Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists


Abstract
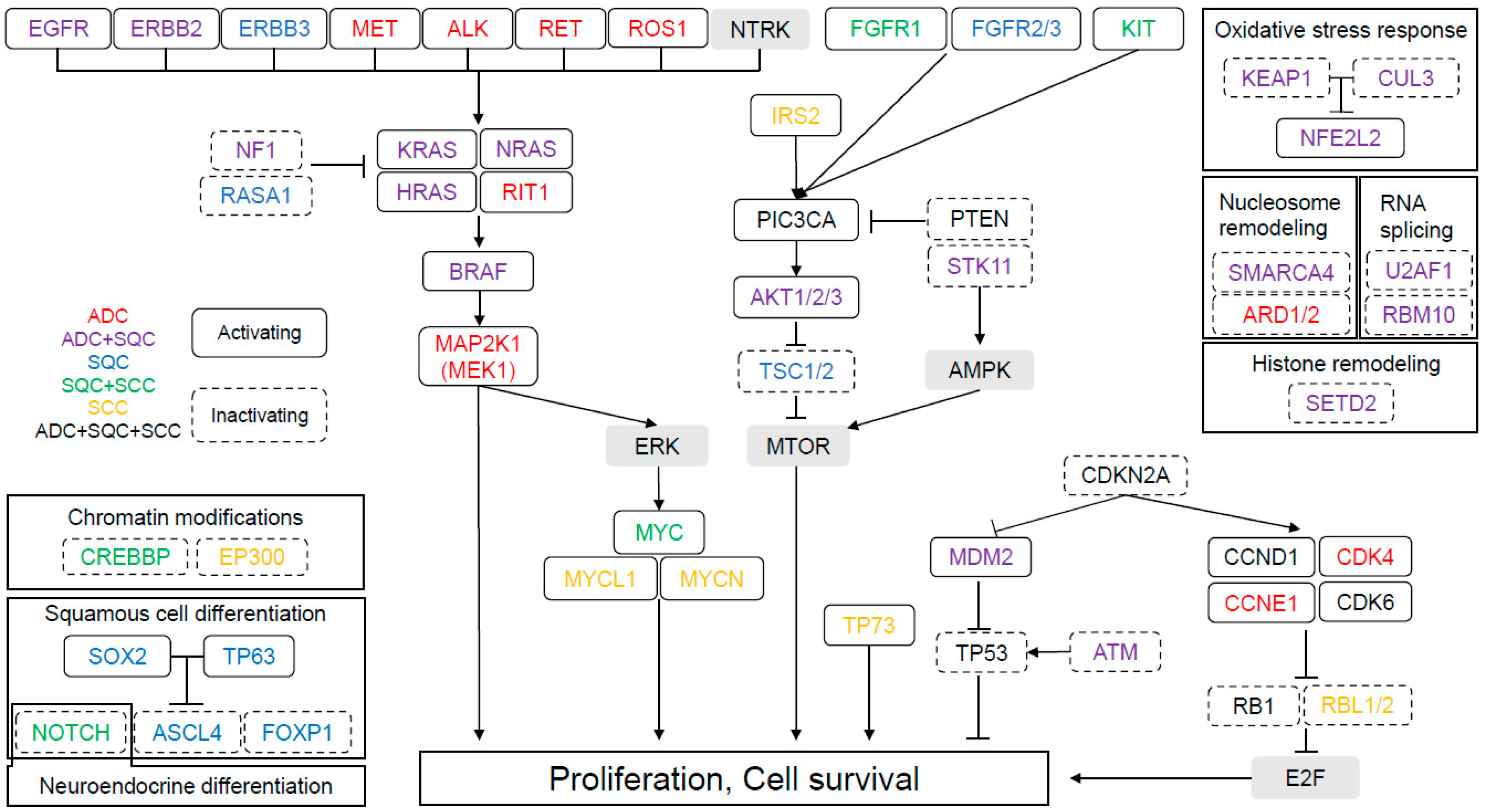
:1. Introduction
2. Adenocarcinomas and Squamous Cell Carcinomas
2.1. Morphological and Immunohistochemical Diagnosis
2.2. Adenocarcinoma
2.2.1. Histological Subtypes of ADC
2.2.2. Molecular Abnormalities in ADC Confirmed by TCGA
2.2.3. Molecular Abnormalities and Histological Pattern in ADC
2.3. Squamous Cell Carcinoma
2.3.1. Morphological Subtypes
2.3.2. Molecular Abnormalities in SQC Confirmed by TCGA
3. Molecular Alterations as Diagnostic Markers between ADC and SQC
4. Neuroendocrine Tumor
4.1. Morphological Definition
4.2. Molecular Abnormalities
4.2.1. Typical and Atypical Carcinoids (Low-Grade NET)
4.2.2. SCC and LCNEC (High-Grade NET)
Molecular Abnormalities of SCC Confirmed by TCGA
Molecular Alterations of LCNEC
Transformed SCC from NSCLC after Treatment with EGFR-Tyrosine Kinase Inhibitors (TKI)
4.2.3. Genetic Differences Between Low- and High-Grade NET
5. Other Histological Subtypes
5.1. Adenosquamous Carcinoma
5.2. Sarcomatoid Carcinoma
5.3. Salivary Gland-Type Tumor
5.4. Lymphoepithelioma-Like Carcinoma
5.5. NUT Carcinoma
6. Targeted Treatment
6.1. Targets and Therapies for Non-Small Cell Lung Cancer
6.1.1. EGFR Mutations
6.1.2. ALK Rearrangements
6.1.3. ROS1 Rearrangements
6.1.4. BRAF Mutations
6.1.5. Other Genetic Targets and Reagents for NSCLC
6.2. Biomarkers of Immunotherapy for NSCLC without Targetable Gene Alterations
6.3. Targets and Therapies for NET
7. Future Perspectives
8. Conclusions
Funding
Conflicts of Interest
References
- Noone, A.M.; Howlader, N.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER cancer statistics review (CSR) 1975–2015. Available online: https://seer.cancer.gov/csr/1975_2015/ (accessed on 1 November 2018).
