Histopathological Imaging–Environment Interactions in Cancer Modeling


Abstract
:1. Introduction
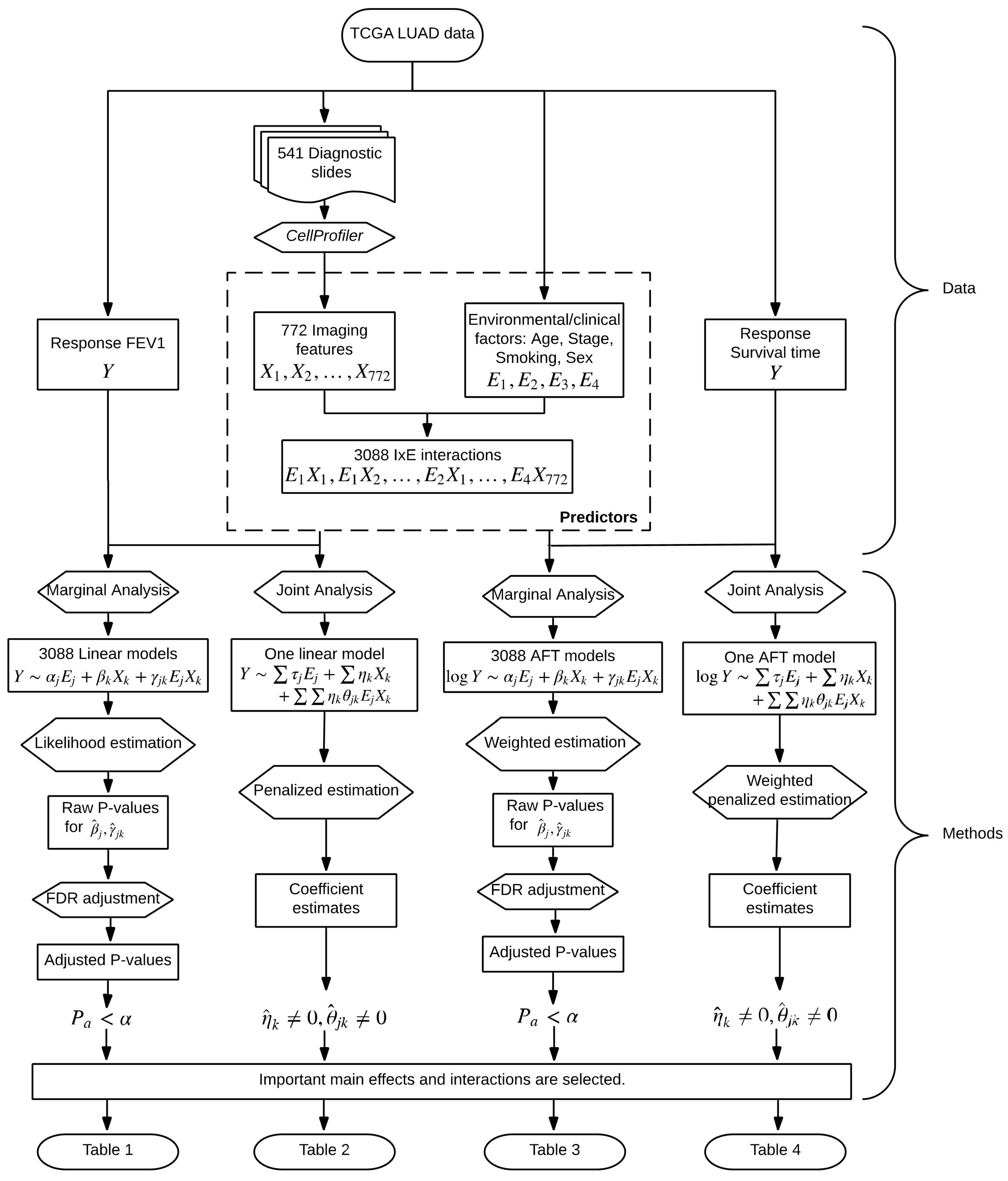
2. Data
3. Methods
3.1. Marginal Analysis
- (a)
- For and , consider the linear regression modelwhere and respectively represent the main effects of the jth clinical/environmental factor and the kth imaging feature, is the interactive effect, and ϵ is the random error. A total of J × K models are built.
