TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity


Abstract
1. Introduction
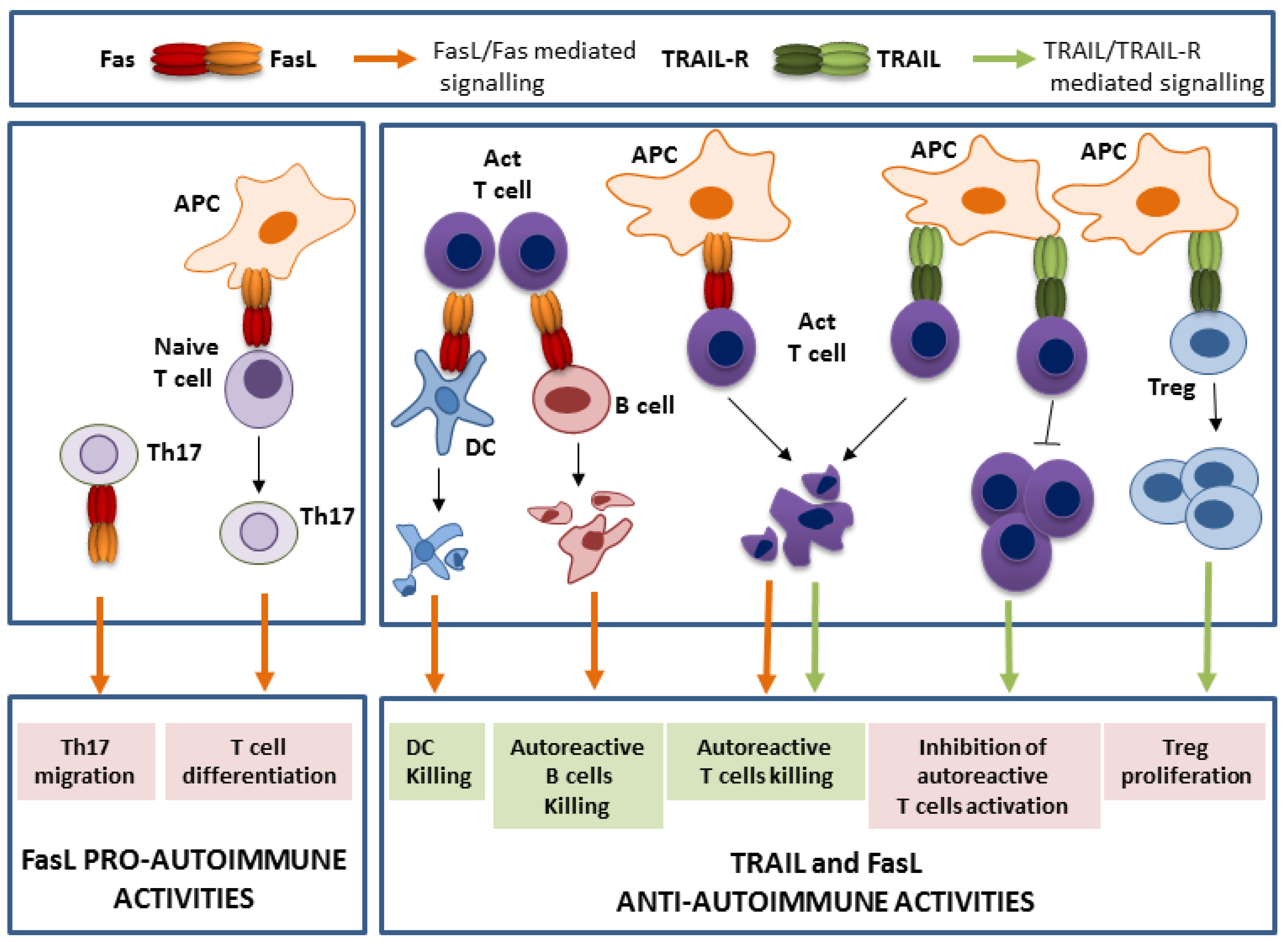
2. TRAIL and FasL Functions in the Control of Autoimmunity
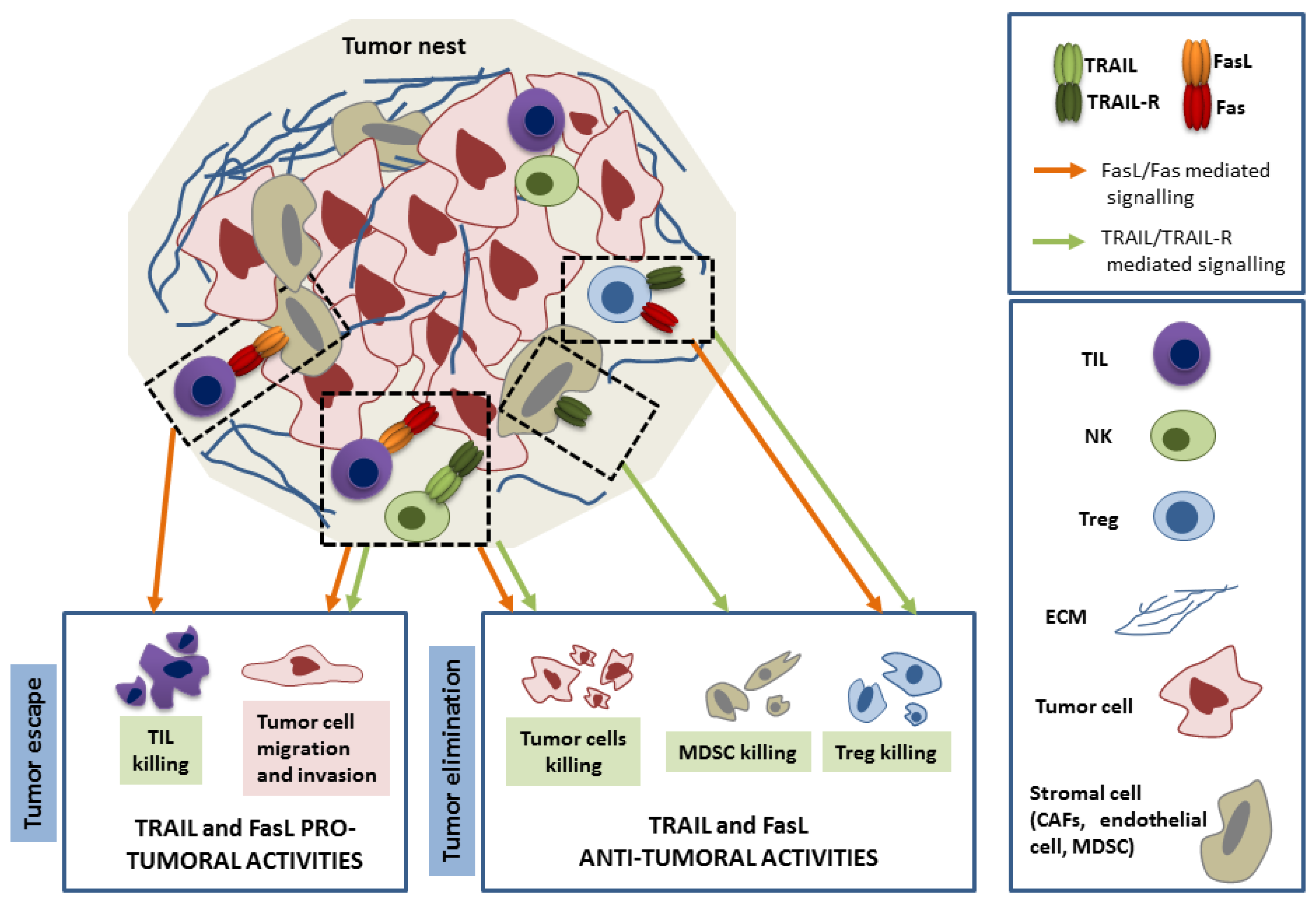
3. TRAIL and FasL Functions in Cancer Immunoediting
3.1. Role of TRAIL and FasL in the Elimination Phase
3.2. Role of TRAIL and FasL in Immune Escape
4. TRAIL and FasL in Clinical Interventions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Mariani, S.M.; Krammer, P.H. Differential regulation of TRAIL and CD95 ligand in transformed cells of the T and B lymphocyte lineage. Eur. J. Immunol. 1998, 28, 973–982. [Google Scholar] [CrossRef]
- Tanaka, M.; Itai, T.; Adachi, M.; Nagata, S. Downregulation of Fas ligand by shedding. Nat. Med. 1998, 4, 31–36. [Google Scholar] [CrossRef]
- Kawakubo, T.; Okamoto, K.; Iwata, J.; Shin, M.; Okamoto, Y.; Yasukochi, A.; Nakayama, K.I.; Kadowaki, T.; Tsukuba, T.; Yamamoto, K. Cathepsin E prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble TRAIL from tumor cell surface. Cancer Res. 2007, 67, 10869–10878. [Google Scholar] [CrossRef]
- O’ Reilly, L.A.; Tai, L.; Lee, L.; Kruse, E.A.; Grabow, S.; Fairlie, W.D.; Haynes, N.M.; Tarlinton, D.M.; Zhang, J.G.; Belz, G.T.; et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 2009, 461, 659–663. [Google Scholar] [CrossRef]
- Schneider, P.; Holler, N.; Bodmer, J.L.; Hahne, M.; Frei, K.; Fontana, A.; Tschopp, J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med. 1998, 187, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Moosmayer, D.; Wüest, T.; Bartke, T.; Gerlach, E.; Schönherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [PubMed]
- Tauzin, S.; Chaigne-Delalande, B.; Selva, E.; Khadra, N.; Daburon, S.; Contin-Bordes, C.; Blanco, P.; Le Seyec, J.; Ducret, T.; Counillon, L.; et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol. 2011, 9, e1001090. [Google Scholar] [CrossRef]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef]
- Smulski, C.R.; Decossas, M.; Chekkat, N.; Beyrath, J.; Willen, L.; Guichard, G.; Lorenzetti, R.; Rizzi, M.; Eibel, H.; Schneider, P.; et al. Hetero-oligomerization between the TNF receptor superfamily members CD40, Fas and TRAILR2 modulate CD40 signalling. Cell Death Dis. 2017, 8, e2601. [Google Scholar] [CrossRef]
- Phillips, T.A.; Ni, J.; Pan, G.; Ruben, S.M.; Wei, Y.F.; Pace, J.L.; Hunt, J.S. TRAIL (Apo-2L) and TRAIL receptors in human placentas: Implications for immune privilege. J. Immunol. 1999, 162, 6053–6059. [Google Scholar]
