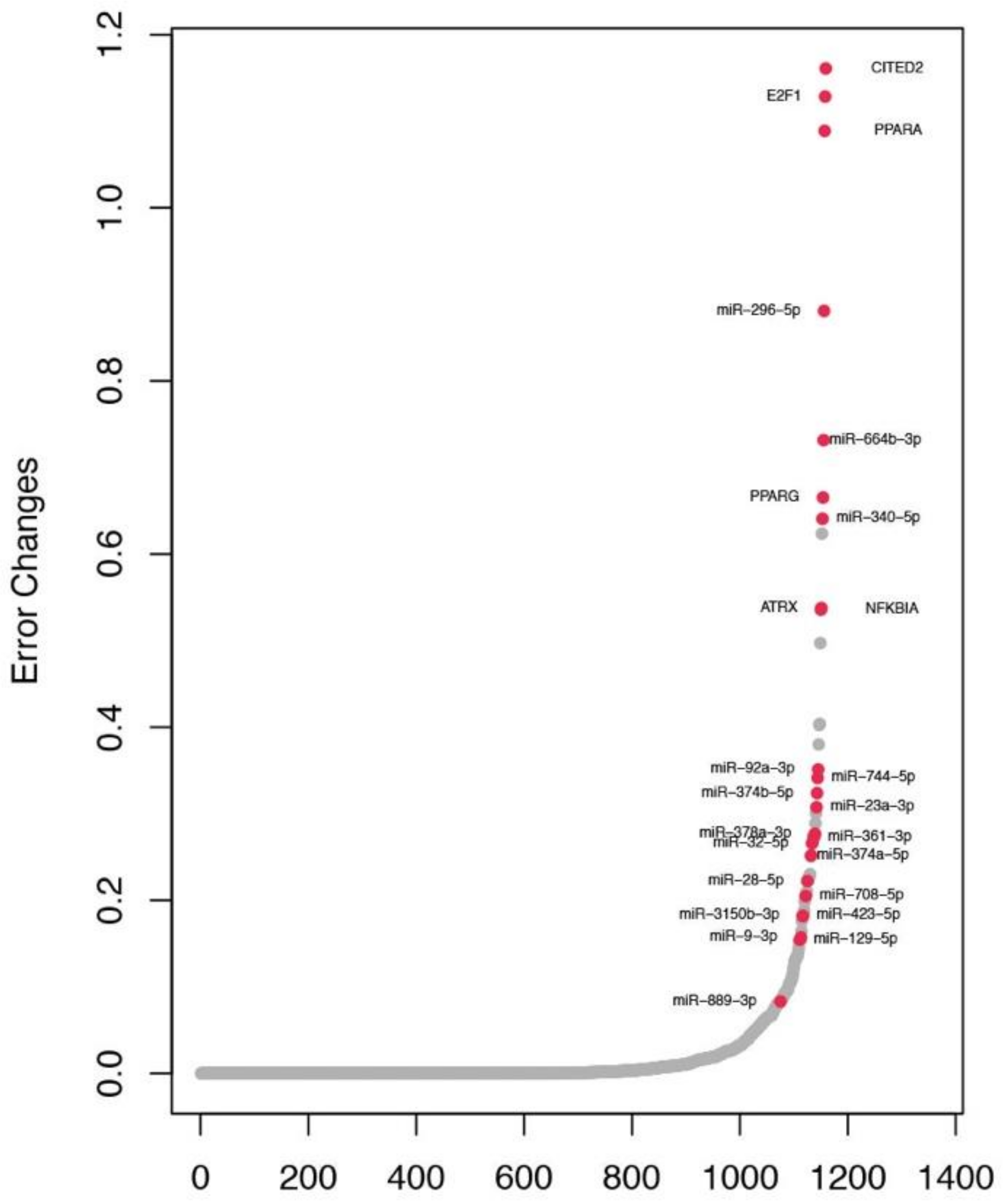
2.4. Identification of Key Regulators in Subtypes
2.5. Identification of Key Regulators in TN Subtype
2.6. Regulatory Network Involving The TN Regulators
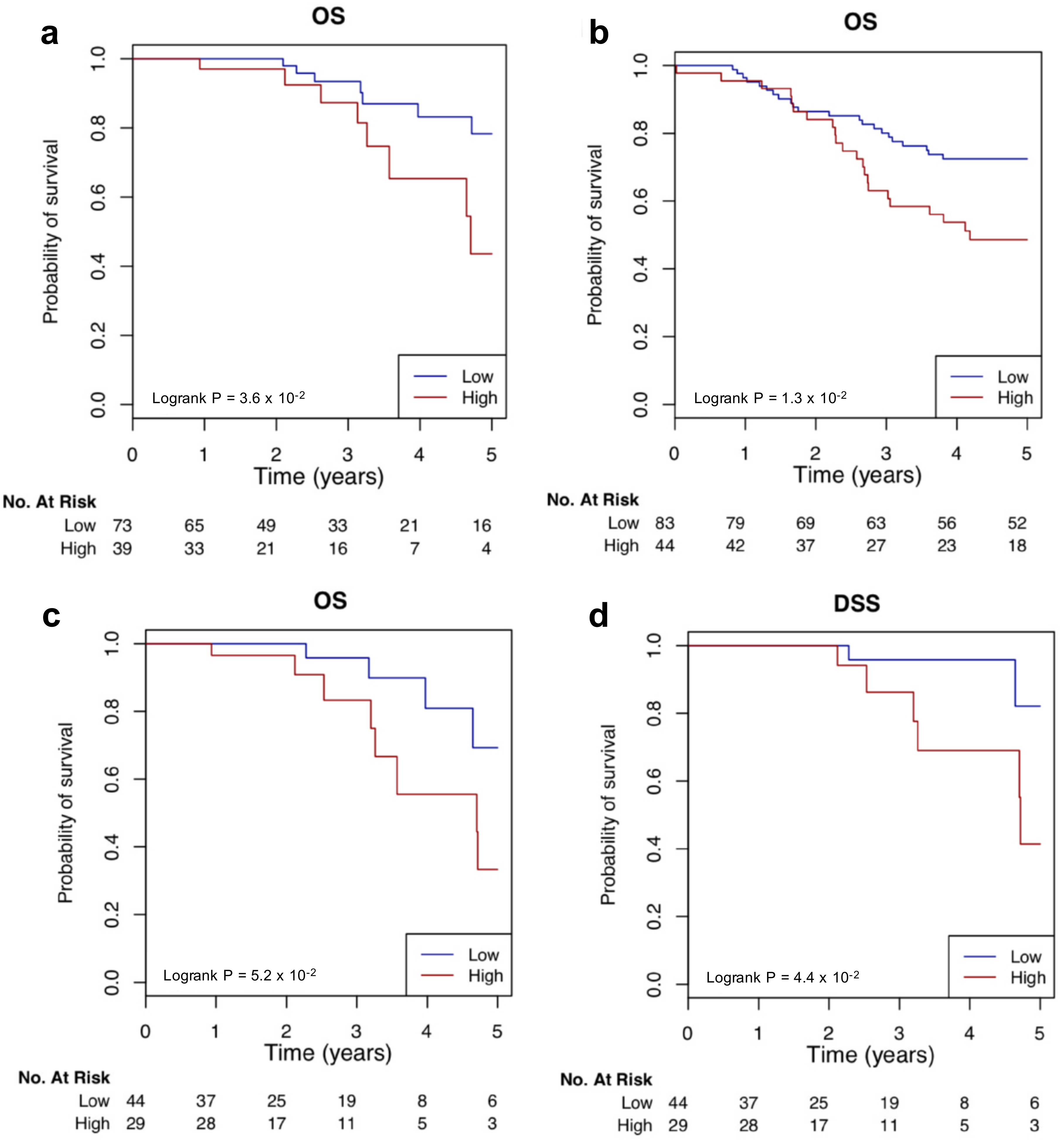
2.7. Clinical Relevance of The TN Regulators
3. Discussion
4. Materials and Methods
4.1. TCGA Data Collection
4.2. Target Prediction for miRNAs and TFs
4.3. Identification of Yin and Yang Genes
4.4. Tumor-Specific Lasso Regression Models
4.5. Feature Selection to Identify the Key Regulators
4.6. Network Construction
4.7. Gene Annotation
4.8. Potential Clinical Outcome Association Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Alizart, M.; Saunus, J.; Cummings, M.; Lakhani, S.R. Molecular classification of breast carcinoma. Diagn. Histopathol. 2012, 18, 97–103. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A. Molecular portraits of human breast tumours. Nature 2000, 406, 747. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef]
- Cava, C.; Bertoli, G.; Castiglioni, I. Integrating genetics and epigenetics in breast cancer: Biological insights, experimental, computational methods and therapeutic potential. BMC Syst. Biol. 2015, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.; Murphy, L.C.; Hu, P. Recurrent copy number alterations in young women with breast cancer. Oncotarget 2018, 9, 11541. [Google Scholar] [CrossRef] [PubMed]
- Vaquerizas, J.M.; Kummerfeld, S.K.; Teichmann, S.A.; Luscombe, N.M. A census of human transcription factors: Function, expression and evolution. Nat. Rev. Genet. 2009, 10, 252. [Google Scholar] [CrossRef]
- Xu, T.; Le, T.D.; Liu, L.; Wang, R.; Sun, B.; Li, J. Identifying Cancer Subtypes from miRNA-TF-mRNA Regulatory Networks and Expression Data. PLoS ONE 2016, 11, e0152792. [Google Scholar] [CrossRef] [PubMed]
- ÖZtemur IslakoĞLu, Y.; Noyan, S.; GÜR DedeoĞLu, B. hsa-miR-301a- and SOX10-dependent miRNA-TF-mRNA regulatory circuits in breast cancer. Turk. J. Biol. 2018, 42. [Google Scholar] [CrossRef]
- Setty, M.; Helmy, K.; Khan, A.A.; Silber, J.; Arvey, A.; Neezen, F.; Agius, P.; Huse, J.T.; Holland, E.C.; Leslie, C.S. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 2012, 8, 605. [Google Scholar] [CrossRef]
- Huang, S.; Kurubanjerdjit, N.; Xu, W. Two opposing effects (Yin and Yang) determine cancer progression. Criti. Rev. Oncog. 2017, 22, 143–155. [Google Scholar] [CrossRef]