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Yoshizawa, A.; Motoi, N.; Riely, G.J.; Sima, C.S.; Gerald, W.L.; Kris, M.G.; Park, B.J.; Rusch, V.W.; Travis, W.D. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: Prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod. Pathol. 2011, 24, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, H.; Hiramatsu, M.; Inamura, K.; Nomura, K.; Okui, M.; Miyoshi, T.; Okumura, S.; Satoh, Y.; Nakagawa, K.; Nishio, M.; et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas: Significance of the micropapillary pattern and the hobnail cell type. Lung Cancer 2009, 63, 235–240. [Google Scholar] [CrossRef]
- Haneda, H.; Sasaki, H.; Lindeman, N.; Kawano, O.; Endo, K.; Suzuki, E.; Shimizu, S.; Yukiue, H.; Kobayashi, Y.; Yano, M.; et al. A correlation between EGFR gene mutation status and bronchioloalveolar carcinoma features in Japanese patients with adenocarcinoma. Jpn. J. Clin. Oncol. 2006, 36, 69–75. [Google Scholar] [CrossRef]
- Matsumoto, S.; Iwakawa, R.; Kohno, T.; Suzuki, K.; Matsuno, Y.; Yamamoto, S.; Noguchi, M.; Shimizu, E.; Yokota, J. Frequent EGFR mutations in noninvasive bronchioloalveolar carcinoma. Int. J. Cancer 2006, 118, 2498–2504. [Google Scholar] [CrossRef]
- Hsieh, R.-K.; Lim, K.-H.; Kuo, H.-T.; Tzen, C.-Y.; Huang, M.-J. Female Sex and Bronchioloalveolar Pathologic Subtype Predict EGFR Mutations in Non-small Cell Lung Cancer. Chest 2005, 128, 317–321. [Google Scholar] [CrossRef]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Nomura, K.; Ninomiya, H.; Okui, M.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Soda, M.; et al. EML4-ALK Fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol. 2008, 3, 13–17. [Google Scholar] [CrossRef]
- Inamura, K.; Takeuchi, K.; Togashi, Y.; Hatano, S.; Ninomiya, H.; Motoi, N.; Mun, M.; Sakao, Y.; Okumura, S.; Nakagawa, K.; et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod. Pathol. 2009, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non–small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef]
- Hwang, D.H.; Szeto, D.P.; Perry, A.S.; Bruce, J.L.; Sholl, L.M. Pulmonary large cell carcinoma lacking squamous differentiation is clinicopathologically indistinguishable from solid-subtype adenocarcinoma. Arch. Pathol. Lab. Med. 2013, 138, 626–635. [Google Scholar] [CrossRef]
- Sholl, L.M.; Sun, H.; Butaney, M.; Zhang, C.; Lee, C.; Jänne, P.A.; Rodig, S.J. ROS1 Immunohistochemistry for detection of ROS1-rearranged lung adenocarcinomas. Am. J. Surg. Pathol. 2013, 37, 1441–1449. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, J.; Zheng, J.; Kong, M.; Sun, K.; Wang, B.; Chen, X.; Ding, W.; Zhou, J. A prediction model for ROS1-rearranged lung adenocarcinomas based on histologic features. PLoS ONE 2016, 11, 1–12. [Google Scholar] [CrossRef]
- Wang, R.; Hu, H.; Pan, Y.; Li, Y.; Ye, T.; Li, C.; Luo, X.; Wang, L.; Li, H.; Zhang, Y.; et al. RET fusions define a unique molecular and clinicopathologic subtype of non–small-cell lung cancer. J. Clin. Oncol. 2012, 30, 4352–4359. [Google Scholar] [CrossRef]
- Nadal, E.; Zhong, J.; Lin, J.; Reddy, R.M.; Ramnath, N.; Orringer, M.B.; Chang, A.C.; Beer, D.G.; Chen, G. A MicroRNA cluster at 14q32 drives aggressive lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 3107–3117. [Google Scholar] [CrossRef]
- Kadota, K.; Yeh, Y.-C.; D’Angelo, S.P.; Moreira, A.L.; Kuk, D.; Sima, C.S.; Chang, A.C.; Beer, D.G.; Chen, G. Associations between mutations and histologic patterns of mucin in lung adenocarcinoma: Invasive mucinous pattern and extracellular mucin are associated with KRAS mutation. Am. J. Surg. Pathol. 2014, 38, 1118–1127. [Google Scholar] [CrossRef]
- Righi, L.; Vatrano, S.; Di Nicolantonio, F.; Massa, F.; Rossi, G.; Cavazza, A.; Volante, M.; Votta, A.; Izzo, S.; Iacono, M.L.; et al. Retrospective multicenter study investigating the role of targeted next-generation sequencing of selected cancer genes in mucinous adenocarcinoma of the lung. J. Thorac. Oncol. 2016, 11, 504–515. [Google Scholar] [CrossRef]
- Nakaoku, T.; Tsuta, K.; Ichikawa, H.; Shiraishi, K.; Sakamoto, H.; Enari, M.; Furuta, K.; Shimada, Y.; Ogiwara, H.; Watanabe, S.I.; et al. Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 3087–3093. [Google Scholar] [CrossRef]
