1. Introduction
Glioblastomas (GBMs) belong to the group of high-grade gliomas that are among the most invasive and malignant brain tumors in adults, representing about 15% of all diagnosed brain tumors and affecting three individuals per 100,000 per year [
1]. Primary brain tumors are classified into several different types and subtypes and since the publication of the World Health Organization classification of central nervous system tumors in 2016, both histological and molecular parameters are used to define a tumor entity [
2]. GBMs have poor prognoses and may be markedly resistant to both radiotherapy and chemotherapy, and no curative treatment is yet available. Standard treatment consists of combinations of maximal neurosurgical resection, reducing tumor burden and pressure on the brain, radiation therapy, and concurrent and continual chemotherapy with temozolomide. Without treatment, the average survival time is just four months from diagnosis. Even with treatment, relapse is anticipated, and with maximal, existing treatment, patient survival is still only about 15 months [
3,
4,
5]. Thus, there is a significant need to improve and find new treatment strategies targeting this form of aggressive tumor.
Genetic and phenotypic heterogeneity is a common feature of GBM, which constitutes a major challenge for the development of an effective therapy [
6,
7,
8]. In recent years, increasing evidence suggests that the major culprit for cancer recurrence and treatment resistance is cancer stem-like cells. Similar to normal stem cells, cancer stem-like cells have self-renewal, anchorage-independent growth, and pluripotent differentiation abilities [
9]. Cancer stem-like cells have been implicated in several human cancers, including gliomas [
10,
11,
12,
13,
14]. The high plasticity of GBM cells has been suggested to be another important driver of tumor heterogeneity and dedifferentiation of tumor cells into a stem-like state [
15]. The glioma stem-like cells are generally believed to express stemness and self-renewal markers, such as Sox2 and Nestin [
16,
17,
18]. Sox2 and Nestin are enriched in glioma stem-like cells and maintain the cells’ stemness property and continued tumorigenicity. Expression of Sox2 identifies patients with the worst clinical outcomes [
19]. With growing evidence that GBM arises, progresses, and recurs from stem-like cells, novel therapies targeting this subpopulation of cancer cells provide new possibilities for extending patient survival.
Rapid invasion of the surrounding normal brain tissue is a main characteristic of malignant and recurrent GBM tumors. The grade of invasiveness is closely related to the prognosis [
3]. Interactions between extracellular matrix (ECM) and integrin receptors on tumor cells have been shown to play a key role in tumor cell processes, such as cell migration and proliferation, as well as in angiogenesis [
20,
21]. Integrins are heterodimeric cell surface receptors, consisting of an α and β chain that link the ECM to the cytoskeleton and connect mechanical cues with intracellular signaling cascades. Integrin α10β1 was discovered by our group and originally identified as a type II collagen-binding receptor on chondrocytes [
22]. The expression of α10β1 in normal tissues is restricted and mainly confined to cartilage-containing tissues [
23,
24]. We have reported that mice lacking the integrin α10β1 have defects in the cartilaginous growth plate and consequently develop growth retardation of the long bones [
25]. We have also reported that integrin α10β1 is present on mesenchymal stem cells (MSCs) and that the expression correlates with the chondrogenic differentiation potential of MSCs [
26]. Recently, we demonstrated that antibodies to integrin α10 identify and select a subfraction of cells from MSC preparations with improved trilineage differentiation potential [
27]. Here, we report that integrin α10β1 is highly expressed in GBM cells, both in cultured GBM cell lines with stem cell characteristics and in patient GBM tumor tissue samples.
Little is known about integrin α10β1 in cancer, but recent studies suggest that integrin α10β1 may play an important role in the development and progression of certain tumors. For example, it has been shown that integrin α10 expression is up-regulated in malignant melanoma cells compared with primary melanocytes [
28]. This study also demonstrated that melanoma cells treated with either an inhibitory antibody to integrin α10 or an antisense construct downregulating α10 expression had reduced migratory potential, thus suggesting a role for integrin α10 in melanoma cell migration. Furthermore, integrin α10β1 has been shown to be an important receptor on α-smooth muscle actin (α-SMA)-expressing stromal cells in human ovarian tumors by binding to the HU177 cryptic collagen epitope [
29]. Additionally, the integrin α10 gene (
ITGA10) has been shown to be the most significant gene associated with disease-specific death and distant metastasis in myxofibrosarcoma [
30]. In the present study, we demonstrate that integrin α10β1 is highly expressed in high-grade glioma tumor tissue and patient-derived GBM cell lines and that it plays a role in the viability and migration of GBM cells. Furthermore, in a xenografted mouse model, we demonstrate that treatment with an antibody–drug conjugate (ADC), composed of the integrin α10 antibody conjugated to the potent cytotoxin saporin [
31], anti-α10-SAP, induces cell death of these tumor cells. Our data indicate that ADC therapy targeting α10β1 may represent a new strategy to treat glioblastoma and other high-grade gliomas.
3. Discussion
We have previously reported that integrin α10β1 is a major collagen binding integrin on chondrocytes [
24]. Moreover, we have also discovered that integrin α10β1 is expressed by MSCs isolated from bone marrow, and that FGF-2 induces the expression of integrin α10β1 [
26] and promotes differentiation of MSCs to chondrocytes [
24,
26]. More recently, we reported that a subfraction of adipose tissue-derived MSCs selected with antibodies directed to integrin α10 have improved differentiation capacity compared with unselected MSCs [
27], further indicating that integrin α10β1 correlates with stemness characteristics. Because several studies have reported that GBM cells have stem cell characteristics that contribute to their resistance to different treatment strategies, we set out to investigate integrin α10β1 expression in GBM. Initial database screening for integrin α10β1 in the human protein atlas (
https://www.proteinatlas.org/) revealed that
ITGA10 mRNA was highly up-regulated in the GBM cell line U138MG, which motivated further investigation. In this study, we have shown that the cell surface protein integrin α10β1 is expressed in tumor tissues from glioma patients and in patient-derived GBM cell lines. We have also demonstrated that the expression of α10β1 is extensively expressed in high-grade gliomas (astrocytoma grade III) and GBM compared with low-grade gliomas (astrocytoma grade II) where expression is more dispersed. These data are in line with previous observations that integrin α10β1 is up-regulated in malignant melanoma and distant metastasis in myxofibrosarcoma [
28,
30]. Moreover, we show that high
ITGA10 expression correlates with a worse overall survival probability in glioma patients and may play a critical role in tumor progression in glioma.