- (b)
- As each model has a low dimension, estimates can be obtained using standard likelihood based approaches and existing software. p-values can be obtained accordingly.
- (c)
- Interactions (and main effects) with small p-values are identified as important. When more definitive conclusions are needed, the false discovery rate (FDR) or Bonferroni approach can be applied.
3.2. Joint Analysis
- (a)
- Consider the joint modelwhere and are the main effects of the jth environmental factor and the kth imaging feature, respectively, and the product of and corresponds to the interaction.
- (b)
- For estimation, consider the Lasso penalizationwhere , and are tuning parameters. In numerical study, we select the tuning parameters using the extended Bayesian information criterion [32].
- (c)
- Interactions (and main effects) with nonzero estimates are identified as being associated with the outcome.
3.3. Accommodating Survival Outcomes
4. Results
4.1. Analysis of FEV1
4.1.1. Marginal Analysis
4.1.2. Joint Analysis
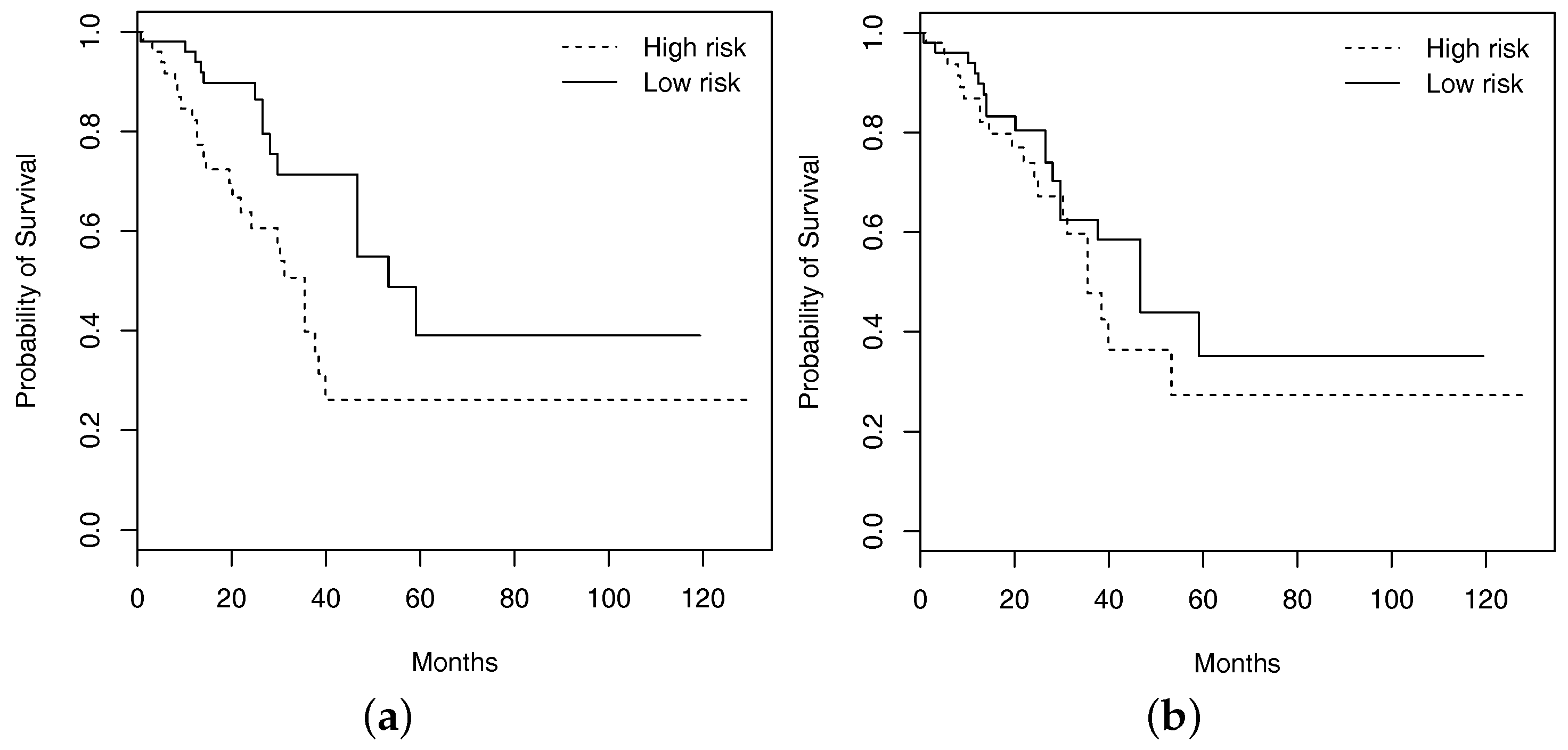
4.2. Analysis of Overall Survival
4.2.1. Marginal Analysis
4.2.2. Joint Analysis
4.3. Simulation
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fass, L. Imaging and cancer: A review. Mol. Oncol. 2008, 2, 115–152. [Google Scholar] [CrossRef] [Green Version]
- Benzaquen, J.; Boutros, J.; Marquette, C.; Delingette, H.; Hofman, P. Lung cancer screening, towards a multidimensional approach: Why and how? Cancers 2019, 11, 212. [Google Scholar] [CrossRef]
- Gurcan, M.N.; Boucheron, L.; Can, A.; Madabhushi, A.; Rajpoot, N.; Yener, B. Histopathological image analysis: A review. IEEE Rev. Biomed. Eng. 2009, 2, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Failmezger, H.; Rueda, O.M.; Ali, H.R.; Gräf, S.; Chin, S.F.; Schwarz, R.F.; Curtis, C.; Dunning, M.J.; Bardwell, H.; et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Sci. Transl. Med. 2012, 4, 157ra143. [Google Scholar] [CrossRef]
- Tabesh, A.; Teverovskiy, M.; Pang, H.Y.; Kumar, V.P.; Verbel, D.; Kotsianti, A.; Saidi, O. Multifeature prostate cancer diagnosis and Gleason grading of histological images. IEEE Trans. Med. Imaging 2007, 26, 1366–1378. [Google Scholar] [CrossRef]