- Soo, R.A.; Stone, E.C.A.; Cummings, K.M.; Jett, J.R.; Field, J.K.; Groen, H.J.M.; Mulshine, J.L.; Yatabe, Y.; Bubendorf, L.; Dacic, S.; et al. Scientific advances in thoracic oncology 2016. J. Thorac. Oncol. 2017, 12, 1183–1209. [Google Scholar] [CrossRef]
- Finberg, K.E.; Sequist, L.V.; Joshi, V.A.; Muzikansky, A.; Miller, J.M.; Han, M.; Behesht, J.; Chirieac, L.R.; Mark, E.J.; Iafrate, A.J. Mucinous differentiation correlates with absence of EGFR mutation and presence of KRAS mutation in lung adenocarcinomas with bronchioloalveolar features. J. Mol. Diagn. 2007, 9, 320–326. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Plenker, D.; Osada, H.; Sun, R.; Menon, R.; Leenders, F.; Ortiz-Cuaran, S.; Peifer, M.; Bos, M.; Daßler, J.; et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014, 4, 415–422. [Google Scholar] [CrossRef]
- Sup, S.H.; Mino-Kenudson, M.; Zheng, Z.; Liebers, M.; Jin, C.Y.; Hoang, H.Q.; Onozato, M.; Phi, L.L.; Heist, R.S.; Iafrate, A.J. Unique genetic and survival characteristics of invasive mucinous adenocarcinoma of the lung. J. Thorac. Oncol. 2015, 10, 1156–1162. [Google Scholar]
- Sonzogni, A.; Bianchi, F.; Fabbri, A.; Cossa, M.; Rossi, G.; Cavazza, A.; Tamborini, E.; Perrone, F.; Busico, A.; Capone, I.; et al. Pulmonary adenocarcinoma with mucin production modulates phenotype according to common genetic traits: A reappraisal of mucinous adenocarcinoma and colloid adenocarcinoma. J. Pathol. Clin. Res. 2017, 3, 139–151. [Google Scholar] [CrossRef]
- Maeda, Y.; Tsuchiya, T.; Hao, H.; Tompkins, D.H.; Xu, Y.; Mucenski, M.L.; Du, L.; Keiser, A.R.; Fukazawa, T.; Naomoto, Y.; et al. Kras(G12D) and Nkx2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J. Clin. Invest. 2012, 122, 4388–4400. [Google Scholar] [CrossRef]
- Hwang, D.H.; Sholl, L.M.; Rojas-Rudilla, V.; Hall, D.L.; Shivdasani, P.; Garcia, E.P.; MacConaill, L.E.; Vivero, M.; Hornick, J.L.; Kuo, F.C.; et al. KRAS and NKX2-1 mutations in invasive mucinous adenocarcinoma of the lung. J. Thorac. Oncol. 2016, 11, 496–503. [Google Scholar] [CrossRef]
- Chen, M.; Liu, P.; Yan, F.; Xu, S.; Jiang, Q.; Pan, J.; He, M.; Shen, P. Distinctive features of immunostaining and mutational load in primary pulmonary enteric adenocarcinoma: Implications for differential diagnosis and immunotherapy. J. Transl. Med. 2018, 16, 81. [Google Scholar] [CrossRef]
- Nottegar, A.; Tabbò, F.; Luchini, C.; Brunelli, M.; Bria, E.; Veronese, N.; Santo, A.; Cingarlini, S.; Gilioli, E.; Ogliosi, C.; et al. Pulmonary adenocarcinoma with enteric differentiation: Immunohistochemistry and molecular morphology. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 383–387. [Google Scholar] [CrossRef]
- Rossi, G.; Murer, B.; Cavazza, A.; Losi, L.; Natali, P.; Marchioni, A.; Migaldi, M.; Capitanio, G.; Brambilla, E. Primary mucinous (so-called colloid) carcinomas of the lung: A clinicopathologic and immunohistochemical study with special reference to CDX-2 homeobox gene and MUC2 expression. Am. J. Surg. Pathol. 2004, 28, 442–452. [Google Scholar] [CrossRef]
- Nakatani, Y.; Miyagi, Y.; Takemura, T.; Oka, T.; Yokoi, T.; Takagi, M.; Yokoyama, S.; Kashima, K.; Hara, K.; Yamada, T.; et al. Aberrant nuclear/cytoplasmic localization and gene mutation of β-catenin in classic pulmonary blastoma: β-catenin immunostaining is useful for distinguishing between classic pulmonary blastoma and a blastomatoid variant of carcinosarcoma. Am. J. Surg. Pathol. 2004, 28, 921–927. [Google Scholar] [CrossRef]
- Fu, Y.; Wu, Q.; Su, F.; Tang, Y.; Lin, Y.; Wang, W.; Jiang, L. Novel gene mutations in well-differentiated fetal adenocarcinoma of the lung in the next generation sequencing era. Lung Cancer 2018, 124, 1–5. [Google Scholar] [CrossRef]
- Suzuki, M.; Yazawa, T.; Ota, S.; Morimoto, J.; Yoshino, I.; Yamanaka, S.; Inayama, Y.; Kawabata, Y.; Shimizu, Y.; Komatsu, M.; et al. High-grade fetal adenocarcinoma of the lung is a tumour with a fetal phenotype that shows diverse differentiation, including high-grade neuroendocrine carcinoma: A clinicopathological, immunohistochemical and mutational study of 20 cases. Histopathology 2015, 67, 806–816. [Google Scholar] [CrossRef]
- Shinmura, K.; Igarashi, H.; Kato, H.; Kawanishi, Y.; Inoue, Y.; Nakamura, S.; Ogawa, H.; Yamashita, T.; Kawase, A.; Funai, K.; et al. CLCA2 as a novel immunohistochemical marker for differential diagnosis of squamous cell carcinoma from adenocarcinoma of the lung. Dis. Markers 2014, 2014, 619273. [Google Scholar] [CrossRef]