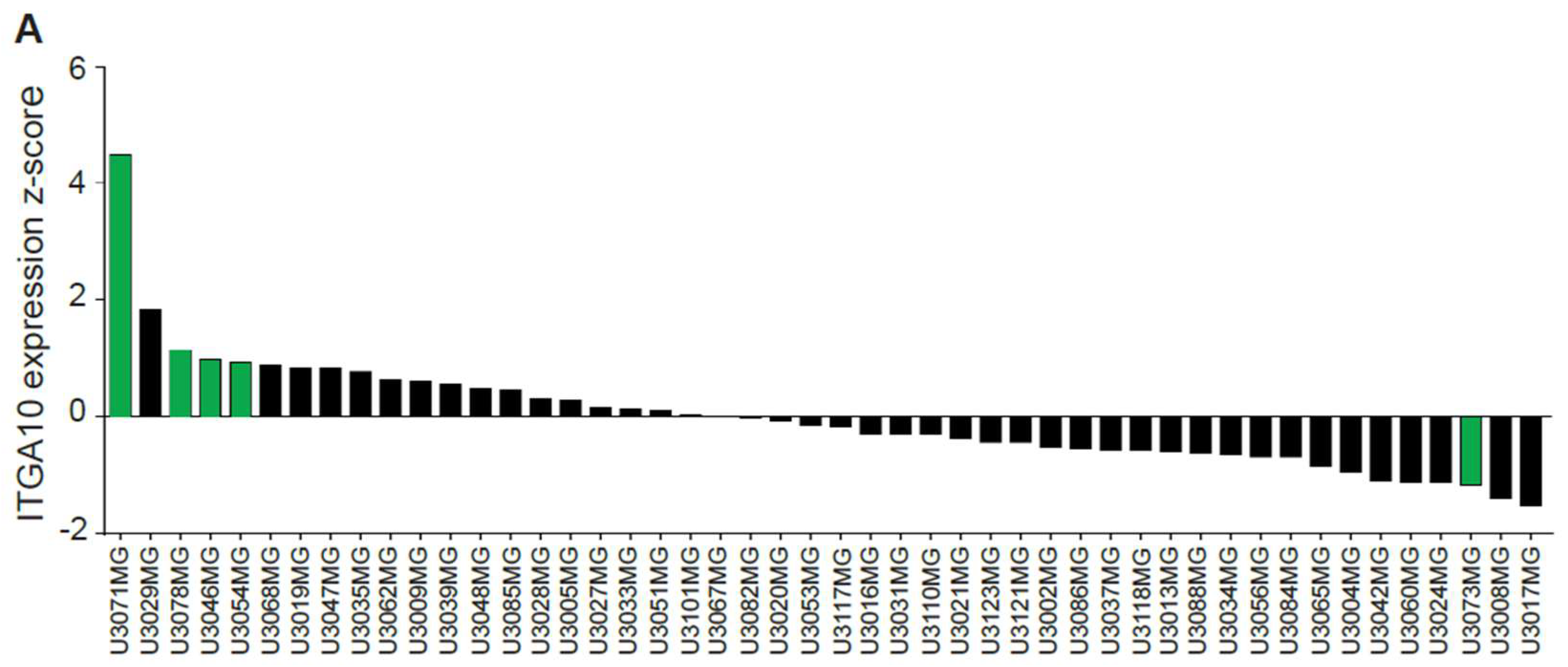
In this study, we used the HGCC cell lines, because these cells are more similar to the original tumor cells than serum-cultured cell lines [
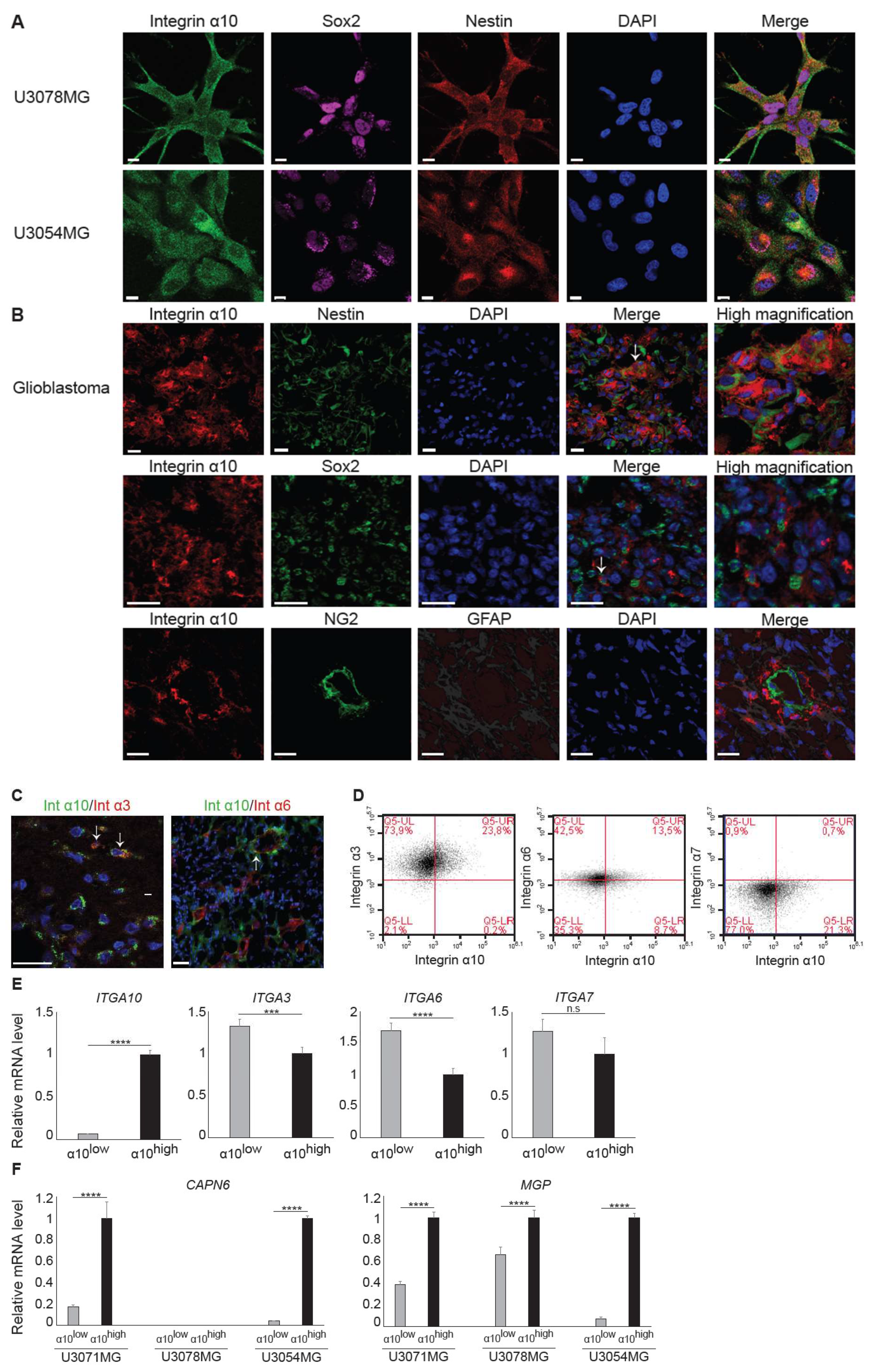
32]. Indeed, we found a higher expression of integrin α10β1 in these GBM cell lines under neural stem cell culture conditions compared with both serum-cultured GBM cells and conventional GBM cell lines, such as U87MG. We also found cellular colocalization of integrin α10β1 and the stemness markers Nestin and Sox2 in the majority of all tested GBM cells (
Figure 3A), suggesting that integrin α10β1 is expressed on GBM cells with a stem/progenitor-like phenotype. When we examined GBM cells cultured as spheres, however, we found that GBM cells with high integrin α10β1 expression had low Nestin expression, and vice versa (
Figure 4B). Additionally, immunohistochemical analysis of GBM tumor tissues demonstrated that only a few integrin α10β1-expressing GBM cells (between 0‒10% in different patient samples) were co-expressed with Nestin, and even fewer of the integrin α10β1-expressing GBM cells were co-expressed with Sox2 (
Figure 3B). The discrepancy between the cells in culture and those examined in the GBM tissues may be because patient-derived GBM cells were cultured under conditions that maintain stem cell characteristics.