- Ferguson, T.A.; Griffith, T.S. A vision of cell death: Fas ligand and immune privilege 10 years later. Immunol. Rev. 2006, 213, 228–238. [Google Scholar] [CrossRef]
- Stenqvist, A.C.; Nagaeva, O.; Baranov, V.; Mincheva-Nilsson, L. Exosomes secreted by human placenta carry functional Fas ligand and TRAIL molecules and convey apoptosis in activated immune cells, suggesting exosome-mediated immune privilege of the fetus. J. Immunol. 2013, 191, 5515–5523. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Takeda, K.; Akiba, H.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Okumura, K.; Yagita, H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999, 163, 1906–1913. [Google Scholar]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef]
- Suda, T.; Okazaki, T.; Naito, Y.; Yokota, T.; Arai, N.; Ozaki, S.; Nakao, K.; Nagata, S. Expression of the Fas ligand in cells of T cell lineage. J. Immunol. 1995, 154, 3806–3813. [Google Scholar]
- Tsutsui, H.; Nakanishi, K.; Matsui, K.; Higashino, K.; Okamura, H.; Miyazawa, Y.; Kaneda, K. IFN-gamma-inducing factor up-regulates Fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J. Immunol. 1996, 157, 3967–3973. [Google Scholar]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef]
- Griffith, T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 1999, 189, 1343–1354. [Google Scholar] [CrossRef]
- Süss, G.; Shortman, K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 1996, 183, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Kiener, P.A.; Davis, P.M.; Starling, G.C.; Mehlin, C.; Klebanoff, S.J.; Ledbetter, J.A.; Liles, W.C. Differential induction of apoptosis by Fas-Fas ligand interactions in human monocytes and macrophages. J. Exp. Med. 1997, 185, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Liles, W.C.; Kiener, P.A.; Ledbetter, J.A.; Aruffo, A.; Klebanoff, S.J. Differential expression of Fas (CD95) and Fas ligand on normal human phagocytes: Implications for the regulation of apoptosis in neutrophils. J. Exp. Med. 1996, 184, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Hara, T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): A novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004, 64, 1037–1043. [Google Scholar] [CrossRef]
- O’Reilly, E.; Tirincsi, A.; Logue, S.E.; Szegezdi, E. The Janus Face of Death Receptor Signaling during Tumor Immunoediting. Front. Immunol. 2016, 7, 446. [Google Scholar] [CrossRef]