- Sun, F.; Yang, X.; Jin, Y.; Chen, L.; Wang, L.; Shi, M.; Zhan, C.; Shi, Y.; Wang, Q. Bioinformatics analyses of the differences between lung adenocarcinoma and squamous cell carcinoma using the Cancer Genome Atlas expression data. Mol. Med. Rep. 2017, 16, 609–616. [Google Scholar] [CrossRef]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Bishitz, Y.; Paul, A.; Krelin, Y.; Nakdimon, I.; Peled, N.; Lavon, A.; Rudoy-Zilberman, E.; Refaely, Y. A molecular signature of lung cancer: Potential biomarkers for adenocarcinoma and squamous cell carcinoma. Oncotarget 2017, 8, 105492–105509. [Google Scholar] [CrossRef]
- Pelosi, G.; Sonzogni, A.; Harari, S.; Albini, A.; Bresaola, E.; Marchiò, C.; Massa, F.; Righi, L.; Gatti, G.; Papanikolaou, N.; et al. Classification of pulmonary neuroendocrine tumors: New insights. Transl. Lung Cancer Res. 2017, 6, 513–529. [Google Scholar] [CrossRef]
- Den Bakker, M.A.; Willemsen, S.; Grünberg, K.; Noorduijn, L.A.; Van Oosterhout, M.F.M.; Van Suylen, R.J.; Timens, W.; Vrugt, B.; Wiersma-van, T.A.; Thunnissen, F.B.J.M. Small cell carcinoma of the lung and large cell neuroendocrine carcinoma interobserver variability. Histopathology 2010, 56, 356–363. [Google Scholar] [CrossRef]
- Den Bakker, M.A.; Thunnissen, F.B.J.M. Neuroendocrine tumours—challenges in the diagnosis and classification of pulmonary neuroendocrine tumours. J. Clin. Pathol. 2013, 66, 862–869. [Google Scholar] [CrossRef]
- Bari, M.F.; Brown, H.; Nicholson, A.G.; Kerr, K.M.; Gosney, J.R.; Wallace, W.A.; Soomro, I.; Muller, S.; Peat, D.; Moore, J.D.; et al. BAI3, CDX2 and VIL1: A panel of three antibodies to distinguish small cell from large cell neuroendocrine lung carcinomas. Histopathology 2013, 64, 547–556. [Google Scholar] [CrossRef]
- Henry Walter, R.F.; Vollbrecht, C.; Christoph, D.; Werner, R.; Schmeller, J.; Flom, E.; Trakada, G.; Rapti, A.; Adamidis, V.; Hohenforst-Schmidt, W.; et al. Massive parallel sequencing and digital gene expression analysis reveals potential mechanisms to overcome therapy resistance in pulmonary neuroendocrine tumors. J. Cancer 2016, 7, 2165–2172. [Google Scholar] [CrossRef]
- Armengol, G.; Sarhadi, V.K.; Rönty, M.; Tikkanen, M.; Knuuttila, A.; Knuutila, S. Driver gene mutations of non-small-cell lung cancer are rare in primary carcinoids of the lung: NGS study by Ion Torrent. Lung 2015, 193, 303–308. [Google Scholar] [CrossRef]
- Rapa, I.; Votta, A.; Felice, B.; Righi, L.; Giorcelli, J.; Scarpa, A.; Speel, E.J.M.; Scagliotti, G.V.; Papotti, M.; Volante, M.; et al. Identification of MicroRNAs differentially expressed in lung carcinoid subtypes and progression. Neuroendocrinology 2015, 101, 246–255. [Google Scholar] [CrossRef]
- Rekhtman, N.; Pietanza, M.C.; Hellmann, M.D.; Naidoo, J.; Arora, A.; Won, H.; Halpenny, D.F.; Wang, H.; Tian, S.K.; Litvak, A.M.; et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin. Cancer Res. 2016, 22, 3618–3629. [Google Scholar] [CrossRef]
- Miyoshi, T.; Umemura, S.; Matsumura, Y.; Mimaki, S.; Tada, S.; Makinoshima, H.; Ishii, G.; Udagawa, H.; Matsumoto, S.; Yoh, K.; et al. Genomic profiling of large-cell neuroendocrine carcinoma of the lung. Clin. Cancer Res. 2017, 23, 757–765. [Google Scholar] [CrossRef]
- Popat, S.; Wotherspoon, A.; Nutting, C.M.; Gonzalez, D.; Nicholson, A.G.; O’Brien, M. Transformation to “high grade” neuroendocrine carcinoma as an acquired drug resistance mechanism in EGFR-mutant lung adenocarcinoma. Lung Cancer 2013, 80, 1–4. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 199–203. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef]
- Meder, L.; König, K.; Ozretic, L.; Schultheis, A.M.; Ueckeroth, F.; Ade, C.P.; Albus, K.; Boehm, D.; Rommerscheidt-Fuss, U.; Florin, A.; et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int. J. Cancer 2016, 138, 927–938. [Google Scholar] [CrossRef]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretić, L.; Seidel, D.; Zander, T.; Leenders, F.; George, J.; Müller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5, 3518. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Mafficini, A.; Sikora, K.O.; Fassan, M.; Barbi, S.; Corbo, V.; Mastracci, L.; Rusev, B.; Grillo, F.; Vicentini, C.; et al. Lung neuroendocrine tumours: Deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J. Pathol. 2017, 241, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Pan, Y.; Li, C.; Zhang, H.; Garfield, D.; Li, Y.; Ye, T.; Hu, H.; Luo, X.; Li, H.; et al. Analysis of major known driver mutations and prognosis in resected adenosquamous lung carcinomas. J. Thorac. Oncol. 2014, 9, 760–768. [Google Scholar] [CrossRef]
