Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
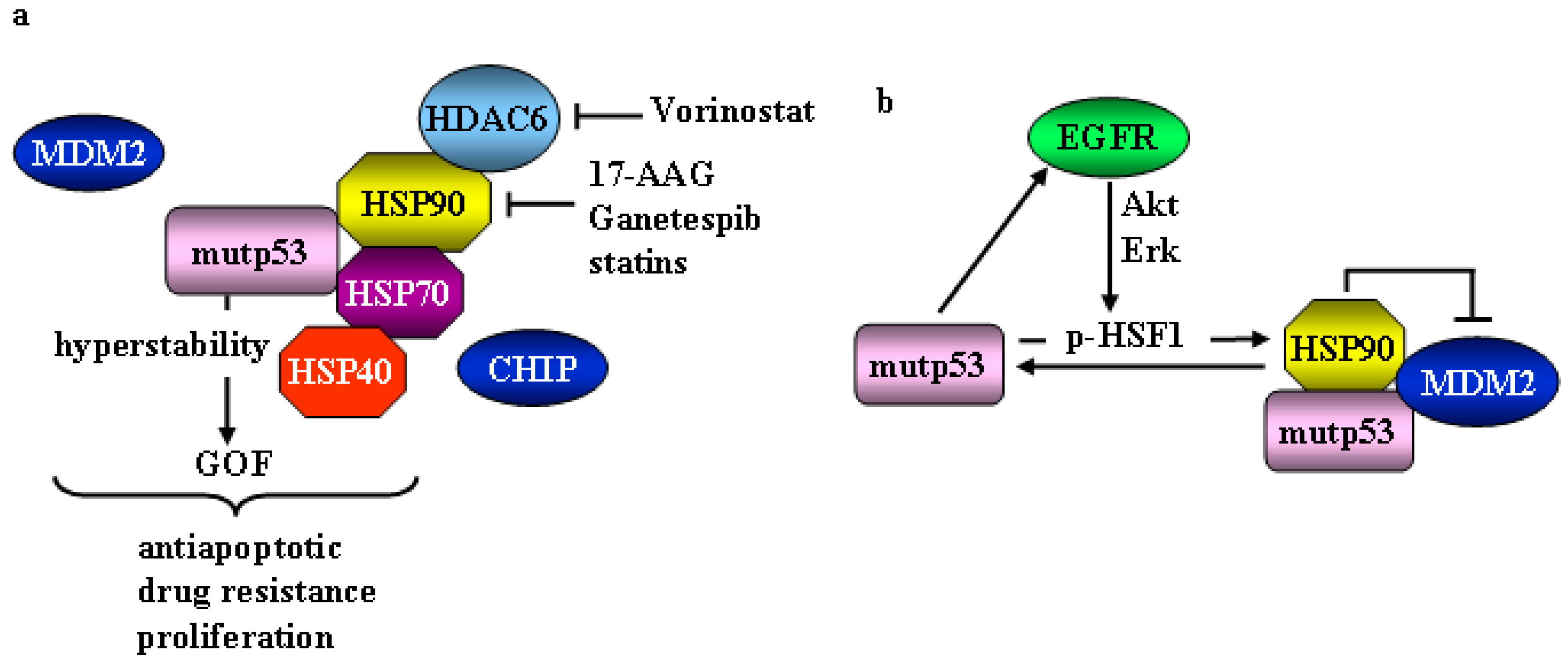
2. Mutant p53 and Heat Shock Factor 1 (HSF1)/Heat Shock Proteins (HSP) Oncogenic Signaling
3. Mutant p53 and the Endoplasmic Reticulum (ER) Stress/Unfolded Protein Response (UPR)
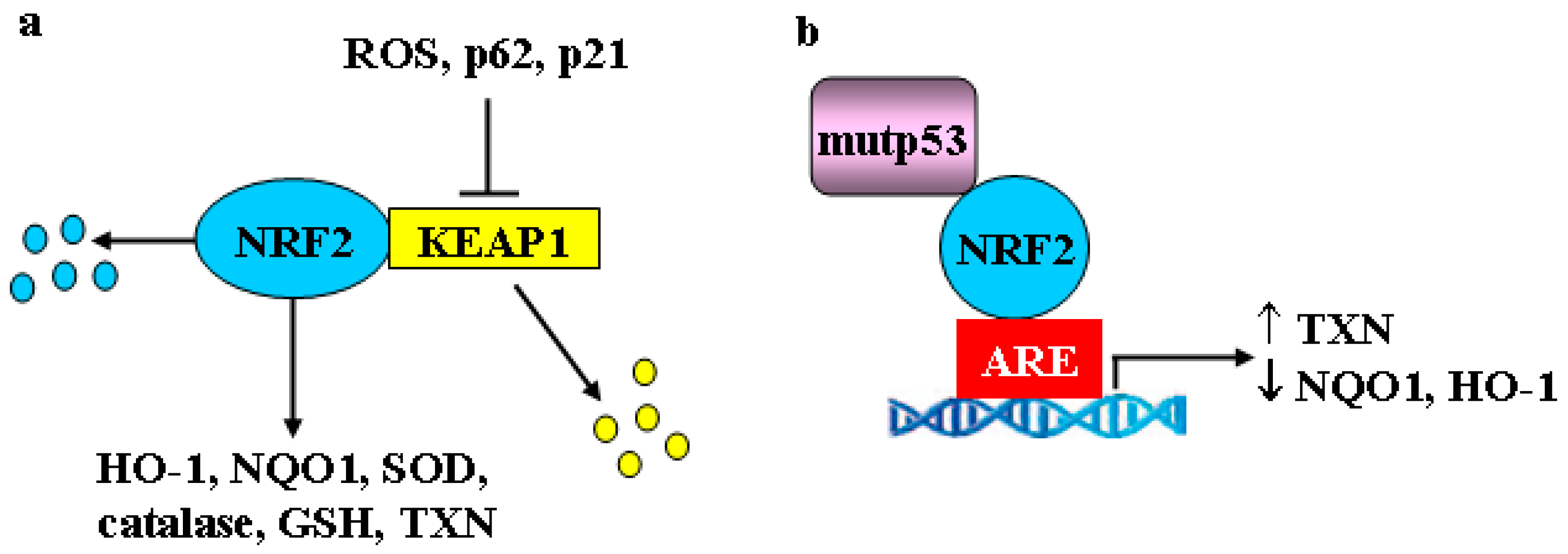
4. Mutant p53, Nuclear Factor Erythroid 2-Related Factor 2 (NRF2), Hypoxia-Inducible Factor-1 (HIF-1) and Estrogen Receptor α (ERα) Oncogenic Signaling
5. Mutant p53 and Autophagy: Linking the Pathways Together
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vousden, K.H. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in ageing and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple mechanisms define biologic activity in cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell. 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.B.; Attardi, L.D. Illuminating p53 function in cancer with genetically engineered mouse models. Semin. Cell. Dev. Biol. 2014, 0, 74–85. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-function (GOF) mutant p53 are actionable therapeutic target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell. Death Diff. 2017, 25, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Pacchiana, R.; Butera, G.; D’Orazi, G.; Scarpa, A.; Donadelli, M. Mutant p53 proteins alter cancer cell secretome and tumor micorenvironmen: Involvement in cancer invasion and metastasis. Cancer Lett. 2016, 376, 303–309. [Google Scholar] [CrossRef]
- Gurtner, A.; Falcone, E.; Garibaldi, F.; Piaggio, G. Dysregulation of microRNA biogenesis in cancer: The impact of mutant p53 on Drosha complex activity. J. Exp. Clin. Cancer Res. 2016, 35, 45. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cells. Cell. Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Riha, R.; Gupta-Saraf, P.; Bhanja, P.; Badkul, S.; Saha, S. Stressed out–– Therapeutic implications of ER stress related cancer research. Oncomedicine 2017, 2, 156–167. [Google Scholar] [CrossRef]
- Sane, S.; Rezvani, K. Essential Roles of E3 ubiquitin ligases in p53 regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell. Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structure-function-rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef]
- Li, D.; Marchencko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell. Death. Diff. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Alexandrova, E.M.; Moll, U.M. Depleting stabilized GOF mutant p53 proteins by inhibiting molecular folding chaperones: A new promise in cancer therapy. Cell. Death Differ. 2017, 24, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by HSP90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 44, 40583–40590. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schultz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepel, J.; Mollapour, M.; Gaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Whitesell, L.; Sutphin, P.D.; Pulcini, E.J.; Martinez, J.D.; Cook, P.H. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol. Cell. Biol. 1998, 18, 1517–1524. [Google Scholar] [CrossRef]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating preclinical hypotheses into clinical promise. Cancer Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Solárová, Z.; Mojžiš, J.; Solár, P. Hsp90 inhibitors as a sensitizer of cancer cells to different therapies. Int. J. Oncol. 2015, 46, 907–926. [Google Scholar] [CrossRef]