- Zhong, T.; Wu, M.; Ma, S. Examination of independent prognostic power of gene expressions and histopathological imaging features in cancer. Cancers 2019, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xing, F.; Su, H.; Stromberg, A.; Yang, L. Novel image markers for non-small cell lung cancer classification and survival prediction. BMC Bioinform. 2014, 15, 310. [Google Scholar] [CrossRef]
- Hunter, D.J. Gene–environment interactions in human diseases. Nat. Rev. Genet. 2005, 6, 287–298. [Google Scholar] [CrossRef]
- Luo, X.; Zang, X.; Yang, L.; Huang, J.; Liang, F.; Rodriguez-Canales, J.; Wistuba, I.I.; Gazdar, A.; Xie, Y.; Xiao, G. Comprehensive computational pathological image analysis predicts lung cancer prognosis. J. Thorac. Oncol. 2017, 12, 501–509. [Google Scholar] [CrossRef]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 12474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bychkov, D.; Linder, N.; Turkki, R.; Nordling, S.; Kovanen, P.E.; Verrill, C.; Walliander, M.; Lundin, M.; Haglund, C.; Lundin, J. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci. Rep. 2018, 8, 3395. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yao, J.; Zhu, F.; Huang, J. Wsisa: Making survival prediction from whole slide histopathological images. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 7234–7242. [Google Scholar]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Boolell, V.; Alamgeer, M.; Watkins, D.; Ganju, V. The evolution of therapies in non-small cell lung cancer. Cancers 2015, 7, 1815–1846. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, A.; Ringner, M.; Lauss, M.; Botling, J.; Micke, P.; Planck, M.; Staaf, J. Genomic and transcriptional alterations in lung adenocarcinoma in relation to smoking history. Clin. Cancer Res. 2014, 20, 4912–4924. [Google Scholar] [CrossRef]
- Li, X.; Shi, Y.; Yin, Z.; Xue, X.; Zhou, B. An eight-miRNA signature as a potential biomarker for predicting survival in lung adenocarcinoma. J. Transl. Med. 2014, 12, 159. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Data Portal Website. Available online: https://portal.gdc.cancer.gov/projects/TCGA-LUAD (accessed on 23 April 2019).