- Fallet, V.; Saffroy, R.; Girard, N.; Mazieres, J.; Lantuejoul, S.; Vieira, T.; Rouquette, I.; Thivolet-Bejui, F.; Ung, M.; Poulot, V.; et al. High-throughput somatic mutation profiling in pulmonary sarcomatoid carcinomas using the LungCartaTM Panel: Exploring therapeutic targets. Ann. Oncol. 2015, 26, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Terra, S.B.S.P.; Jang, J.S.; Bi, L.; Kipp, B.R.; Jen, J.; Yi, E.S.; Boland, J.M. Molecular characterization of pulmonary sarcomatoid carcinoma: Analysis of 33 cases. Mod. Pathol. 2016, 29, 824–831. [Google Scholar] [CrossRef]
- Nakagomi, T.; Goto, T.; Hirotsu, Y.; Shikata, D.; Yokoyama, Y.; Higuchi, R.; Amemiya, K.; Okimoto, K.; Oyama, T.; Mochizuki, H.; et al. New therapeutic targets for pulmonary sarcomatoid carcinomas based on their genomic and phylogenetic profiles. Oncotarget 2018, 9, 10635–10649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, A.C.; García, J.J.; Wehrs, R.N.; Colby, T.V.; Khoor, A.; Leslie, K.O.; Chen, L. Histopathologic, immunophenotypic and cytogenetic features of pulmonary mucoepidermoid carcinoma. Mod. Pathol. 2014, 27, 1479–1488. [Google Scholar] [CrossRef]
- Achcar, R.D.O.D.; Nikiforova, M.N.; Dacic, S.; Nicholson, A.G.; Yousem, S.A. Mammalian mastermind like 2 11q21 gene rearrangement in bronchopulmonary mucoepidermoid carcinoma. Hum. Pathol. 2009, 40, 854–860. [Google Scholar] [CrossRef]
- Salem, A.; Bell, D.; Sepesi, B.; Papadimitrakopoulou, V.; El-Naggar, A.; Moran, C.A.; Kalhor, N. Clinicopathologic and genetic features of primary bronchopulmonary mucoepidermoid carcinoma: The MD Anderson Cancer Center experience and comprehensive review of the literature. Virchows Arch. 2017, 470, 619–626. [Google Scholar] [CrossRef]
- Yu, Y.; Song, Z.; Gao, H.; Zhu, L.; Lu, S.; Zhang, J.; Luo, Q. EGFR L861Q mutation is a frequent feature of pulmonary mucoepidermoid carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 1421–1425. [Google Scholar] [CrossRef]
- Rossi, G.; Sartori, G.; Cavazza, A.; Tamberi, S. Mucoepidermoid carcinoma of the lung, response to EGFR inhibitors, EGFR and K-RAS mutations, and differential diagnosis. Lung Cancer 2009, 63, 159–160. [Google Scholar] [CrossRef]
- Han, S.-W.; Kim, H.-P.; Jeon, Y.K.; Oh, D.-Y.; Lee, S.-H.; Kim, D.-W.; Im, S.-A.; Chung, D.H.; Heo, D.S.; Bang, Y.-J.; et al. Mucoepidermoid carcinoma of lung: Potential target of EGFR-directed treatment. Lung Cancer 2008, 61, 30–34. [Google Scholar] [CrossRef]
- Shang, J.; Shui, Y.; Sheng, L.; Wang, K.; Hu, Q.; Wei, Q. Epidermal growth factor receptor and human epidermal growth receptor 2 expression in parotid mucoepidermoid carcinoma: Possible implications for targeted therapy. Oncol. Rep. 2008, 19, 435–440. [Google Scholar] [CrossRef]
- Nakano, T.; Yamamoto, H.; Hashimoto, K.; Tamiya, S.; Shiratsuchi, H.; Nakashima, T.; Nishiyama, K.; Higaki, Y.; Komune, S.; Oda, Y. HER2 and EGFR gene copy number alterations are predominant in high-grade salivary mucoepidermoid carcinoma irrespective of MAML2 fusion status. Histopathology 2013, 63, 378–392. [Google Scholar] [CrossRef]
- Aubry, M.C.; Heinrich, M.C.; Molina, J.; Lewis, J.E.; Yang, P.; Cassivi, S.D.; Corless, C.L. Primary adenoid cystic carcinoma of the lung: Absence of KIT mutations. Cancer 2007, 110, 2507–2510. [Google Scholar] [CrossRef]
- Macarenco, R.S.; Uphoff, T.S.; Gilmer, H.F.; Jenkins, R.B.; Thibodeau, S.N.; Lewis, J.E.; Molina, J.R.; Yang, P.; Aubry, M.-C. Salivary gland-type lung carcinomas: An EGFR immunohistochemical, molecular genetic, and mutational analysis study. Mod. Pathol. 2008, 21, 1168–1175. [Google Scholar] [CrossRef]
- Huo, Z.; Wu, H.; Li, S.; Liang, Z. Molecular genetic studies on EGFR, KRAS, BRAF, ALK, PIK3CA, PDGFRA, and DDR2 in primary pulmonary adenoid cystic carcinoma. Diagn. Pathol. 2015, 10, 161. [Google Scholar] [CrossRef]