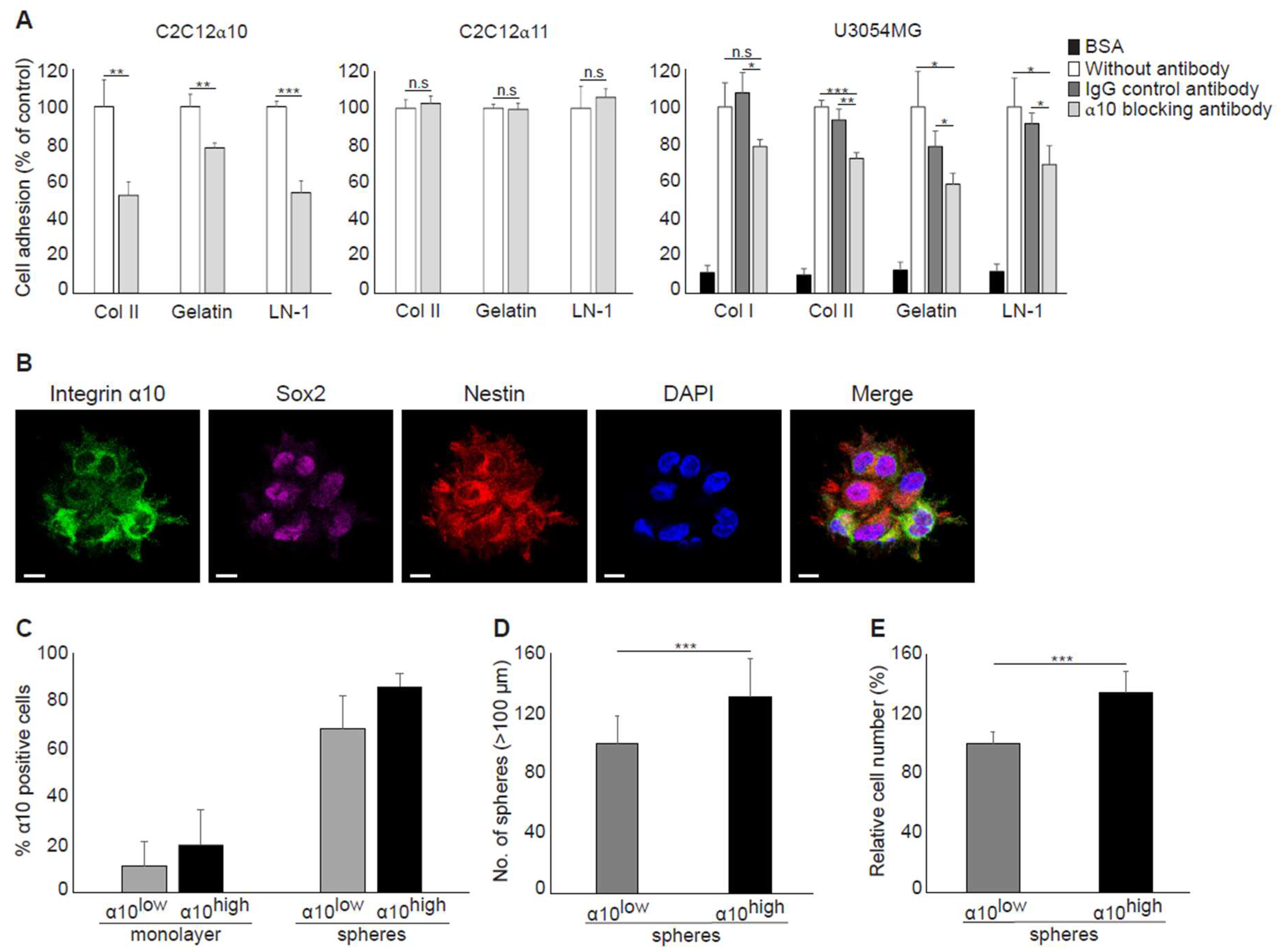
Our findings that cell surface expression of integrin α10β1 is increased during sphere formation compared with monolayer cultures and that integrin α10β1-selected GBM cells have an increased ability to grow as spheres indicate that integrin α10β1 has an important role in glioma growth. Integrins have an important role in the cell matrix and mediating cellular functions, such as adhesion, migration, and proliferation, although their specific role in GBM cells is unclear. A number of integrins have been demonstrated to be expressed by GBM cells [
20,
36] and the integrin subunits α3, α6, and α7 are not only up-regulated in glioma stem-like cells, but increase their tumorigenicity [
33,
34,
35,
37]. Because these integrin subunits are all laminin-binding receptors, we decided to investigate whether integrin α10β1, a collagen receptor [
24], could also mediate the binding of GBM cells to laminin. Previous studies by Tulla et al. have demonstrated binding of the integrin α10 I domain to laminin-111 [
38]; however, this has not been verified with integrin α10β1-expressing cells. Using a function-blocking antibody to integrin α10, we were able to demonstrate that integrin α10β1 can indeed mediate adhesion of GBM cells to laminin-111, indicating that integrin α10β1 on GBM cells may use laminin as a ligand in addition to collagen. It has previously been reported that the integrins α1β1 and α2β1 can both bind laminin [
38]. These integrins, as well as integrins α10β1 and α11β1, are well-characterized collagen-binding integrins involved in cellular functions, such as adhesion and migration, although their biological function in vivo is not fully understood [
39]. A recent publication has shown that integrin α10β1 binds to cartilage collagen molecules, but not to fibrillar collagens [
40], which indicates that integrin α10β1 may interact with matrix structures other than collagen fibrils in the GBM tumors. Our results from GBM tissues have shown that integrin α10β1 expression on single cells was dispersed within the tumor, on cells in the vicinity of blood vessels, and on large cell clusters within the tumor mass. The specific integrin α10β1–matrix interaction in these different areas remains to be elucidated.
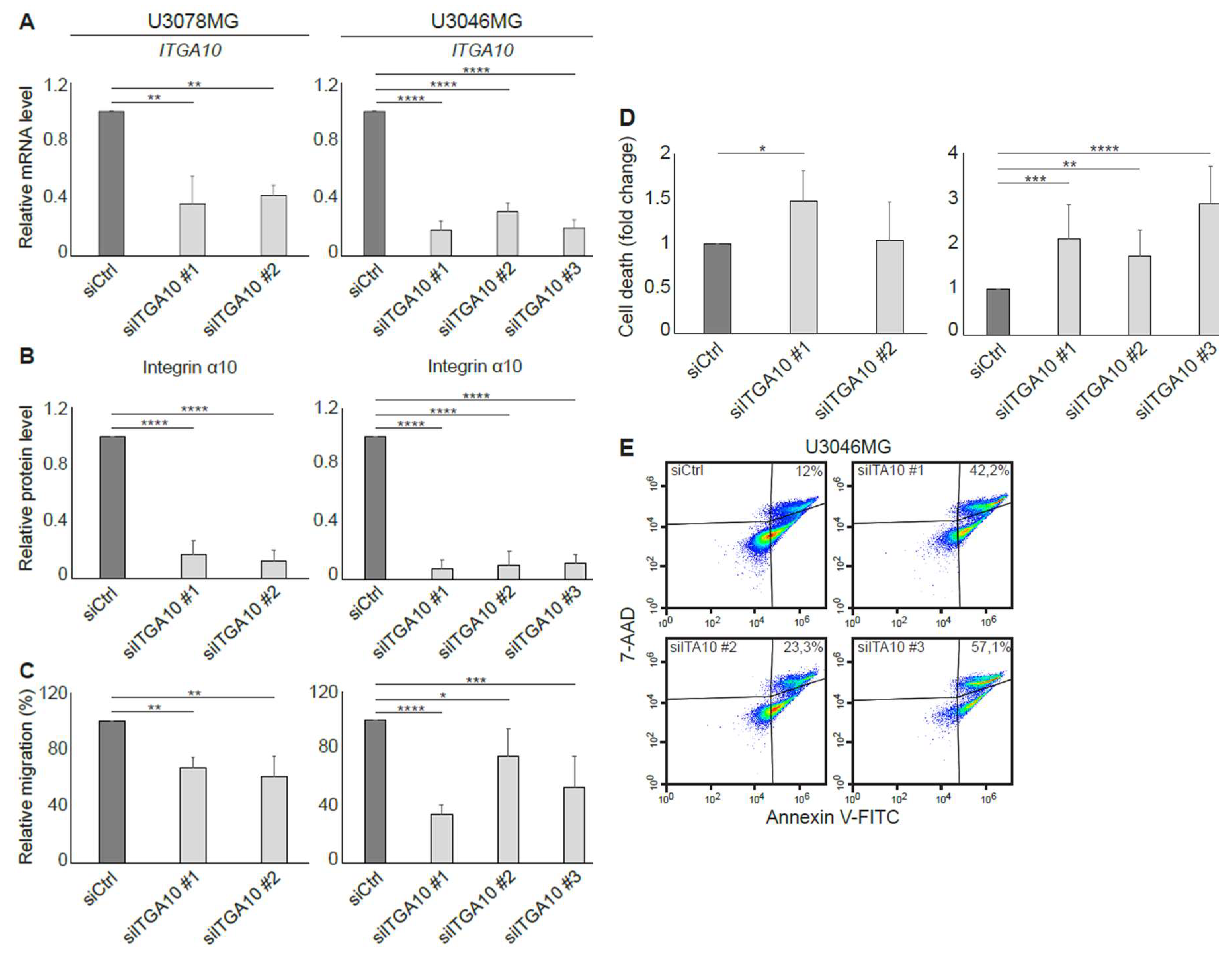
In agreement with a previous study, where integrin α10β1 was shown to be associated with melanoma cell migration [
28], our results suggest that integrin α10β1 is involved in the migration of GBM cells as demonstrated using siRNA and suggested by our gene expression analysis of the integrin α10
high-selected GBM cell population. In the microarray data, eight of the top 20 up-regulated genes in the high α10 intensity populations have previously been shown to be involved in migration and/or invasion. Furthermore, integrin α10β1 on ovarian tumor cells binds to a cryptic epitope, HU177, present within multiple forms of collagen after denaturation [
29]. Blocking this interaction caused diminished migration of α10β1-expressing, fibroblast-like cells on denatured collagen, as well as reduced proliferation of ovarian tumor cells, decreased angiogenesis, and accumulation of α-SMA-expressing stromal cells [
29]. Together, these novel data indicate that integrin α10β1 has an important role in cell proliferation, migration, and survival.