- Yu, K.; Berry, G.J.; Rubin, D.L.; Re, C.; Altman, R.B.; Snyder, M. Association of omics features with histopathology patterns in lung adenocarcinoma. Cell Syst. 2017, 5, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yao, J.; Luo, X.; Xiao, G.; Xie, Y.; Gazdar, A.F.; Huang, J. Lung cancer survival prediction from pathological images and genetic data-an integration study. In Proceedings of the 2016 IEEE 13th International Symposium on Biomedical Imaging, Prague, Czech Republic, 13–16 April 2016. [Google Scholar]
- Sun, D.; Li, A.; Tang, B.; Wang, M. Integrating genomic data and pathological images to effectively predict breast cancer clinical outcome. Comput. Methods Progr. Biomed. 2018, 161, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Soliman, K. CellProfiler: Novel automated image segmentation procedure for super-resolution microscopy. Biol. Proced. Online 2015, 17, 11. [Google Scholar] [CrossRef]
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015, 517, 489–492. [Google Scholar] [CrossRef]
- Nordquist, L.; Simon, G.; Cantor, A.; Alberts, W.; Bepler, G. Improved survival in never-smokers vs current smokers with primary adenocarcinoma of the lung. Chest 2004, 126, 347–351. [Google Scholar] [CrossRef]
- Bryant, A.; Cerfolio, R. Differences in epidemiology, histology, and survival between cigarette smokers and never-smokers who develop non-small cell lung cancer. Chest 2008, 132, 185–192. [Google Scholar] [CrossRef]
- Landi, M.; Dracheva, T.; Rotunno, M.; Figueroa, J.; Liu, H.; Dasgupta, A.; Mann, F.; Fukuoka, J.; Hames, M.; Bergen, A.; et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef]
- Wu, M.; Zang, Y.; Zhang, S.; Huang, J.; Ma, S. Accommodating missingness in environmental measurements in gene–environment interaction analysis. Genet. Epidemiol. 2017, 41, 523–554. [Google Scholar] [CrossRef]
- Wu, M.; Ma, S. Robust genetic interaction analysis. Brief. Bioinform. 2018. [Google Scholar] [CrossRef]
- Zhang, Y.; Dai, Y.; Zheng, T.; Ma, S. Risk factors of non-Hodgkin’s lymphoma. Expert Opin. Med. Diagn. 2011, 5, 539–550. [Google Scholar] [CrossRef]
- Witten, D.M.; Tibshirani, R. Survival analysis with high-dimensional covariates. Stat. Methods Med. Res. 2010, 19, 29–51. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, M.; Zhang, Q.; Ma, S. Robust identification of gene–environment interactions for prognosis using a quantile partial correlation approach. Genomics 2018. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z. Extended Bayesian information criteria for model selection with large model spaces. Biometrika 2008, 95, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ma, S.; Xie, H. Regularized estimation in the accelerated failure time model with high-dimensional covariates. Biometrics 2006, 62, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.H.; Li, W.; Zhu, J. Variable selection with the strong heredity constraint and its oracle property. J. Am. Stat. Assoc. 2010, 105, 354–364. [Google Scholar] [CrossRef]
- Bien, J.; Taylor, J.; Tibshirani, R. A lasso for hierarchical interactions. Ann. Stat. 2013, 41, 1111–1141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Zhang, Y.; Lan, Q.; Rothman, N.; Zheng, T.; Ma, S. Identification of gene–environment interactions in cancer studies using penalization. Genomics 2013, 102, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosny, A.; Parmar, C.; Quackenbush, J.; Schwartz, L.H.; Aerts, H. Artificial intelligence in radiology. Nat. Rev. Cancer 2018, 18, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Thrall, J.; Li, X.; Li, Q.; Cruz, C.; Do, S.; Dreyer, K.; Brink, J. Artificial intelligence and machine learning in radiology: Opportunities, challenges, pitfalls, and criteria for success. J. Am. Coll. Radiol. 2018, 15, 504–508. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Feature Group | Feature Name | Estimate | Pr | |