- Chang, Y.L.; Wu, C.T.; Shih, J.Y.; Lee, Y.C. Unique p53 and epidermal growth factor receptor gene mutation status in 46 pulmonary lymphoepithelioma-like carcinomas. Cancer Sci. 2011, 102, 282–287. [Google Scholar] [CrossRef]
- Wong, D.W.S.; Leung, E.L.H.; So, K.K.T.; Tam, I.Y.S.; Sihoe, A.D.L.; Cheng, L.C.; Ho, K.K.; Au, J.S.K.; Chung, L.P.; Wong, M.P. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 2009, 115, 1723–1733. [Google Scholar] [CrossRef]
- French, C.A.; Kutok, J.L.; Faquin, W.C.; Toretsky, J.A.; Antonescu, C.R.; Griffin, C.A.; Nose, V.; Vargas, S.O.; Moschovi, M.; Tzortzatou-Stathopoulou, F.; et al. Midline carcinoma of children and young adults with NUT rearrangement. J. Clin. Oncol. 2004, 22, 4135–4139. [Google Scholar] [CrossRef]
- French, C.A. Demystified molecular pathology of NUT midline carcinomas. J. Clin. Pathol. 2010, 63, 492–496. [Google Scholar] [CrossRef]
- Mao, N.; Liao, Z.; Wu, J.; Liang, K.; Wang, S.; Qin, S.; Dou, Y.; Lin, H.; Dong, X. Diagnosis of NUT carcinoma of lung origin by next-generation sequencing: Case report and review of the literature. Cancer Biol. Ther. 2018, 11, 1–7. [Google Scholar] [CrossRef]
- Baras, A.S.; Naidoo, J.; Hann, C.L.; Illei, P.B.; Reninger, C.W.; Lauring, J. Rediagnosis of Lung Cancer as NUT Midline Carcinoma Based on Clues From Tumor Genomic Profiling. J. Natl. Compr. Cancer Netw. 2018, 16, 467–472. [Google Scholar] [CrossRef]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non-Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; Porta, R.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M.; et al. Phase III Study of Afatinib or Cisplatin Plus Pemetrexed in Patients With Metastatic Lung Adenocarcinoma With EGFR Mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Hu, C.-P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Wu, Y.-L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.-P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.-H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.-H.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2016, 376, 629–640. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 378, 113–125. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Presented at the ESMO 2018 Congress, Munich, Germany, 20 October 2018. Abstract LBA50. [Google Scholar]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef]
- Nakamura, A.; Inoue, A.; Morita, S.; Hosomi, Y.; Kato, T.; Fukuhara, T.; Gemma, A.; Takahashi, K.; Fujita, Y.; Harada, T.; et al. Phase III study comparing gefitinib monotherapy (G) to combination therapy with gefitinib, carboplatin, and pemetrexed (GCP) for untreated patients (pts) with advanced non-small cell lung cancer (NSCLC) with EGFR mutations (NEJ009). J. Clin. Oncol. 2018, 36, 9005. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.S.K.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; Ou, S.-H.I.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20–selective kinase inhibitor in non–small cell lung cancer. Nat. Med. 2018, 24, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Jänne, P.A.; Costa, D.B.; et al. Anaplastic Lymphoma Kinase Inhibition in Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Peters, S.; Mok, T.; Gadgeel, S.M.; Cheema, P.K.; Pavlakis, N.; De Marinis, F.; Stroyakovskiy, D.L.; Cho, B.C.; Zhang, L.; et al. Updated efficacy and safety data from the global phase III ALEX study of alectinib (ALC) vs crizotinib (CZ) in untreated advanced ALK+ NSCLC. J. Clin. Oncol. 2018, 36, 9043. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Zhou, C.; Lu, Y.; Kim, S.-W.; Reungwetwattana, T.; Zhou, J.; Zhang, Y.; He, J.; Yang, J.-J.; Cheng, Y.; Lee, S.H.; et al. Primary results of ALESIA: A randomised, phase III, open-label study of alectinib vs crizotinib in Asian patients with treatment-naïve ALK+ advanced NSCLC. Ann. Oncol. 2018, 29, doi. [Google Scholar]