- Xu, W.; Jia, G.; Cai, N.; Huang, S.; Davie, J.R.; Pitz, M.; Banerji, S.; Murphy, L. A 16 Y in Y ang gene expression ratio signature for ER+/node− breast cancer. Int. J. Cancer 2017, 140, 1413–1424. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef]
- Cheneby, J.; Gheorghe, M.; Artufel, M.; Mathelier, A.; Ballester, B. ReMap 2018: An updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2018, 46, D267–D275. [Google Scholar] [CrossRef]
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2. 0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2013, 42, D92–D97. [Google Scholar] [CrossRef]
- Denechaud, P.-D.; Fajas, L.; Giralt, A. E2F1, a novel regulator of metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef]
- Tien, E.S.; Davis, J.W.; Heuvel, J.P.V. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor α coregulator. J. Biol. Chem. 2004, 279, 24053–24063. [Google Scholar] [CrossRef]
- Lau, W.M.; Doucet, M.; Huang, D.; Weber, K.L.; Kominsky, S.L. CITED2 modulates estrogen receptor transcriptional activity in breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 437, 261–266. [Google Scholar] [CrossRef]
- Minemura, H.; Takagi, K.; Sato, A.; Takahashi, H.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Sasano, H.; Suzuki, T. CITED 2 in breast carcinoma as a potent prognostic predictor associated with proliferation, migration and chemoresistance. Cancer Sci. 2016, 107, 1898–1908. [Google Scholar] [CrossRef]
- Unterbruner, K.; Matthes, F.; Schilling, J.; Nalavade, R.; Weber, S.; Winter, J.; Krauß, S. MicroRNAs miR-19, miR-340, miR-374 and miR-542 regulate MID1 protein expression. PLoS ONE 2018, 13, e0190437. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, X.; Shen, Y.; Chen, L.; Xu, C.; Zhao, H.; Wu, Y.; Zhang, Q.; Zhong, J.; Tang, Z. MicroRNA-32 promotes calcification in vascular smooth muscle cells: Implications as a novel marker for coronary artery calcification. PLoS ONE 2017, 12, e0174138. [Google Scholar] [CrossRef]
- Chang, J.T.; Wang, F.; Chapin, W.; Huang, R.S. Identification of MicroRNAs as breast cancer prognosis markers through the cancer genome atlas. PLoS ONE 2016, 11, e0168284. [Google Scholar] [CrossRef]