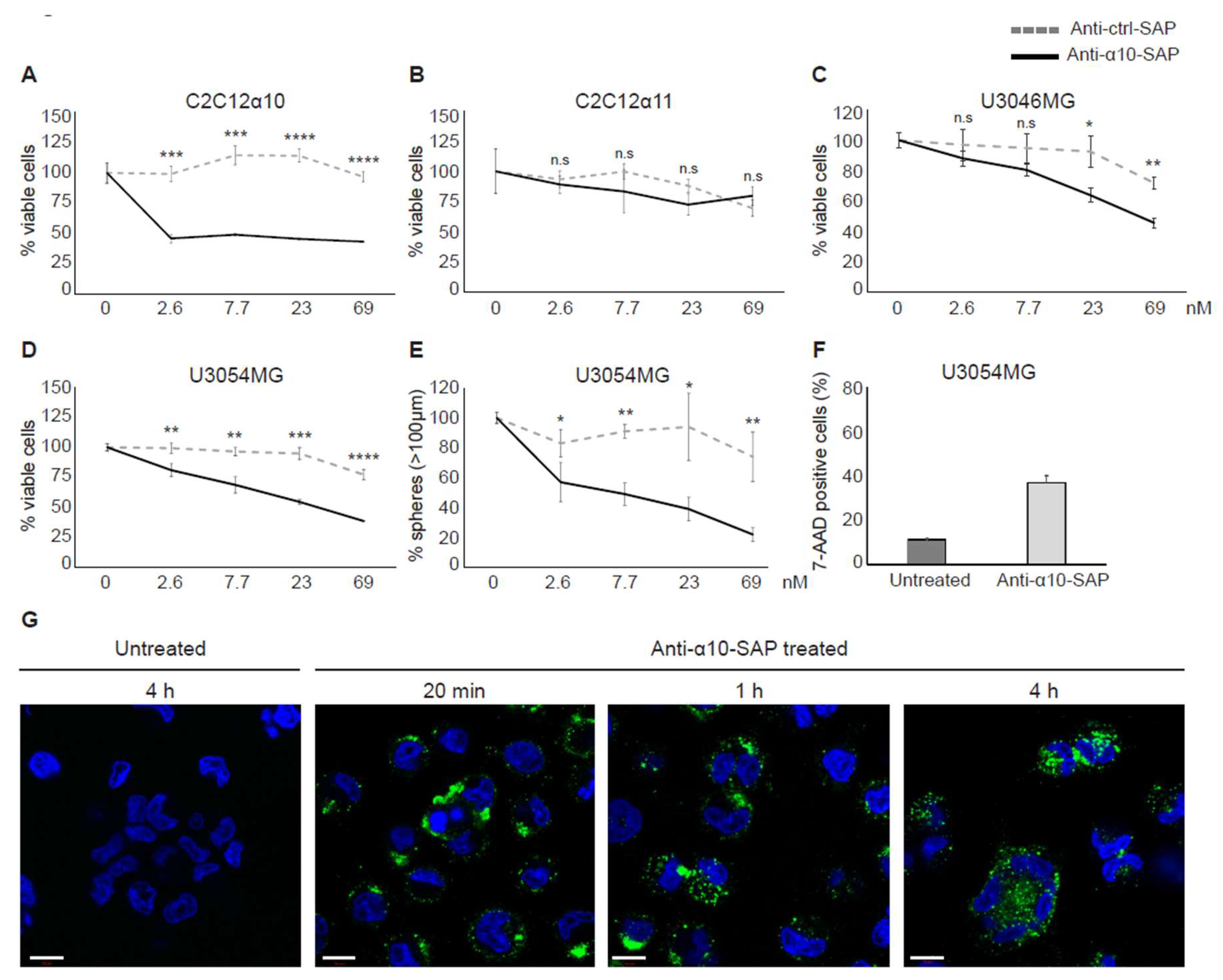
Our finding that integrin α10β1 is highly expressed by tumor cells in high-grade gliomas, in combination with the restricted expression of integrin α10β1 in normal tissues, suggests that it is both a novel marker and an attractive target for therapeutic intervention in GBM. We employed an ADC approach using antibodies against α10β1 to target invasive cells by specifically delivering saporin to the integrin α10β1-expressing tumor cells. Saporin is a ribosome inactivating protein (RIP) that in minute quantities can stop protein synthesis, thereby causing cell death [
31]. We have shown that our ADC complex (anti-α10-SAP) binds, internalizes, and subsequently kills the targeted cells.
Today, several ADC therapies are in development for the treatment of different tumor types, including glioma [
41]. The ADCs are an attractive treatment approach because of their specific and targeted delivery of a highly potent cytotoxic drug into tumor cells. Our study shows that integrin α10β1 represents a promising target for ADC treatment of GBM due to its localization at the cell surface, its involvement in important tumor cell functions, such as cell migration and viability, and its overexpression in glioma tissue and restricted expression in normal tissue. In addition, we demonstrated that our ADC complex binds specifically to integrin α10β1-expressing cells and reduces the GBM cell viability and sphere formation in a dose-dependent manner. To ensure the ADC specificity for integrin α10β1, α11β1-expressing C2C12 cells were used as a negative control, because α11 is the integrin subunit most closely related to α10 and both are collagen binding integrins.
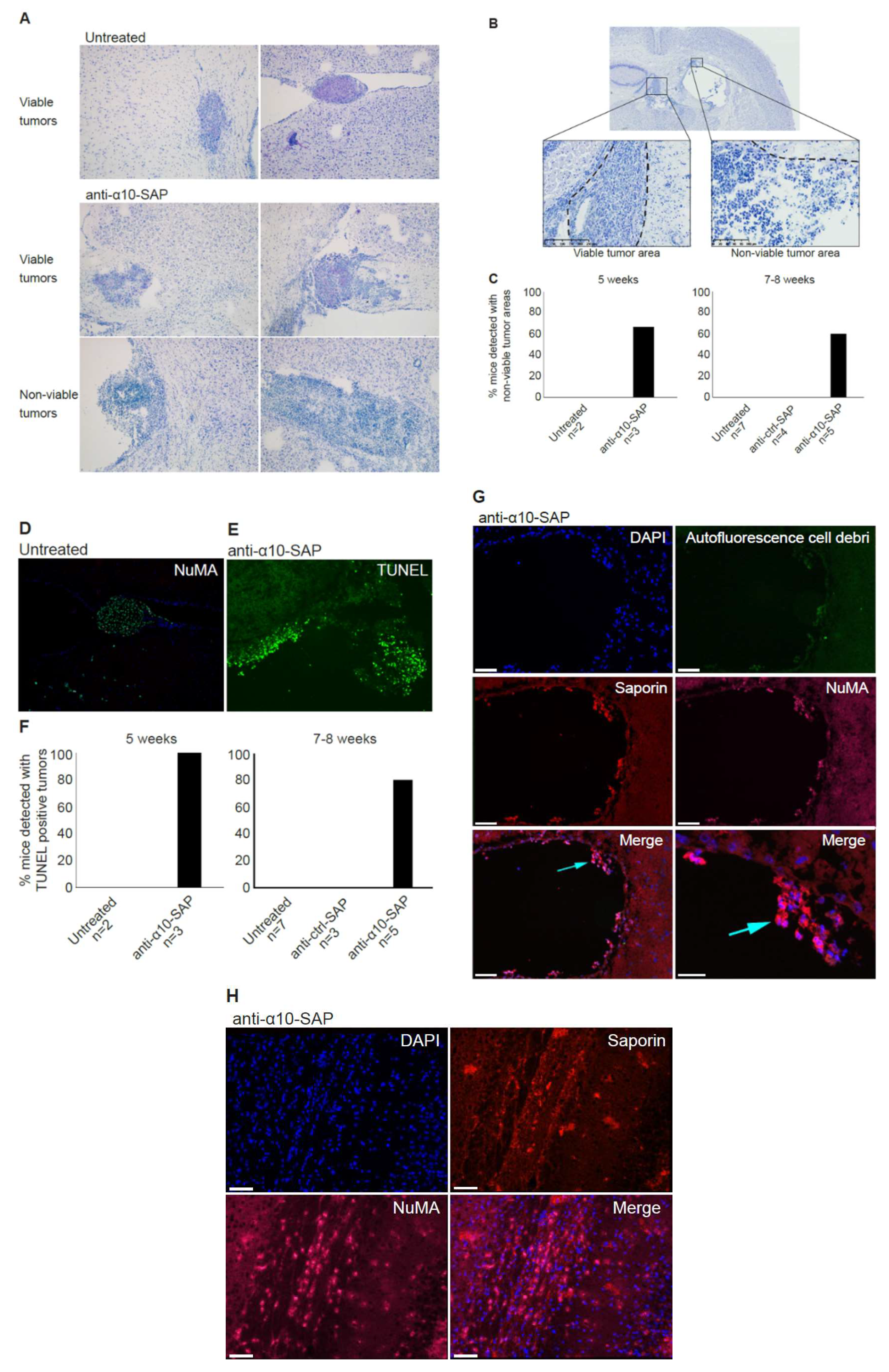
In the in vivo study, non-viable tumor tissue was only detected in the anti-α10-SAP-treated mice, demonstrating a targeted anti-tumor effect of anti-α10-SAP by its ability to cause specific tumor cell death. We performed a histological evaluation in combination with a TUNEL assay to demonstrate tumor cell death. The TUNEL assay confirmed the histopathological, non-viable tumor identification, but also detected additional non-viable tumors. A likely explanation is that TUNEL staining is a more sensitive method of detecting cell death compared with histopathological evaluation.