- Wang, Y.; Tian, P.-W.; Wang, W.-Y.; Wang, K.; Zhang, Z.; Chen, B.-J.; He, Y.-Q.; Li, L.; Liu, H.; Chuai, S.; et al. Noninvasive genotyping and monitoring of anaplastic lymphoma kinase (ALK) rearranged non-small cell lung cancer by capture-based next-generation sequencing. Oncotarget 2016, 7, 65208–65217. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, B.; et al. Crizotinib in ROS1-Rearranged Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Yang, J.C.-H.; Kim, D.-W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.-J.; Yamamoto, N.; Ahn, M.-J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients With ROS1-Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Shaw, A.T.; Riely, G.J.; Chiari, R.; Bauman, J.R.; Clancy, J.S.; Thurm, H.; Peltz, G.; Abbattista, A.; Solomon, B.J. Clinical Activity of Lorlatinib in Patients with ROS1+ Advanced Non-Small Cell Lung Cancer: Phase 2 Study Cohort EXP-6. In International Association for the Study of Lung Cancer; Canada, September 23–26; Elsevier: Amsterdam, The Netherlands, 2018; OA02.02. [Google Scholar]
- Doebele, R.C.; Ahn, M.-J.; Siena, S.; Drilon, A.; Krebs, M.G.; Lin, C.-C.; De Braud, F.G.; John, T.; Tan, D.S.W.; Seto, T.; et al. Efficacy and Safety of Entrectinib in Locally Advanced or Metastatic ROS1 Fusion-Positive Non-Small Cell Lung Cancer (NSCLC). In International Association for the Study of Lung Cancer; Canada, September 23–26; Elsevier: Amsterdam, The Netherlands, 2018; OA02.01. [Google Scholar]
- Song, A.; Kim, T.M.; Kim, D.-W.; Kim, S.; Keam, B.; Lee, S.-H.; Heo, D.S. Molecular Changes Associated with Acquired Resistance to Crizotinib in ROS1–Rearranged Non–Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 2379–2387. [Google Scholar] [CrossRef]
- Katayama, R.; Kobayashi, Y.; Friboulet, L.; Lockerman, E.L.; Koike, S.; Shaw, A.T.; Engelman, J.A.; Fujita, N. Cabozantinib Overcomes Crizotinib Resistance in ROS1 Fusion–Positive Cancer. Clin. Cancer Res. 2015, 21, 166–174. [Google Scholar] [CrossRef]
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Planchard, D.; Kim, T.M.; Mazieres, J.; Quoix, E.; Riely, G.; Barlesi, F.; Souquet, P.-J.; Smit, E.F.; Groen, H.J.M.; Kelly, R.J.; et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: A single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 642–650. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: An open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef]
- Velcheti, V.; Hida, T.; Reckamp, K.L.; Yang, J.C.; Nokihara, H.; Sachdev, P.; Feit, K.; Kubota, T.; Nakada, T.; Dutcus, C.E.; et al. Phase 2 study of lenvatinib (LN) in patients (Pts) with RET fusion-positive adenocarcinoma of the lung. Ann. Oncol. 2016, 27, 1204PD. [Google Scholar] [CrossRef]
- Yoh, K.; Seto, T.; Satouchi, M.; Nishio, M.; Yamamoto, N.; Murakami, H.; Nogami, N.; Matsumoto, S.; Kohno, T.; Tsuta, K.; et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): An open-label, multicentre phase 2 trial. Lancet Respir. Med. 2017, 5, 42–50. [Google Scholar] [CrossRef]
- Drilon, A.E.; Subbiah, V.; Oxnard, G.R.; Bauer, T.M.; Velcheti, V.; Lakhani, N.J.; Besse, B.; Park, K.; Patel, J.D.; Cabanillas, M.E.; et al. A phase 1 study of LOXO-292, a potent and highly selective RET inhibitor, in patients with RET-altered cancers. J. Clin. Oncol. 2018, 36, 102. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non–Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef]
- Wolf, J.; Han, J.; Nishio, M.; Souquet, P.; Paz-Ares, L.; De Marinis, F.; Seto, T.; De Jonge, M.; Kim, T.M.; Vansteenkiste, J.; et al. PS04.06 GEOMETRY Mono-1: Phase II, Multicenter Study of MET Inhibitor Capmatinib (INC280) in EGFR wt, MET-dysregulated Advanced NSCLC: Topic: Medical Oncology. J. Thorac. Oncol. 2017, 12, S1578–S1579. [Google Scholar] [CrossRef]
- Paik, P.K.; Drilon, A.; Fan, P.-D.; Yu, H.; Rekhtman, N.; Ginsberg, M.S.; Borsu, L.; Schultz, N.; Berger, M.F.; Rudin, C.M.; et al. Response to MET Inhibitors in Patients with Stage IV Lung Adenocarcinomas Harboring MET Mutations Causing Exon 14 Skipping. Cancer Discov. 2015, 5, 842–849. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Wang, Y.; Xu, Y.; Tian, Y.; Huang, M.; Lu, Y. Response to Crizotinib Observed in Lung Adenocarcinoma with MET Copy Number Gain but without a High-Level MET/CEP7 Ratio, MET Overexpression, or Exon 14 Splicing Mutations. J. Thorac. Oncol. 2016, 11, e59–e62. [Google Scholar] [CrossRef]
- Caparica, R.; Yen, C.T.; Coudry, R.; Ou, S.-H.I.; Varella-Garcia, M.; Camidge, D.R.; de Castro, G. Responses to Crizotinib Can Occur in High-Level MET-Amplified Non-Small Cell Lung Cancer Independent of MET Exon 14 Alterations. J. Thorac. Oncol. 2017, 12, 141–144. [Google Scholar] [CrossRef]