- Lee, K.-H.; Lin, F.-C.; Hsu, T.-I.; Lin, J.-T.; Guo, J.-H.; Tsai, C.-H.; Lee, Y.-C.; Lee, Y.-C.; Chen, C.-L.; Hsiao, M. MicroRNA-296-5p (miR-296-5p) functions as a tumor suppressor in prostate cancer by directly targeting Pin1. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 2055–2066. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Narrandes, S.; Huang, S.; Murphy, L.; Xu, W. The exploration of contrasting pathways in Triple Negative Breast Cancer (TNBC). BMC Cancer 2018, 18, 22. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346. [Google Scholar] [CrossRef]
- Li, Y.; Liang, M.; Zhang, Z. Regression analysis of combined gene expression regulation in acute myeloid leukemia. PLoS Comput. Biol. 2014, 10, e1003908. [Google Scholar] [CrossRef]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K. The role of PPARs in cancer. PPAR Research 2008, 2008, 102737. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181. [Google Scholar] [CrossRef]
- Lianggeng, X.; Baiwu, L.; Maoshu, B.; Jiming, L.; Youshan, L. Impact of Interaction Between PPAR Alpha and PPAR Gamma on Breast Cancer Risk in the Chinese Han Population. Clin. Breast Cancer 2017, 17, 336–340. [Google Scholar] [CrossRef]
- Bredel, M.; Kim, H.; Nanda, T.; Scholtens, D.; Robe, P.; Branimir, S.; Bonner, J. Deletion of the tumor suppressor NFKBIA in triple-negative breast cancer. Int. J.Radiat. Oncol. Biol. Phys. 2013, 87, S98. [Google Scholar] [CrossRef]
- Zhong, S.; Chen, X.; Wang, D.; Zhang, X.; Shen, H.; Yang, S.; Lv, M.; Tang, J.; Zhao, J. MicroRNA expression profiles of drug-resistance breast cancer cells and their exosomes. Oncotarget 2016, 7, 19601. [Google Scholar] [CrossRef]
- Nilsson, S.; Möller, C.; Jirström, K.; Lee, A.; Busch, S.; Lamb, R.; Landberg, G. Downregulation of miR-92a is associated with aggressive breast cancer features and increased tumour macrophage infiltration. PLoS ONE 2012, 7, e36051. [Google Scholar] [CrossRef]
- Luan, Q.; Zhang, B.; Li, X.; Guo, M. MiR-129-5p is downregulated in breast cancer cells partly due to promoter H3K27m3 modification and regulates epithelial-mesenchymal transition and multi-drug resistance. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4257–4265. [Google Scholar]
- Meng, R.; Fang, J.; Yu, Y.; Hou, L.; Chi, J.; Chen, A.; Zhao, Y.; Cao, X. miR-129-5p suppresses breast cancer proliferation by targeting CBX4. Neoplasma 2018, 65, 572. [Google Scholar] [CrossRef]
- Meng, Q.; Xiang, L.; Fu, J.; Chu, X.; Wang, C.; Yan, B. Transcriptome profiling reveals miR-9-3p as a novel tumor suppressor in gastric cancer. Oncotarget 2017, 8, 37321. [Google Scholar] [CrossRef]