In this study, the ADC was delivered by injection into the cerebrospinal fluid (CSF) of the ventricle instead of local delivery to the tumor injection site, because GBM is not a localized disease and can affect and recur throughout the entire brain. Thus, CSF delivery via Ommaya intracerebroventricular or lumbar puncture is a promising treatment for GBM, both upon its initial presentation and after cellular tumor spread through the CSF following tissue disruption caused by resection neurosurgery. Notably, intracerebroventricular and lumbar intrathecal injections of chemotherapy drugs, such as methotrexate and other, newer agents, into the CSF on a weekly basis by neuro-oncologists is a standard patient treatment for CNS lymphoma. To ensure extensive dissemination of anti-α10-SAP in the mouse brain tissue and that it reached tumor cells throughout the entire CNS via the CSF pathways, including the brain parenchyma via the Virchow–Robin perivascular spaces [
42], we chose to inject anti-α10-SAP through a guide screw in the ventricle. Staining the mouse brain tissue with an antibody to saporin allowed us to demonstrate that the anti-α10-SAP did indeed reach the intraparenchymal brain tissue.
Taken together, the ADC experiments, demonstrating the anti-α10-SAP-induced cell death of GBM cells both in vitro and in vivo, further support the potential of integrin α10β1 as a novel therapeutic target in GBMs.
4. Materials and Methods
4.1. Cell Lines
Glioblastoma cells from the HGCC (
www.hgcc.se) resource at Uppsala University were used. The HGCC cells are derived from patient material obtained from brain tumor tissue samples collected as surgical biopsies [
32]. The cells were cultured in serum-free, defined neural stem cell (NSC) medium containing Dulbecco’s Modified Eagle Medium (DMEM)/F12 w/Glutamax and Neurobasal media (1:1) (Gibco, Scotland, UK), supplemented with B27 (Gibco), N2 (Gibco), 100 U/mL Antibiotic-Antimycotic (Gibco), bFGF (10 ng/mL) (Miltenyi, Bergisch Gladbach, Germany), and EGF (10 ng/mL) (Gibco) at 37 °C with 5% CO
2. The adherent cell lines were cultured and expanded on Corning Primaria cell culture dishes coated with laminin-111 (1:100, L2020, Sigma Aldrich, St. Louis, MO, USA). The mouse myoblast cell line C2C12 transduced with integrin α10 vector (C2C12α10) or integrin α11 vector (C2C12α11) was cultured in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% Fetal bovine serum (FBS, Biological Industries, Beit Haemek, Israel) and 100 U/mL Antibiotic-Antimycotic (Gibco). The C2C12α10 and C2C12α11 cells were selected using G418 (1 µg/µL) and puromycin (10 µg/mL), respectively. As part of our laboratory routine, all cell lines were regularly tested for mycoplasma and replaced on a tri-monthly basis.
4.2. Analysis of TCGA and HGCC Gene Expression Data
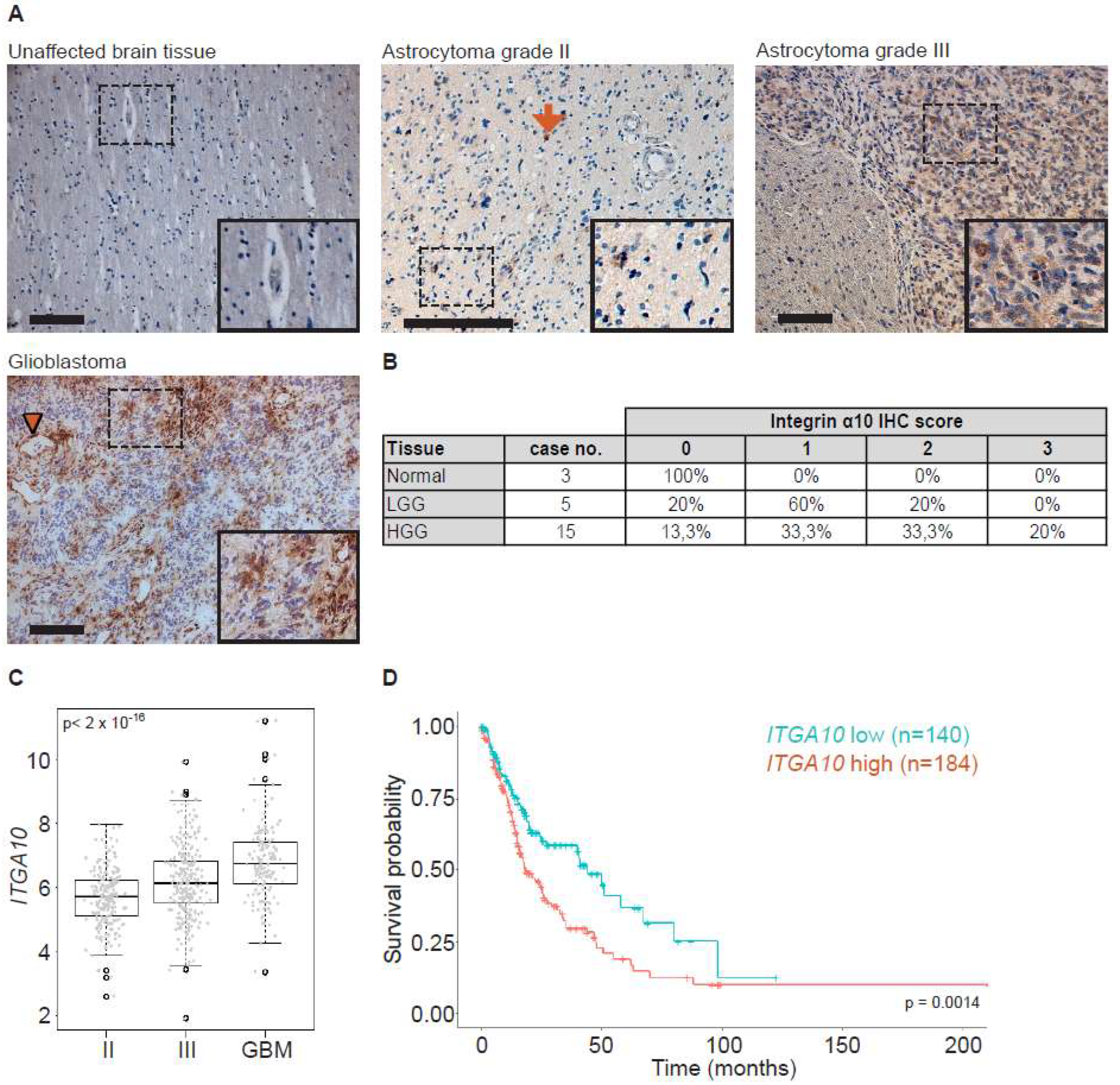
Normalized TCGA gene expression data (IlluminaHiSeq, San Diego, CA, USA) was downloaded using the UCSC Cancer Browser (genome-cancer.ucsc.edu) for low-grade glioma and GBM together with corresponding clinical information. Clinical information was used to stratify patients into astrocytoma grade II, astrocytoma grade III, and GBM. One-way ANOVA test and the non-parametric alternative (Kruskal–Wallis test) were used to test for a difference in expression between the three groups.