- Krämer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ozog, L.; Marchenko, N. A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis. 2014, 5, e1194. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heath shock transcription factors and their role in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Home, T.; Jensen, R.A.; Rap, R. Heat shock factor 1 in protein homeostasis and oncogenic signal integration. Cancer Res. 2015, 75, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Seo, Y.H. Small molecule inhibitors of HSF1-activated pathways as potential next-generation anticancer therapeutics. Molecules 2018, 23, 2757. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Santagata, S.; Tang, Z.; Shi, J.; Cao, J.; Kwon, H.; Bronson, R.T.; Whitesell, L.; Lindquist, S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J. Clin. Investig. 2012, 122, 3742–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. the ERBB network at last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell. Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signaling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by yje mevalonate pathway. Nat. Cell. Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell. Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P., IV; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 2019, 176, 564–580.e19. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Marchenko, N.D. Mutant p53-heat shock response oncogenic cooperation: A new mechanism of cancer cell survival. Front. Endocrinol. 2015, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Gilmore, R.; Blobel, G. Protein translocation across the endoplasmic reticulum. Cell 1984, 38, 5–8. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decision under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Storchov, Z. Aneuploidy and proteotoxic stress in cancer. Mol. Cell. Oncol. 2015, 2, e976491. [Google Scholar] [CrossRef] [Green Version]
- Deegan, S.; Saveljeva, S.; Gorman, A.M.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2013, 70, 2425–2541. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, L.; Chen, Z.; Nice, E.C.; Huang, C. Stress management by autophagy: Implications for chemoresistance. Int. J. Cancer 2016, 139, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [Green Version]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–697. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Char, J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell. 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015, 3, 141–148. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetx, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Shajahan, A.N.; Riggings, R.B.; Clarke, R. The role of X-box binding protein-1 in tumorigenicity. Drug News Perspect. 2009, 22, 241–246. [Google Scholar] [CrossRef]
- Namba, T.; Chu, K.; Kodama, R.; Byun, S.; Wan Yoon, K.; Hiraki, M.; Mandinova, A.; Lee, S.W. Loss of p53 enhances the function of the endoplasmic reticulum through activation of the IRE1α/XBP1 pathway. Oncotarget 2015, 6, 19990–20001. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Lee, A.S. The critical role of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef]