- Barbano, R.; Pasculli, B.; Rendina, M.; Fontana, A.; Fusilli, C.; Copetti, M.; Castellana, S.; Valori, V.M.; Morritti, M.; Graziano, P. Stepwise analysis of MIR9 loci identifies miR-9-5p to be involved in Oestrogen regulated pathways in breast cancer patients. Sci. Rep. 2017, 7, 45283. [Google Scholar] [CrossRef]
- Krist, B.; Florczyk, U.; Pietraszek-Gremplewicz, K.; Józkowicz, A.; Dulak, J. The role of miR-378a in metabolism, angiogenesis, and muscle biology. Int. J. Endocrinol. 2015, 2015, 281756. [Google Scholar] [CrossRef]
- Monteleone, N.J.; Lutz, C.S. miR-708-5p: A microRNA with emerging roles in cancer. Oncotarget 2017, 8, 71292. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wei, W.; Sarkar, F.H. miR-23a, a critical regulator of “migR” ation and metastasis in colorectal cancer. Cancer Discov. 2012, 2, 489–491. [Google Scholar] [CrossRef]
- Fackler, M.J.; McVeigh, M.; Mehrotra, J.; Blum, M.A.; Lange, J.; Lapides, A.; Garrett, E.; Argani, P.; Sukumar, S. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. 2004, 64, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Nimmrich, I.; Sieuwerts, A.M.; Meijer-van Gelder, M.E.; Schwope, I.; Bolt-de Vries, J.; Harbeck, N.; Koenig, T.; Hartmann, O.; Kluth, A.; Dietrich, D. DNA hypermethylation of PITX2 is a marker of poor prognosis in untreated lymph node-negative hormone receptor-positive breast cancer patients. Breast Cancer Res. Treat. 2008, 111, 429–437. [Google Scholar] [CrossRef]
- Lu, Y.; Harada, M.; Kamijo, Y.; Nakajima, T.; Tanaka, N.; Sugiyama, E.; Kyogashima, M.; Gonzalez, F.J.; Aoyama, T. Peroxisome proliferator-activated receptor α attenuates high-cholesterol diet-induced toxicity and pro-thrombotic effects in mice. Arch. Toxicol. 2019, 93, 149–161. [Google Scholar] [CrossRef]
- Ruan, B.; Zhang, B.; Chen, A.; Yuan, L.; Liang, J.; Wang, M.; Zhang, Z.; Fan, J.; Yu, X.; Zhang, X. Cholesterol inhibits entotic cell-in-cell formation and actomyosin contraction. Biochem. Biophys. Res. Commun. 2018, 495, 1440–1446. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, N.; Kokuryo, T.; Oda, K.; Senga, T.; Yokoyama, Y.; Nagino, M.; Nimura, Y.; Hamaguchi, M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009, 100, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Cappello, P.; Blaser, H.; Gorrini, C.; Lin, D.; Elia, A.; Wakeham, A.; Haider, S.; Boutros, P.; Mason, J.; Miller, N. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene 2014, 33, 2375. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619. [Google Scholar] [CrossRef]