- Zheng, L.; Li, J.; Lenardo, M. Restimulation-induced cell death: New medical and research perspectives. Immunol. Rev. 2017, 277, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Wang, D.; Shi, J.; Xiang, Y.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induces chemotactic migration of monocytes via a death receptor 4-mediated RhoGTPase pathway. Mol. Immunol. 2010, 47, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gülcüler, G.S.; Golbach, L.; Block, H.; Zarbock, A.; Martin-Villalba, A. Endothelial cell-derived CD95 ligand serves as a chemokine in induction of neutrophil slow rolling and adhesion. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Park, D.R.; Thomsen, A.R.; Frevert, C.W.; Pham, U.; Skerrett, S.J.; Kiener, P.A.; Liles, W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 2003, 170, 6209–6216. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, M.; Tang, H.; Cao, X. Fas signal links innate and adaptive immunity by promoting dendritic-cell secretion of CC and CXC chemokines. Blood 2005, 106, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Clancy, L.; Chan, F.K. Lipopolysaccharide-induced expression of TRAIL promotes dendritic cell differentiation. Immunology 2010, 130, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Guégan, J.P.; Legembre, P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. 2018, 285, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Lafont, E.; Hartwig, T.; Walczak, H. Paving TRAIL’s Path with Ubiquitin. Trends Biochem. Sci. 2018, 43, 44–60. [Google Scholar] [CrossRef]
- Siegmund, D.; Lang, I.; Wajant, H. Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J. 2017, 284, 1131–1159. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Sedger, L.M.; Glaccum, M.B.; Schuh, J.C.; Kanaly, S.T.; Williamson, E.; Kayagaki, N.; Yun, T.; Smolak, P.; Le, T.; Goodwin, R.; et al. Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur. J. Immunol. 2002, 32, 2246–2254. [Google Scholar] [CrossRef]
- Diehl, G.E.; Yue, H.H.; Hsieh, K.; Kuang, A.A.; Ho, M.; Morici, L.A.; Lenz, L.L.; Cado, D.; Riley, L.W.; Winoto, A. TRAIL-R as a negative regulator of innate immune cell responses. Immunity 2004, 21, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Cretney, E.; McQualter, J.L.; Kayagaki, N.; Yagita, H.; Bernard, C.C.; Grewal, I.S.; Ashkenazi, A.; Smyth, M.J. TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L suppresses experimental autoimmune encephalomyelitis in mice. Immunol. Cell Biol. 2005, 83, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Hirata, S.; Fukushima, S.; Matsunaga, Y.; Ito, T.; Uchino, M.; Nishimura, Y.; Senju, S. Dual effects of TRAIL in suppression of autoimmunity: The inhibition of Th1 cells and the promotion of regulatory T cells. J. Immunol. 