Furthermore, normalized RNA-seq data for
ITGA10 together with clinical data from a curated, published dataset [
43] originating from the TCGA-LGG and TCGA-GBM cohorts, was downloaded from the Gliovis website (
http://gliovis.bioinfo.cnio.es/) [
44]. Patients diagnosed with oligodendroglioma and oligoastrocytoma were excluded from the study; remaining patients with an assigned grade (
n = 59, 116, and 149 patients for grades II and III and GBM, respectively) were divided into groups according to high and low
ITGA10 expression based on a median cut-off for the subsequent survival analysis by the Kaplan–Meier method and log-rank test.
Gene expression z-scores for ITGA10 from the 48 published HGCC cell lines were downloaded from hgcc.se.
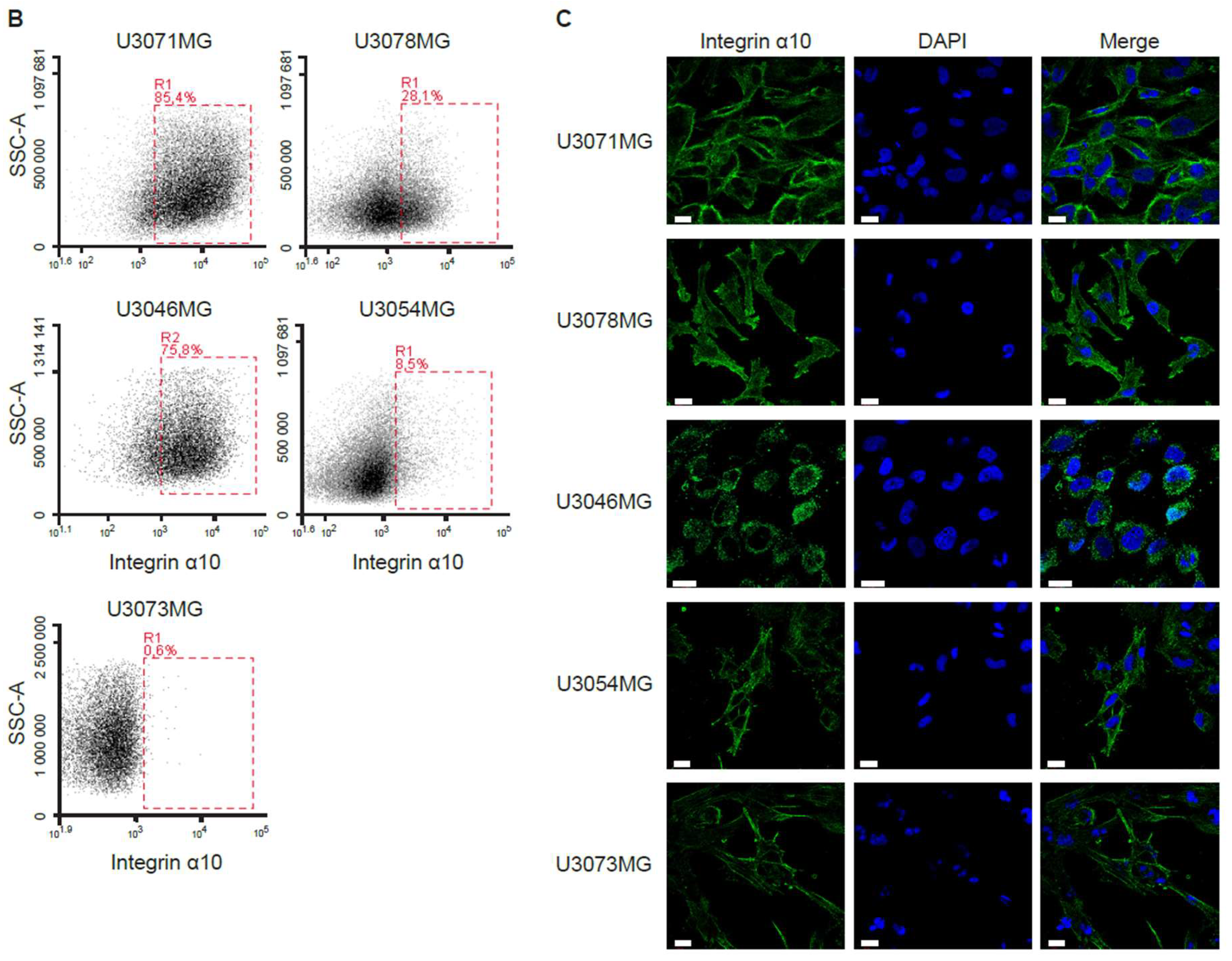
4.3. Flow Cytometry Analysis and Fluorescence-Activated Cell Sorting
Immunostaining of GBM cells was performed by incubating cells with antibodies against integrin α10 (Alexa Fluor 647 conjugate), α3, α6, and α7 (see
Table S2) for 30 min in the dark at 4 °C prior to flow cytometry analysis using a BD Accuri C6 flow cytometer. After a 30-min incubation with a primary antibody, cells were washed twice with Dulbecco’s phosphate-buffered saline (DPBS, SH3002802, Hyclone, Logan, UT, USA) containing 1% FBS and 0.1% sodium azide and incubated with a secondary antibody (
Table S3) for 20 min in the dark at 4 °C. For co-staining integrin α3 and α7 with integrin α10, cells were then incubated with integrin α10 Alexa Fluor 647 antibody for 30 min in the dark at 4 °C before subsequent analysis using flow cytometry.
GBM cells with high expression of integrin α10β1 (α10high) or low expression of integrin α10β1 (α10low) were sorted by fluorescence-activated cell sorting (FACSAria, BD Biosciences, San Jose, CA, USA). Discrimination of live/dead cells was accompanied by 7-AAD staining (BioLegend, San diego, CA, USA). Sorted cells were washed in medium and re-seeded for recovery and expansion, and some were immediately frozen for RNA extraction.
4.4. Gene Expression Microarray Analysis in Sorted Cells
The RNA concentration and purity were determined using Nanodrop and Bioanalyzer. Total gene expression analysis was performed by SCIBLU Genomics at Lund University using Human Gene 2.0 ST arrays. Basic Affymetrix chip and experimental quality analysis were performed using Expression Console Software v1.1.2 (Thermo Fisher Scientific, Santa Clara, CA, USA). Probe summarization and data normalization were done by robust multi-array analysis (RMA) [
45]. Probe sets without signal intensity above the median of negative control intensity signals in at least 80% of samples were excluded, and controls and probe-sets with no gene symbol were filtered. Signals were log 2-transformed. A linear model was applied to adjust for the paired sample design using the limma package in R. The fitted values were used for the empirical Bayes method to calculate the
t-statistics. Genes were sorted by fold change after excluding those that were not expressed differently in all three cell lines, and a
p-value > 0.05 was used to estimate the top, differentially expressed genes.
4.5. RNA Extraction
Total RNA was extracted using QIAzol lysis reagent (Qiagen) for the samples used in the microarray analysis and for qRT-PCR. Total RNA was purified with a RNeasy Plus Mini kit (Qiagen, Hilden, Germany) according to the manufacturer´s instructions. The cDNA was synthesized with SuperScript VILO (Invitrogen, Carlsbad, CA, USA) and then amplified using qRT-PCR for evaluation of relative mRNA expression levels in the Applied Biosystems StepOne Plus Real Time PCR System device using the TaqMan Universal Master Mix II (Applied Biosystems, Carlsbad, CA, USA). The comparative Ct method was used to quantify relative mRNA expression, and
GAPDH was used as a reference gene; each reaction was performed in duplicate. The TaqMan primers are listed in
Table S4.