- O’Regan, R.M.; Nahta, R. Targeting forkhead box M1 transcription factor in breast cancer. Biochemical pharmacology 2018. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Hamurcu, Z.; Delibaşı, N.; Nalbantoglu, U.; Sener, E.F.; Nurdinov, N.; Tascı, B.; Taheri, S.; Özkul, Y.; Donmez-Altuntas, H.; Canatan, H. FOXM1 plays a role in autophagy by transcriptionally regulating Beclin-1 and LC3 genes in human triple-negative breast cancer cells. J. Mol. Med. 2019, 97, 491–508. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef]
- Gerstein, M.B.; Kundaje, A.; Hariharan, M.; Landt, S.G.; Yan, K.-K.; Cheng, C.; Mu, X.J.; Khurana, E.; Rozowsky, J.; Alexander, R. Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2014, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47–e47. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef]
- Schröder, M.S.; Culhane, A.C.; Quackenbush, J.; Haibe-Kains, B. survcomp: An R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics 2011, 27, 3206–3208. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.Y.; Wei, L.J. The robust inference for the Cox proportional hazards model. J. Am. Stat. Assoc. 1989, 84, 1074–1078. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Platform/Category | Database | All Data | Common Data |
|---|---|---|---|---|
| Subtype annotation | IHC | TCGA | 694 tumors: 424 LumA, 121 LumB, 37 Her2+, 112 TN | 391 tumors: 239 LumA, 74 LumB, 21 Her2+, 57 TN |
| mRNA expression 1 | Illumina Hiseq 2000 | TCGA | 17675 genes × 1104 tumors | 3008 genes × 391 tumors |
| miRNA expression 2 | Illumina Hiseq 2000 | TCGA | 600 miRNAs × 756 tumors | 362 miRNAs × 391 tumors |
| Copy number variation (CNV) | Affymetrix SNP 6.0 | TCGA | 24776 genes × 1080 tumors | 3008 genes × 391 tumors |
| DNA methylation (DM) | Illumina Infinium HumanMethylation450 | TCGA | 26586 genes × 790 tumors | 3008 genes × 391 tumors |
| TF-target | Sequence-based | TRRUST v2.0 | 2492 genes × 795 TFs | 3008 genes × 795 TFs |
| miRNA-target | Sequence-based | starBase v3.0 | 15168 targets × 618 miRNAs | 3008 genes × 362 miRNAs |
| Subtype | Number of Regulators | Regulator |
|---|---|---|
| Her2+, LumA, LumB, TN | 9 | E2F1, CITED2, hsa-miR-374b-5p, hsa-miR-32-5p, hsa-miR-3150b-3p, hsa-miR-361-3p, hsa-miR-340-5p, hsa-miR-664b-3p, hsa-miR-296-5p |