|---|---|---|---|---|
| Geometry | AreaShape_Zernike_2_2 | Main | 0.270 | 0.002 |
| Geometry | AreaShape_Zernike_5_3 | Main | −0.319 | 0.001 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_9_9 | Main | −0.259 | 0.004 |
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_7_1 | Main | −0.249 | 0.005 |
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_8_6 | Main | −0.272 | 0.003 |
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_01 | Main | 0.280 | 0.002 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_8 | Main | −0.251 | 0.005 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_9_1 | Main | −0.259 | 0.004 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Center_Y | Sex | 0.291 | 0.002 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_2 | Sex | 0.304 | 0.001 |
| Geometry | StDev_Identifyhemasub2_Location_Center_Y | Sex | 0.294 | 0.002 |
| Feature Group | Feature Name | Main | Age | Stage | Smoking | Sex |
|---|---|---|---|---|---|---|
| −0.049 | −0.052 | −0.002 | 0.006 | |||
| Geometry | AreaShape_Zernike_2_2 | 0.163 | 0.040 | −0.014 | −0.185 | |
| Geometry | AreaShape_Zernike_5_3 | −0.053 | ||||
| Geometry | AreaShape_Zernike_6_0 | −0.034 | ||||
| Texture | Granularity_10_ImageAfterMath | 0.137 | 0.110 | −0.020 | 0.064 | |
| Geometry | Location_Center_X | 0.002 | ||||
| Geometry | Mean_Identifyeosinprimarycytoplasm_Location_Center_X | 0.005 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_7_1 | −0.127 | −0.073 | 0.072 | 0.003 | |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_2 | −0.170 | −0.083 | 0.188 | ||
| Texture | StDev_Identifyhemasub2_Granularity_6_ImageAfterMath | −0.029 | ||||
| Texture | Texture_AngularSecondMoment_ImageAfterMath_3_00 | −0.044 | ||||
| Texture | Texture_AngularSecondMoment_ImageAfterMath_3_03 | −0.010 |
| Feature Group | Feature Name | Estimate | Pr | Pa | |
|---|---|---|---|---|---|
| Holistic | Threshold_FinalThreshold_Identifyeosinprimarycytoplasm | Main | −0.301 | 0 | 0.095 |
| Holistic | Threshold_OrigThreshold_Identifyeosinprimarycytoplasm | Main | −0.301 | 0 | 0.095 |
| Holistic | Threshold_WeightedVariance_identifyhemaprimarynuclei | Main | −0.360 | 0 | 0.077 |
| Geometry | AreaShape_Area | Smoking | 0.253 | 0.004 | 0.078 |
| Geometry | AreaShape_MaximumRadius | Smoking | 0.266 | 0.004 | 0.074 |
| Geometry | AreaShape_MeanRadius | Smoking | 0.265 | 0.005 | 0.079 |
| Geometry | AreaShape_MedianRadius | Smoking | 0.266 | 0.005 | 0.079 |
| Geometry | AreaShape_MinFeretDiameter | Smoking | 0.257 | 0.003 | 0.073 |
| Geometry | AreaShape_MinorAxisLength | Smoking | 0.264 | 0.002 | 0.07 |
| Geometry | AreaShape_Zernike_4_4 | Smoking | −0.241 | 0.005 | 0.079 |
| Geometry | AreaShape_Zernike_7_3 | Smoking | −0.308 | 0 | 0.027 |
| Geometry | AreaShape_Zernike_8_4 | Smoking | −0.242 | 0.007 | 0.096 |
| Geometry | AreaShape_Zernike_8_6 | Smoking | −0.252 | 0.005 | 0.079 |
| Geometry | AreaShape_Zernike_9_1 | Smoking | −0.303 | 0 | 0.027 |
| Texture | Granularity_13_ImageAfterMath.1 | Smoking | −0.317 | 0.001 | 0.054 |
| Texture | Mean_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_03 | Smoking | 0.232 | 0.005 | 0.079 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Area | Smoking | 0.297 | 0.001 | 0.049 |
| Geometry | Mean_Identifyhemasub2_AreaShape_MaximumRadius | Smoking | 0.318 | 0.001 | 0.049 |
| Geometry | Mean_Identifyhemasub2_AreaShape_MeanRadius | Smoking | 0.318 | 0.001 | 0.049 |
| Geometry | Mean_Identifyhemasub2_AreaShape_MedianRadius | Smoking | 0.308 | 0.002 | 0.054 |