- Farago, A.F.; Taylor, M.S.; Doebele, R.C.; Zhu, V.W.; Kummar, S.; Spira, A.I.; Boyle, T.A.; Haura, E.B.; Arcila, M.E.; Benayed, R.; et al. Clinicopathologic Features of Non-Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis. Oncol. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Lange, A.M.; Lo, H.-W. Inhibiting TRK Proteins in Clinical Cancer Therapy. Cancers 2018, 10, 105. [Google Scholar] [CrossRef]
- Okuma, Y.; Wakui, H.; Utsumi, H.; Sagawa, Y.; Hosomi, Y.; Kuwano, K.; Homma, S. Soluble Programmed Cell Death Ligand 1 as a Novel Biomarker for Nivolumab Therapy for Non-Small-cell Lung Cancer. Clin. Lung Cancer 2018, 19, 410–417. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Presley, C.; Tang, D.; Soulos, P.; Chiang, A.C.; Longtine, J.A.; Adelson, K.B.; Herbst, R.S.; Zhu, W.; Nussbaum, N.C.; Sorg, R.A.; et al. Association of broad-based genomic sequencing with survival among patients with advanced non–small cell lung cancer in the community oncology setting. JAMA 2018, 320, 469–477. [Google Scholar] [CrossRef]
- Coudray, N.; Ocampo, P.S.; Sakellaropoulos, T.; Narula, N.; Snuderl, M.; Fenyö, D.; Moreira, A.L.; Razavian, N.; Tsirigos, A. Classification and mutation prediction from non–small cell lung cancer histopathology images using deep learning. Nat. Med. 2018, 24, 1559–1567. [Google Scholar] [CrossRef]
- Yu, K.H.; Zhang, C.; Berry, G.J.; Altman, R.B.; Ré, C.; Rubin, D.L.; Snyder, M. Predicting non-small cell lung cancer prognosis by fully automated microscopic pathology image features. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Histology | Targeted Molecule | Reagents | FDA-Approved (April 2019) | Note |
|---|---|---|---|---|
| NSCLC | EGFR (ADC 14%, SQC 9%) | Gefitinib (First-generation) | Yes | Selective and reversible TKI. |
| Erlotinib (First-generation) | Yes | Selective and reversible TKI. | ||
| Afatinib (Second-generation) | Yes | Irreversible ErbB family blocker. | ||
| Dacomitinib (Second-generation) | Yes | Irreversible TKI. | ||
| Osimertinib (Third-generation) | Yes | Irreversible and also active against the resistance mutation (T790M). | ||
| Poziotinib | No | Irreversible and active against exon20 mutation and HER2 mutation. | ||
| TAK-788 | No | Active against exon20 mutation and HER2 mutation. | ||
| TAS6417 | No | Selective against exon 20 insertion mutation. | ||
| ALK (ADC 5%) | Crizotinib(First-generation) | Yes | Multi-targeted TKI. | |
| Alectinib (Second-generation) | Yes | Highly selective inhibitor for ALK. | ||
| Ceritinib (Second-generation) | Yes | Highly selective inhibitor for ALK. | ||
| Brigatinib (Second-generation) | Yes | ALK/ROS1 inhibitor. | ||
| Lorlatinib (Third-generation) | Yes | ALK/ROS1 inhibitor. | ||
| ROS1 (ADC 1%) | Crizotinib | Yes | ||
| Lorlatinib | No | |||
| Entrectinib | No | Inhibits ROS1, TRK and ALK. | ||
| BRAF (ADC 7%, SQC 4%) | Dabrafenib/trametinib | Yes | Reversible ATP-competitive kinase inhibitor. | |
| RET (ADC 1%) | Cabozantinib | No | Multi-targeted TKI. | |
| Lenvatinib | No | Multi-targeted TKI. | ||
| Vandetanib | Yes | Multi-targeted TKI. | ||
| LOXO-292 | No | Selective RET inhibitor | ||
| MET (ADC 2–4%) | Crizotinib | No | ||
| Capmatinib | No | Reversible MET inhibitor. | ||
| Tepotinib | No | Reversible MET inhibitor. | ||
| NTRK (<1%) | Entrectinib | No | ||
| Larotrectinib | Yes | TRK inhibitor. | ||
| PD-1 | Nivolumab | Yes | IHC: 28-8 | |
| Pembrolizumab | Yes | IHC: 22C3 | ||
| PD-L1 | Atezolizumab | Yes | IHC: SP142 | |
| Duvalumab | Yes | IHC: SP263 | ||
| SCC | DLL3 (80%) | Rovalpituzumab tesirine | No | Antibody against DLL3 conjugated with cytotoxic reagent. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kashima, J.; Kitadai, R.; Okuma, Y. Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists. Cancers 2019, 11, 599. https://doi.org/10.3390/cancers11050599
Kashima J, Kitadai R, Okuma Y. Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists. Cancers. 2019; 11(5):599. https://doi.org/10.3390/cancers11050599
Chicago/Turabian StyleKashima, Jumpei, Rui Kitadai, and Yusuke Okuma. 2019. "Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists" Cancers 11, no. 5: 599. https://doi.org/10.3390/cancers11050599
APA StyleKashima, J., Kitadai, R., & Okuma, Y. (2019). Molecular and Morphological Profiling of Lung Cancer: A Foundation for “Next-Generation” Pathologists and Oncologists. Cancers, 11(5), 599. https://doi.org/10.3390/cancers11050599