- Dai, C.; Dai, S.; Cao, J. Proteotoxic stress of cancer: Implication of the heat-shock response in oncogenesis. Cell. Physiol. 2012, 227, 2982–2987. [Google Scholar] [CrossRef]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1 alpha. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Schultze, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef] [Green Version]
- Donze, O.; Abbas-Terki, T.; Picard, D. The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J. 2001, 20, 3771–3780. [Google Scholar] [CrossRef]
- Xu, W.; Mimmaugh, E.; Rosser, M.F.; Nicchitta, C.; Marcu, M.; Yarden, Y.; Neckers, L. Sensitivity of mature Erbb2 to geldanamicyn is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem. 2001, 276, 3702–3708. [Google Scholar] [CrossRef]
- Vogiatzi, F.; Brandt, D.T.; Schneikert, J.; Fuchs, J.; Grikscheit, K.; Wanzel, M.; Pavlakis, E.; Charles, J.P.; Timofeev, O.; Nist, A.; et al. Mutant p53 promotes tumor progression and metastasis by the endoplasmic reticulum UDPase ENTPD5. Proc. Natl. Acad. Sci. USA 2016, 113, E8433–E8442. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and kallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Kostov, R.V.; Dinkova-Kostova, A.T. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol. Sci. 2015, 36, 6–14. [Google Scholar] [CrossRef]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancers. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of Nrf2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- No, J.H.; Kim, Y.B.; Song, Y.S. Targeting Nrf2 signaling to combat chemoresistance. J. Cancer Prev. 2014, 19, 111–117. [Google Scholar] [CrossRef]
- Glorieux, V.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase down-regulation in cancer cells exposed to arsenic trioxide is involved in their increased sensitivity to a pro-oxidant treatment. Cancer Cell. Int. 2018, 18, 24. [Google Scholar] [CrossRef]
- Zeekpudsa, P.; Kukongviriyapan, V.; Senggunprai, L.; Sripa, B.; Prawan, A. Suppression of NAD(P)H-quinone oxidoreductase 1 enhanced the susceptibility of cholangiocarcinoma cells to chemotherapeutic agents. J. Exp. Clin. Cancer Res. 2014, 3, 11. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wu, Q.; Cui, X.; Lin, Z.; Liu, S.; Chen, L. Clinical implications of high NQO1 expression in breast cancers. J. Exp. Clin. Cancer Res. 2014, 33, 14. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrcke, E.H. Life, death and autophagy. Nat. Cell. Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhand, D.D. p62 links autophagy and Nrf2 signaling. Free Rad. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhangm, Y.; Bertolottim, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [Green Version]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Taguchi, K.; Masamume, A.; Yamamoto, M.; Shimosegawa, T. Nrf2 promotes mutant R-ras/p53 driven pancreatic carcinogenesis. Carcinogenesis 2017, 38, 661–670. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell. Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Yu, T.; Tang, B.; Sun, X. Development of inhibitors targeting Hypoxia-inducible Factor 1 and 2 for cancer therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. P53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Ye, W.; Huang, C.; Lou, B.; Zhang, J.; Yu, D.; Huang, X.; Chen, B.; Zhou, M. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-κb/Stat3/Bcl-2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 10, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, W.; Huang, C.; Yu, D.; Chen, H.; Deng, T.; Zhang, F.; Lou, B.; Zhang, J.; Shi, K. Brusatol enhances the chemotherapy efficacy of gemcitabine in pancreatic cancer via the Nrf2 signalling pathway. Oxid. Med. Cell. Long. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, Y.; Wang, S.; Guo, X.; Shi, P.; Wang, W.; Xu, B. Triptolide, a Chinese herbal extract, enhances drug sensitivity of resistant myeloid leukemia cell lines through downregulation of HIF-1α and Nrf2. Pharmacogenom 2013, 14, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohash, H. Nrf2 Redirects Glucose and Glutamine into Anabolic Pathways in Metabolic Reprogramming. Cancer Cell. 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Wolpaw, A.J.; Dang, C.V. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell. Biol. 2018, 201–212. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell. Biol. 2019, 1–9. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.; Jones, D.T.; Buffa, F.M.; Bridges, E.; Interiano, R.B.; Quc, C.; Vogt, N.; Lia, J.-L.; Baban, D.; et al. Estrogen receptor-α directly regulates the hypoxia inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15172–15177. [Google Scholar] [CrossRef]
- Shanle, E.K.; Xu, W. Selectively targeting estrogen receptors for cancer treatment. Adv. Drug Deliv. Rev. 2010, 62, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. Estrogen receptor acting in cis enhances WT and mutant p53 transactivation at canonical and noncanonical p53 target sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 1500–1505. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Clarke, P.A.G.; Parmar, J.; Hu, R.; Schwartz-Roberts, J.L.; Abu-Asab, M.; Warri, A.; Baumann, W.T.; Clarke, R. Knockdown of estrogen receptor-α induces autophagy and inhibits antiestrogen-mediated unfolded protein response activation, promoting ROS-induced breast cancer cell death. FASEB J. 2014, 28, 3891–3905. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Trisciuoglio, D.; Porru, M.; Leonetti, C.; Stoppacciaro, A.; D’Orazi, V.; Avantaggiati, M.; Crispini, A.; Pucci, D.; D’Orazi, G. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R174H and R273H) p53 in cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 72. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell. Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Garufi, A.; D’Orazi, V.; Crispini, A.; D’Orazi, G. Zn(II)-curc targets p53 in thyroid cancer cells. Int. J. Oncol. 2015, 47, 1241–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reactivation of mutant p53 by capsaicin, the major constituent of peppers. J. Exp. Clin. Cancer Res. 2016, 351, 136. [Google Scholar] [CrossRef]
- Garufi, A.; Pistritto, G.; Baldari, S.; Toietta, G.; Cirone, M.; D’Orazi, G. p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions. J. Exp. Clin. Cancer Res. 2017, 36, 126. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cirone, M.; University ‘G. d’Annunzio’, Chieti, Italy. Unpublished work. 2019.