| Geometry | Mean_Identifyhemasub2_AreaShape_MinFeretDiameter | Smoking | 0.299 | 0.001 | 0.049 |
| Geometry | Mean_Identifyhemasub2_AreaShape_MinorAxisLength | Smoking | 0.310 | 0.001 | 0.045 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_4_4 | Smoking | −0.263 | 0.003 | 0.07 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_5_1 | Smoking | −0.268 | 0.002 | 0.07 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_8_2 | Smoking | −0.277 | 0.003 | 0.073 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_8_8 | Smoking | −0.290 | 0.003 | 0.073 |
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_9_1 | Smoking | −0.226 | 0.004 | 0.074 |
| Texture | Mean_Identifyhemasub2_Granularity_13_ImageAfterMath | Smoking | −0.325 | 0.001 | 0.054 |
| Texture | Mean_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_01 | Smoking | 0.330 | 0 | 0.039 |
| Texture | Mean_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_02 | Smoking | 0.297 | 0.002 | 0.07 |
| Texture | Mean_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_03 | Smoking | 0.397 | 0 | 0.01 |
| Texture | Mean_Identifyhemasub2_Texture_SumVariance_ImageAfterMath_3_02 | Smoking | 0.258 | 0.007 | 0.093 |
| Texture | Median_Identifyeosinprimarycytoplasm_Texture_Correlation_maskosingray_3_03 | Smoking | 0.233 | 0.004 | 0.079 |
| Geometry | Median_Identifyhemasub2_AreaShape_Area | Smoking | 0.344 | 0 | 0.027 |
| Geometry | Median_Identifyhemasub2_AreaShape_MaxFeretDiameter | Smoking | 0.242 | 0.005 | 0.079 |
| Geometry | Median_Identifyhemasub2_AreaShape_MaximumRadius | Smoking | 0.323 | 0.001 | 0.049 |
| Geometry | Median_Identifyhemasub2_AreaShape_MeanRadius | Smoking | 0.323 | 0.001 | 0.049 |
| Geometry | Median_Identifyhemasub2_AreaShape_MedianRadius | Smoking | 0.266 | 0.005 | 0.079 |
| Geometry | Median_Identifyhemasub2_AreaShape_MinFeretDiameter | Smoking | 0.346 | 0 | 0.027 |
| Geometry | Median_Identifyhemasub2_AreaShape_MinorAxisLength | Smoking | 0.342 | 0 | 0.027 |
| Geometry | Median_Identifyhemasub2_AreaShape_Perimeter | Smoking | 0.247 | 0.006 | 0.085 |
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_4_4 | Smoking | −0.242 | 0.002 | 0.059 |
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_5_1 | Smoking | −0.256 | 0.003 | 0.073 |
| Texture | Median_Identifyhemasub2_Granularity_13_ImageAfterMath | Smoking | −0.311 | 0.001 | 0.049 |
| Texture | Median_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_01 | Smoking | 0.319 | 0.001 | 0.049 |
| Texture | Median_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_02 | Smoking | 0.274 | 0.005 | 0.081 |
| Texture | Median_Identifyhemasub2_Texture_Correlation_ImageAfterMath_3_03 | Smoking | 0.394 | 0 | 0.01 |
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_SumAverage_maskosingray_3_00 | Smoking | 0.272 | 0.003 | 0.073 |
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_SumAverage_maskosingray_3_01 | Smoking | 0.273 | 0.003 | 0.073 |
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_SumAverage_maskosingray_3_02 | Smoking | 0.270 | 0.004 | 0.074 |
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_SumAverage_maskosingray_3_03 | Smoking | 0.275 | 0.003 | 0.073 |
| Geometry | StDev_identifyhemaprimarynuclei_Location_Center_Y | Smoking | −0.245 | 0.007 | 0.093 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_4 | Smoking | −0.280 | 0.001 | 0.045 |
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_8 | Smoking | −0.236 | 0.007 | 0.094 |
| Texture | StDev_Identifyhemasub2_Texture_SumVariance_ImageAfterMath_3_01 | Smoking | 0.266 | 0.007 | 0.096 |
| Texture | StDev_Identifyhemasub2_Texture_SumVariance_ImageAfterMath_3_02 | Smoking | 0.283 | 0.005 | 0.079 |
| Texture | StDev_Identifyhemasub2_Texture_SumVariance_ImageAfterMath_3_03 | Smoking | 0.283 | 0.006 | 0.084 |