4.6. siRNA Transfections
The U3078MG and U3046MG GBM cells were seeded in antibiotic-free medium in 60 mm plates 24 h before transfection. Thereafter, cells were transfected for 96 h using Lipofectamine RNAiMAX (Invitrogen) and 2.25 pmol siRNA in Opti-MEM (Gibco) according to the supplier’s protocol. Then, 5 h after transfection, 15 ng/mL bFGF2 was additionally added to the plates. Non-targeting siRNA (ThermoFisher Scientific, Carlsbad, CA, USA) was used as a negative control. The oligonucleotides used were silencer select control no. 2 (4390846; ThermoFisher Scientific) and three different silencer select ITGA10 oligos (s16180, s16181, and s16182; ThermoFisher Scientific).
4.7. Antibody–Saporin Conjugation
Conjugation of saporin to the monoclonal mouse integrin α10 antibody (Xintela AB, Lund, Sweden) to generate anti-α10-SAP was performed by Advanced Targeting Systems (San Diego, CA, USA). As a control for the anti-α10-SAP targeting, we used two different anti-ctrl-SAP preparations (Advanced Targeting Systems and Innovagen AB, Lund, Sweden).
4.8. Migration Assay
The trans-well inserts (CytoSelect Cell Migration Assay kit, Cell Biolabs, San Diego, CA, USA) were coated with gelatin (Sigma Aldrich, Saint Louis, MO, USA) for approximately 2‒4 h at 37 °C. The ITGA10-knocked-down U3078MG and U3046MG cells were seeded to the upper compartment of the CytoSelect Insert. The lower compartment was filled with NSC medium supplemented with 5% FBS. After incubation for 72 h at 37 °C, the inserts were collected, and the cells adhering to the lower surface were fixed, stained with the cell stain solution, and the extraction was quantified by measuring the absorbance at 560 nm in a SpectraMax i3 multi-mode plate reader (Molecular Devices, San Jose, CA, USA) according to the manufacturer’s recommendation.
4.9. Sphere Formation, Cell Proliferation, and Apoptosis Assay
The U3054MG GBM cells were seeded at a density of 2500 cells/well in an uncoated 96-well plate, and treated with anti-ctrl-SAP and anti-α10-SAP antibody for 7 d. Whole microscopy images of each well were taken and spheres that had reached a diameter of 100 µm were counted in the digital images using ImageJ software (ImageJ 1.51k, LOCI, University of Wisconsin, Madison, WI, USA) [
46].
The cell viability was determined by using the WST-1 assay (Roche, Mannheim, Germany) according to the manufacturer´s instructions. All conditions were done in triplicate.
To analyze apoptosis, cells were stained with the Annexin V-FITC apoptosis detection kit with 7-AAD (BioLegend, San Diego, CA, USA) according to the supplier’s protocol, and the amount of bound Annexin V-FITC and 7-AAD was quantified with a BD Accuri C6 flow cytometer.
4.10. Cell Adhesion
First, 48-well dishes were coated with 0.1% gelatin (G1393, Sigma Aldrich), 10 µg/mL collagen type I from rat tail (C7661, Sigma Aldrich), 10 µg/mL collagen type II from chicken sternal cartilage (C9301, Sigma Aldrich), and 1 mg/mL laminin-111 (L2020, Sigma Aldrich) solution and blocked with 0.25% BSA (Sigma Aldrich) in PBS. BSA-coated wells were used as a negative and non-specific control. The wells were rinsed with PBS before the experiment. Cells were suspended in PBS (+Ca/Mg, Gibco) and incubated with antibodies for 30 min before plating the cells. The isotype control IgG2a antibody (401504, Biolegend) and blocking monoclonal integrin α10 antibody were used at a concentration of 3‒5 µg/mL. The C2C12α10 and C2C12α11 cells were added to the wells at a concentration of 65,000/well and U3054MG cells at a concentration of 100,000/well, and cells were allowed to adhere for 1 h at 37 °C. After incubation, cells were gently rinsed with PBS (+Ca/Mg) to remove non-adherent cells. Adherent cells were fixed with ethanol, stained with 0.09% crystal violet (Sigma Aldrich), and the absorbed dye was extracted by 10% acetic acid (Sigma Aldrich). The extraction was quantified by measuring the absorbance at 560 nm in the SpectraMax i3 multi-mode plate reader (Molecular Devices).
4.11. In Vivo Study
Six-week-old female, NOD-SCID mice (NOD.CB17/AlhnRj-Prkdcscid, Janvier Labs, Le Genest St Isle, France) were housed in a controlled environment, and all procedures were carried out in accordance with an ethical permit approved by the regional ethical committee for animal research (approval no. C41/14). Mice were inoculated with 5 × 105 U3054MG cells intracranially (into the striatum, injection site: AP 0, ML 1.5 (R), DV −2.5 (cells); AP 0.1, ML 0.8 (R), DV −4 (ICV)) and the tumor cells were allowed to grow in either the absence (n = 11) or presence of anti-α10-SAP (n = 11), or the isotype control ADC anti-ctrl-SAP (n = 8). The ADCs were first administered 1 week after cell injection. For administration, guide screws (Guide Screw 1.6 mm length 26 GA, Bilaney Consultants GmbH, Düsseldorf, Germany) were stereotactically implanted, and infusions of anti-α10-SAP and anti-ctrl-SAP were made intracerebroventricularly. Three untreated and three anti-α10-SAP treated mice were sacrificed 5 weeks after cell injection to investigate the potential effect at an early time point. The mice sacrificed after 5 weeks had received treatments once per week for 5 weeks, and the other mice had received one additional treatment (1 µg in 2 µL per infusion). The mice were monitored at least every second day. The brain tissues were snap-frozen and cryo-sectioned for further analysis.
In total, 30 mice were utilized for this study; however, four mice were excluded in the analysis. One mouse in the untreated group had to be euthanized after cell injection due to weight loss following surgery, and one mouse in the anti-ctrl-SAP group was unable to wake up after blood sampling at week 4. In the anti-α10-SAP group, two mice were excluded because of technical failure during animal preparation.
4.12. Immunolabelings of Human and Mouse Brain Tissue and GBM Cell Lines
4.12.1. Immunohistochemistry
Human brain tissues were obtained from archival neurosurgical tissue biopsies embedded in paraffin blocks (Lund University). Ethical permission was obtained from the Regional Ethics Committee at Lund University (Dnr 2019-00598). Tissue samples contained regions with both normal tissue and tumor histopathology, as determined by a clinical pathologist (E.E.). Tissues were sectioned at 5 µm and collected on microscope slides (SuperFrost plus, Thermo Scientific). Sections were then re-hydrated and de-paraffinized by immersion in xylene (100% × 2) followed by immersion in a graded alcohol series (100% ethanol 1 min × 2, 95% ethanol 1 min, 70% ethanol 1 min × 2), ending with distilled water. Heat-induced antigen retrieval was performed in citrate buffer, pH 6.0 (10 mM sodium citrate) containing 0.05% Tween 20 (Sigma-Aldrich, St. Louis, MO, USA) for 10 min at 90 °C, followed by immersion in distilled water for 10 min and in PBS (3 × 3 min). Sections were then incubated in PBS containing 0.3% H2O2 for 10 min, followed by rinses in PBS (3 × 3 min). Sections were incubated in PBS containing 0.05% Triton X-100 (AppliChem, Darmstadt, GmbH) and 1% bovine serum albumin (BSA; Sigma-Aldrich, PBS-TX-BSA) for 30 min at room temperature (RT). Incubation was performed in primary antibodies made in rabbit against α10 (Xintela AB, diluted in PBS-TX-BSA) for 16 h at 4 °C. After rinses in PBS (3 × 3 min), sections were incubated with horseradish peroxidas (HRP)-conjugated secondary antibodies (goat anti-rabbit, DAKO Envision-HRP, Denmark) for 45 min at RT. Following rinses in PBS (3 × 3 min), sections were incubated for 10 min in PBS containing 3,3′-di-aminobenzidine (DAB, 25 mg/mL) and 0.05% H2O2. Sections were then rinsed in PBS (3 × 3 min) and counterstained with hematoxylin (Mayers, Histolab Gothenburg, Sweden). Sections were then dehydrated in a graded alcohol series (70%, 96% and 100%) ending with xylene (100%). Sections were mounted in Pertex (Histolab Gothenburg, Sweden) and cover-slipped. The grading of integrin α10 expression in brain tissues from patients with different clinical diagnoses was performed by scoring labeling intensities for integrin α10 immunoreactive cells. The labeling intensity was scored as 0 (negative), 1 (weak), 2 (moderate), and 3 (strong).