| Her2+, LumA, LumB | 1 | CEBPZ |
| LumA, LumB, TN | 5 | ATRX, PPARG, hsa-miR-374a-5p, hsa-miR-92a-3p, hsa-miR-744-5p |
| Her2+, LumA, TN | 3 | PPARA, hsa-miR-889-3p, hsa-miR-423-5p |
| LumA, LumB | 1 | hsa-miR-429 |
| LumA, TN | 5 | NFKBIA, hsa-miR-23a-3p, hsa-miR-378a-3p, hsa-miR-129-5p, hsa-miR-28-5p |
| Her2+, LumB | 1 | hsa-miR-760 |
| Her2+, TN | 1 | hsa-miR-708-5p |
| LumA | 1 | hsa-miR-181b-5p |
| LumB | 3 | hsa-miR-655-3p, hsa-miR-654-5p, hsa-miR-625-5p |
| TN | 1 | hsa-miR-9-3p |
| Regulators | FDR | Expression 1 | Targets 2 | Enriched Pathways 3 | Ratio 4 | Enrichment FDR 5 |
|---|---|---|---|---|---|---|
| E2F1 | 0.00 | Up | 48 | REACTOME_CELL_CYCLE | 19/241 | 4.43 × 10–12 |
| PID_FOXM1_PATHWAY | 9/40 | 1.70 × 10–11 | ||||
| KEGG_PATHWAYS_IN_CANCER | 9/328 | 2.09 × 10–5 | ||||
| CITED2 | 0.00 | Down | 2 | REACTOME_EXTRACELLULAR_MATRIX_ORGANIZATION | 2/87 | 8.03 × 10–4 |
| REACTOME_DEGRADATION_OF_THE_EXTRACELLULAR_MATRIX | 2/29 | 1.74 × 10–4 | ||||
| NABA_ECM_REGULATORS | 2/238 | 4.04 × 10–3 | ||||
| PPARA | 0.00 | Down | 20 | REACTOME_METABOLISM_OF_LIPIDS_AND_LIPOPROTEINS | 11/478 | 8.87 × 10–8 |
| BIOCARTA_PPARA_PATHWAY | 4/58 | 2.08 × 10–4 | ||||
| KEGG_PATHWAYS_IN_CANCER | 4/328 | 3.76 × 10–2 | ||||
| PPARG | 0.02 | Down | 29 | KEGG_PATHWAYS_IN_CANCER | 9/328 | 5.73 × 10–5 |
| REACTOME_HORMONE_SENSITIVE_LIPASE_HSL_MEDIATED_TRIACYLGLYCEROL_HYDROLYSIS | 3/13 | 5.48 × 10–4 | ||||
| REACTOME_METABOLISM_OF_LIPIDS_AND_LIPOPROTEINS | 8/478 | 3.77 × 10–3 | ||||
| BIOCARTA_PPARA_PATHWAY | 3/58 | 1.92 × 10–2 | ||||
| REACTOME_DEGRADATION_OF_THE_EXTRACELLULAR_MATRIX | 2/29 | 4.81 × 10–2 | ||||
| ATRX | 0.05 | Down | 2 | BIOCARTA_AHSP_PATHWAY | 2/13 | 1.97 × 10–6 |
| NFKBIA | 0.05 | Down | 3 | REACTOME_DEGRADATION_OF_THE_EXTRACELLULAR_MATRIX | 3/29 | 9.65 × 10–7 |
| REACTOME_EXTRACELLULAR_MATRIX_ORGANIZATION | 3/87 | 1.40 × 10–5 | ||||
| KEGG_PATHWAYS_IN_CANCER | 2/328 | 1.37 × 10–2 | ||||
| hsa-miR-374b-5p | 0.00 | Down | 589 | KEGG_PATHWAYS_IN_CANCER | 28/328 | 4.84 × 10–2 |
| hsa-miR-32-5p | 0.00 | Up | 523 | KEGG_FOCAL_ADHESION | 19/201 | 4.45 × 10–2 |
| NABA_CORE_MATRISOME | 22/275 | 4.74 × 10–2 | ||||
| hsa-miR-361-3p | 0.00 | - | 488 | KEGG_FOCAL_ADHESION | 23/201 | 8.25 × 10–5 |
| KEGG_PATHWAYS_IN_CANCER | 27/238 | 2.41 × 10–3 | ||||
| PID_AVB3_INTEGRIN_PATHWAY | 11/75 | 6.61 × 10–3 | ||||