| Geometry | StDev_identifytissueregion_Location_Center_Y | Smoking | −0.289 | 0.002 | 0.059 |
| Texture | Texture_Correlation_ImageAfterMath_3_01 | Smoking | 0.252 | 0.004 | 0.078 |
| Texture | Texture_Correlation_ImageAfterMath_3_03 | Smoking | 0.329 | 0 | 0.027 |
| Texture | Texture_Correlation_maskosingray_3_03 | Smoking | 0.237 | 0.004 | 0.074 |
| Texture | Texture_Entropy_ImageAfterMath_3_01 | Smoking | 0.220 | 0.007 | 0.093 |
| Texture | Texture_Entropy_ImageAfterMath_3_03 | Smoking | 0.233 | 0.004 | 0.074 |
| Feature Group | Feature Name | Main | Age | Stage | Smoking | Sex |
|---|---|---|---|---|---|---|
| −0.024 | −0.317 | −0.038 | −0.088 | |||
| Geometry | AreaShape_Zernike_6_0 | −0.038 | ||||
| Geometry | AreaShape_Zernike_6_4 | −0.019 | ||||
| Geometry | AreaShape_Zernike_6_6 | 0.052 | ||||
| Geometry | AreaShape_Zernike_9_3 | 0.027 | ||||
| Geometry | AreaShape_Zernike_9_5 | 0.153 | ||||
| Texture | Granularity_10_ImageAfterMath.1 | −0.033 | ||||
| Texture | Granularity_9_ImageAfterMath | 0.081 | ||||
| Geometry | Mean_Identifyhemasub2_AreaShape_Center_X | 0.002 | ||||
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_5_1 | 0.013 | ||||
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_6_2 | −0.002 | ||||
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_6_4 | −0.010 | ||||
| Geometry | Mean_Identifyhemasub2_AreaShape_Zernike_9_9 | −0.146 | ||||
| Geometry | Mean_Identifyhemasub2_Location_Center_X | 0.002 | ||||
| Geometry | Mean_identifytissueregion_Location_Center_X | 0.056 | ||||
| Geometry | Median_Identifyeosinprimarycytoplasm_Location_Center_X | −0.071 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_4_0 | 0.023 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_7_3 | 0.083 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_8_4 | −0.120 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_8_6 | −0.098 | ||||
| Geometry | Median_Identifyhemasub2_AreaShape_Zernike_9_1 | −0.044 | ||||
| Geometry | Median_identifytissueregion_Location_Center_Y | −0.063 | ||||
| Holistic | Neighbors_SecondClosestDistance_Adjacent | −0.170 | −0.072 | 0.002 | ||
| Geometry | StDev_Identifyeosinprimarycytoplasm_Location_Center_Y | 0.095 | ||||
| Texture | StDev_Identifyeosinprimarycytoplasm_Texture_DifferenceVariance_maskosingray_3_00 | 0.036 | ||||
| Geometry | StDev_Identifyhemasub2_AreaShape_Orientation | −0.159 | ||||
| Geometry | StDev_Identifyhemasub2_AreaShape_Zernike_8_8 | −0.146 | ||||
| Texture | StDev_Identifyhemasub2_Granularity_12_ImageAfterMath | −0.101 | ||||
| Texture | StDev_Identifyhemasub2_Granularity_13_ImageAfterMath | 0.327 | 0.130 | 0.072 | −0.189 | 0.174 |
| Texture | StDev_Identifyhemasub2_Granularity_9_ImageAfterMath | 0.003 | ||||
| Texture | StDev_Identifyhemasub2_Texture_SumVariance_ImageAfterMath_3_01 | −0.034 | ||||
| Geometry | StDev_identifytissueregion_Location_Center_Y | 0.016 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhong, T.; Wu, M.; Ma, S. Histopathological Imaging–Environment Interactions in Cancer Modeling. Cancers 2019, 11, 579. https://doi.org/10.3390/cancers11040579
Xu Y, Zhong T, Wu M, Ma S. Histopathological Imaging–Environment Interactions in Cancer Modeling. Cancers. 2019; 11(4):579. https://doi.org/10.3390/cancers11040579
Chicago/Turabian StyleXu, Yaqing, Tingyan Zhong, Mengyun Wu, and Shuangge Ma. 2019. "Histopathological Imaging–Environment Interactions in Cancer Modeling" Cancers 11, no. 4: 579. https://doi.org/10.3390/cancers11040579
APA StyleXu, Y., Zhong, T., Wu, M., & Ma, S. (2019). Histopathological Imaging–Environment Interactions in Cancer Modeling. Cancers, 11(4), 579. https://doi.org/10.3390/cancers11040579