4.12.2. Immunofluorescence and Confocal Microscopy
Immunofluorescence labeling, including multiple labeling for simultaneous visualization of epitopes, was performed in cryosections from human and mouse tissues, and in GBM cell lines.
The U3071MG, U3078MG, U3046MG, U3054MG, and U3073MG cells were seeded on 8-well, glass bottom, microscope chamber slides (ibiTreat, Ibidi, Germany). For single and multi-labeling, cells were briefly rinsed once in cold PBS directly followed by fixation in cold 4% paraformaldehyde (Fisher Scientific, Loughborough, UK) for 10 min. Cells were washed and permeabilized via incubation in PBS containing 0.05% Triton X-100 (AppliChem, Darmstadt, Germany) for 2 × 5 min at RT and then incubated in PBS-TX-BSA at RT for 20 min. Cells were incubated with primary mouse antibodies made against integrin α10 (Xintela AB) either alone or together with antibodies against investigated epitopes (made in other species, see
Table S2), for 90 min at RT. Following rinses in PBS, cells were incubated with a mixture of fluorophore-conjugated species-specific secondary antibodies (
Table S3). Following rinses in PBS, cells were incubated in 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Molecular Probes, Invitrogen, 0.1 µM in PBS)) solution (Molecular Probes, Invitrogen) for 10 min.
Fresh frozen human brain tissues were embedded and frozen in optimal cutting temperature (OCT) compound (at approx. −60 °C). Frozen tissue was sectioned at 8 or 10 µm thickness and collected on SuperFrost slides (Thermo Scientific). Slides were air-dried at 37 °C for 20 min, rinsed in PBS and post-fixed in either acetone (100%) for 5‒10 min at −20 °C or paraformaldehyde (4% in PBS at 4‒8 °C, indicated optimal for NuMA labeling) for 20 min. Following rinses in PBS, sections were incubated in PBS-TX-BSA for 30 min at RT. Sections were incubated with primary rabbit antibodies made against integrin α10 (Xintela AB) alone or together with antibodies against investigated epitopes made in other species (See
Table S2). For the fluorescence multilabeling, sections were incubated with a mixture of primary antibodies (diluted in PBS-TX-BSA) for 16 h at 4 °C. Following rinses in PBS, sections were incubated with fluorophore-conjugated secondary antibodies (see
Table S3) for 45 min at RT. Following rinses in PBS-TX and once in PBS, sections were incubated in DAPI solution for 15 min, rinsed in PBS, and mounted in “anti-fade solution” ProLong Gold (Invitrogen, USA).
Immunofluorescence analysis and cell and tissue imaging were performed in a laser confocal scanning system (LSM 800 or 710, Zeiss, Germany) equipped for specific detection of the emission wavelengths for the used fluorophores. The pinhole aperture was set at one airy unit or was optimized for simultaneous detection of all visualized channels in multilabelings. The laser power and detection levels were optimized for each channel. Sequential scanning and detection of individual fluorophores was performed at magnifications provided by ×40 or ×63 magnification oil immersion lenses. For illustrations, one optical section was obtained for each fluorophore (0.5‒1 µm thick depending on the lens used), and image acquisition was performed through as many cells as possible in the scanned region. For analyses of the cellular relationship between labeled epitopes (i.e., co-labeling or not), individual channels of the imaged fluorophores were merged using the LSM software. Brightness and contrast were optimized for representative illustrations.
4.13. Histochemistry and Histopathological Evaluation of Mouse Brains
Mouse brain cryosections (12 µm) collected on microscope slides (SuperFrost plus, Thermo Scientific) were air-dried at RT for 30 min and then stained with 0.5% toluidine blue (Sigma Aldrich, cat. no. 198161) for 2 min. The sections were dehydrated stepwise with 70% ethanol for 30 sec, 96% ethanol for 20 sec and two changes of 100% ethanol for 20 sec and 10 sec, respectively, followed by two changes in xylene (100%) for 3 min each. Labeled sections were analyzed in a bright-field transmission microscope (Leica DMRE, Germany) and in images from whole scanned sections (40× magnification, Hamamatsu Nanozoomer 2.0ht, C900-12, Hamamatsu, Japan).
In scanned images, areas with findings of tumors consisting of 10 or more tumor cells with or without signs of necrosis, borders were delineated and individually marked in the images (Hamamatsu NPD.view 2.6.13 software, Hamamatsu City, Japan). Assessment and scoring of findings were performed by two blinded experts in histology/histopathology (B.H./J.F.). Areas defined as tumor areas without signs of apoptosis or necrosis (see below) were morphologically defined by the following: (1) typical cancerous neoplastic cells with a large and irregular shaped nucleus, prominent nucleoli and predominantly with a pale cytoplasmatic toluidine blue staining; or (2) cell populations with a high grade of mitosis, including atypical mitotic forms. Tumor areas with signs of apoptosis/necrosis (non-viable tumor areas), indicating treatment effects, were defined by the following morphological criteria: tumor areas (as above) containing inflammatory cells, cell membrane and/or nuclear debris.
4.14. Immunofluorescence Labelings of Mouse Brains
For immunofluorescence labelings of saporin and NuMA, mouse brain cryosections (12 µm), collected on microscope slides (SuperFrost plus, Thermo Scientific) were air-dried at RT for 30 min. Sections were then post-fixed in either acetone (100%, at −20 °C for 10 min) or paraformaldehyde (4%, at RT for 20 min). Sections were rinsed in PBS and incubated with primary antibodies against saporin and/or NUMA (see
Table S2) for 16 h at 4 °C. Sections were incubated with fluorescens conjugated secondary antibodies and processed further as described above (for human frozen tissues). Analyses and imaging were performed in a wide-field epifluorescence microscope (Olympus IX73, Tokyo, Japan). Illustrations were adjusted for brightness and contrast.
For labeling of apoptotic and/or dead cells, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed according to the process recommended by the manufacturer (Roche, cat no 11 684 795 910). All labelling experiments included negative control sections (by excluding terminal transferase from the protocol) and positive control sections (pretreatment of sections with DNAse I). Nuclear labeling was achieved by mounting in Fluoroshield mounting media containing DAPI (Abcam, Cambridge, UK). Labeling was analyzed and documented in an epi-fluorescence microscope (Leica DMRE, Germany) and evaluated for the presence or absence of a signal (detected in the 489 nm/FITC channel) in discernable tumor areas (using nuclear morphology visualized in the 405 nm/DAPI channel). Signal presence or absence was documented as positive or negative, respectively. Brain tissue from one of the mice in the anti-ctrl-SAP-treated group was not TUNEL stained due to a lack of sufficient tissue sections.
4.15. Anti-α10-SAP Targeting Analysis
Cells were fixed in cold 4% paraformaldehyde, followed by rinsing in PBS (2 × 2 min) and incubation in PBS-BSA-TX (15 min at RT). Anti-α10-SAP distribution was determined by labeling the integrin α10 antibody (made in mouse) with a secondary antibody against mouse IgG (mAb α10) conjugated with Alexa Fluor 488 (Jackson ImmunoResearch Inc., USA). Incubation of cells was performed at dilutions of 1:150‒200 for 30 min at RT. After 2 × 5 min rinses in PBS, cells were incubated in DAPI solution (Molecular Probes, Invitrogen) for 10 min. Analysis and digital imaging documentation were performed in a confocal laser scanning system (Zeiss LSM 800 or 710, as described above).
4.16. Statistical Analyses
Statistical analyses were performed using unpaired two-tailed Student t-test, as well as ANOVA followed by Dunnett, as stated in the figure legends. Four levels of significance were used, where * p < 0.05, ** p < 0.01, *** p < 0.001, and p < 0.0001.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