| hsa-miR-340-5p | 0.00 | Up | 927 | KEGG_PATHWAYS_IN_CANCER | 54/328 | 5.10 × 10–9 |
| PID_AVB3_INTEGRIN_PATHWAY | 19/75 | 2.64 × 10−5 | ||||
| KEGG_FOCAL_ADHESION | 31/201 | 1.47 × 10–4 | ||||
| BIOCARTA_PPARA_PATHWAY | 15/58 | 1.47 × 10–4 | ||||
| REACTOME_EXTRACELLULAR_MATRIX_ORGANIZATION | 13/87 | 4.14 × 10–2 | ||||
| PID_FOXM1_PATHWAY | 8/40 | 4.41 × 10–2 | ||||
| hsa-miR-664b-3p | 0.00 | Down | 431 | KEGG_FOCAL_ADHESION | 18/201 | 9.09 × 10–3 |
| BIOCARTA_PPARA_PATHWAY | 9/58 | 9.09 × 10–3 | ||||
| KEGG_CYTOKINE_CYTOKINE_RECEPTOR_INTERACTION | 19/267 | 4.57 × 10–2 | ||||
| hsa-miR-296-5p | 0.00 | Down | 353 | KEGG_LEUKOCYTE_TRANSENDOTHELIAL_MIGRATION | 12/118 | 6.14 × 10–3 |
| hsa-miR-423-5p | 0.00 | - | 590 | KEGG_PATHWAYS_IN_CANCER | 30/328 | 1.43 × 10–3 |
| PID_AVB3_INTEGRIN_PATHWAY | 11/75 | 1.94 × 10–2 | ||||
| REACTOME_GROWTH_HORMONE_RECEPTOR_SIGNALING | 6/24 | 3.31 × 10–2 | ||||
| hsa-miR-92a-3p | 0.00 | Down | 524 | KEGG_FOCAL_ADHESION | 19/201 | 4.47 × 10–2 |
| hsa-miR-129-5p | 0.02 | Down | 775 | KEGG_PATHWAYS_IN_CANCER | 37/328 | 1.90 × 10–4 |
| KEGG_FOCAL_ADHESION | 26/201 | 5.83 × 10–4 | ||||
| REACTOME_EXTRACELLULAR_MATRIX_ORGANIZATION | 13/87 | 1.41 × 10–2 | ||||
| PID_AVB3_INTEGRIN_PATHWAY | 12/75 | 1.41 × 10–2 | ||||
| BIOCARTA_PPARA_PATHWAY | 10/58 | 1.92 × 10–2 | ||||
| hsa-miR-744-5p | 0.03 | - | 221 | KEGG_PATHWAYS_IN_CANCER | 15/328 | 1.26 × 10–2 |
| KEGG_FOCAL_ADHESION | 12/201 | 1.20 × 10–2 | ||||
| REACTOME_CELL_SURFACE_INTERACTIONS_AT_THE_VASCULAR_WALL | 7/91 | 2.57 × 10–2 | ||||
| hsa-miR-9-3p | 0.04 | - | 236 | KEGG_PATHWAYS_IN_CANCER | 14/328 | 2.98 × 10–2 |
| BIOCARTA_PPARA_PATHWAY | 6/58 | 2.98 × 10–2 | ||||
| KEGG_MAPK_SIGNALING_PATHWAY | 12/267 | 3.19 × 10–2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Xu, W.; Hu, P.; Lakowski, T.M. Integrative Analysis Reveals Subtype-Specific Regulatory Determinants in Triple Negative Breast Cancer. Cancers 2019, 11, 507. https://doi.org/10.3390/cancers11040507
Huang S, Xu W, Hu P, Lakowski TM. Integrative Analysis Reveals Subtype-Specific Regulatory Determinants in Triple Negative Breast Cancer. Cancers. 2019; 11(4):507. https://doi.org/10.3390/cancers11040507
Chicago/Turabian StyleHuang, Shujun, Wayne Xu, Pingzhao Hu, and Ted M. Lakowski. 2019. "Integrative Analysis Reveals Subtype-Specific Regulatory Determinants in Triple Negative Breast Cancer" Cancers 11, no. 4: 507. https://doi.org/10.3390/cancers11040507