- Thongrakard, V.; Titone, R.; Follo, C.; Morani, F.; Suksamrarn, A.; Tencomanao, T.; Isidoro, C. Turmeric toxicity in A431 epidermoid cancer cells associates with autophagy degradation of anti-apoptotic and anti-autophagic p53 mutant. Phytother. Res. 2014, 28, 1761–1769. [Google Scholar] [CrossRef]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Monti, P.; Degan, P.; Fronza, G. Gambogic acid counteracts mutant p53 stability by inducing autophagy. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 382–392. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, Q.; Qi, Q.; Gu, H.Y.; Rong, J.J.; Mu, R. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J. Cell. Biochem. 2011, 112, 509–519. [Google Scholar] [CrossRef]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Berry, D.; Kallakury, B.; Mueller, S.C.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell. Death Diff. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; Gilardini Montani, M.S.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; Avantaggiati, M.L. Dietary downregulation of mutant p53 levels via glucose restriction: Mechanisms and implications for tumor therapy. Cell. Cycle 2012, 11, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; Yuan, J. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldari, S.; Ubertini, V.; Garufi, A.; D’Orazi, G.; Bossi, G. Targeting MKK3 as a novel anticancer strategy: Molecular mechanisms and therapeutical implications. Cell. Death Dis. 2015, 6, e1621. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Cordani, M.; Oppici, E.; Dando, I.; Buttirini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [Green Version]
- Dando, I.; Cordani, M.; Donadelli, M. Mutant p53 and mTOR/PKM2 regulation in cancer cells. IUBMB Life 2016, 68, 722–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.G.; Buel, G.R.; Blenis, J. Nutrient Regulation of the mTOR Complex 1 Signaling Pathway. Mol. Cell 2013, 35, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef]
- Catena, V.; Fanciulli, M. Deptor: Not only a mTOR inhibitor. J. Exp. Clin. Cancer Res. 2017, 36, 12. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 13, 2163–2170. [Google Scholar] [CrossRef]
- Granato, M.; Rizzello, C.; Romeo, M.A.; Yadav, S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Concomitant reduction of c-Myc expression and PI3K/AKT/mTOR signaling by quercetin induces a strong cytotoxic effect against Burkitt’s lymphoma. Int. J. Biochem. Cell Biol. 2016, 79, 393–400. [Google Scholar] [CrossRef]
- Masuelli, L.; Granato, M.; Benvenuto, M.; Mattera, R.; Bernardini, R.; Mattei, M.; d’Amati, G.; D’Orazi, G.; Faggioni, A.; Bei, R.; Cirone, M. Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overepressing breast cancer cells in vitro and in vivo in nude mice while counteracts it in immune competent mice. OncoImmunology 2017, 6, e1356151. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Gurpinar, E.; Vousden, K.H. Hitting cancers’ weak spots: Vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015, 25, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Orazi, G.; Cirone, M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers 2019, 11, 614. https://doi.org/10.3390/cancers11050614
D’Orazi G, Cirone M. Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers. 2019; 11(5):614. https://doi.org/10.3390/cancers11050614
Chicago/Turabian StyleD’Orazi, Gabriella, and Mara Cirone. 2019. "Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression" Cancers 11, no. 5: 614. https://doi.org/10.3390/cancers11050614
APA StyleD’Orazi, G., & Cirone, M. (2019). Mutant p53 and Cellular Stress Pathways: A Criminal Alliance That Promotes Cancer Progression. Cancers, 11(5), 614. https://doi.org/10.3390/cancers11050614