2010, 185, 5259–5267. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Tsai, H.F.; Wu, C.S.; Sung, C.C.; Hsu, P.N. TRAIL-Mediated Suppression of T Cell Receptor Signaling Inhibits T Cell Activation and Inflammation in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 15. [Google Scholar] [CrossRef]
- Lamhamedi-Cherradi, S.E.; Zheng, S.J.; Maguschak, K.A.; Peschon, J.; Chen, Y.H. Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL-/- mice. Nat. Immunol. 2003, 4, 255–260. [Google Scholar] [CrossRef]
- Wang, S.H.; Cao, Z.; Wolf, J.M.; Van Antwerp, M.; Baker, J.R. Death ligand tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis. Endocrinology 2005, 146, 4721–4726. [Google Scholar] [CrossRef][Green Version]
- Wang, S.H.; Chen, G.H.; Fan, Y.; Van Antwerp, M.; Baker, J.R. Tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis by the expansion of CD4+CD25+ regulatory T cells. Endocrinology 2009, 150, 2000–2007. [Google Scholar] [CrossRef]
- Hilliard, B.; Wilmen, A.; Seidel, C.; Liu, T.S.; Göke, R.; Chen, Y. Roles of TNF-related apoptosis-inducing ligand in experimental autoimmune encephalomyelitis. J. Immunol. 2001, 166, 1314–1319. [Google Scholar] [CrossRef]
- Song, K.; Chen, Y.; Göke, R.; Wilmen, A.; Seidel, C.; Göke, A.; Hilliard, B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J. Exp. Med. 2000, 191, 1095–1104. [Google Scholar] [CrossRef]
- Mi, Q.S.; Ly, D.; Lamhamedi-Cherradi, S.E.; Salojin, K.V.; Zhou, L.; Grattan, M.; Meagher, C.; Zucker, P.; Chen, Y.H.; Nagle, J.; et al. Blockade of tumor necrosis factor-related apoptosis-inducing ligand exacerbates type 1 diabetes in NOD mice. Diabetes 2003, 52, 1967–1975. [Google Scholar] [CrossRef]
- Hirata, S.; Senju, S.; Matsuyoshi, H.; Fukuma, D.; Uemura, Y.; Nishimura, Y. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programMed. death-1 ligand. J. Immunol. 2005, 174, 1888–1897. [Google Scholar] [CrossRef]
- Hirata, S.; Matsuyoshi, H.; Fukuma, D.; Kurisaki, A.; Uemura, Y.; Nishimura, Y.; Senju, S. Involvement of regulatory T cells in the experimental autoimmune encephalomyelitis-preventive effect of dendritic cells expressing myelin oligodendrocyte glycoprotein plus TRAIL. J. Immunol. 2007, 178, 918–925. [Google Scholar] [CrossRef]
- Waldner, H.; Sobel, R.A.; Howard, E.; Kuchroo, V.K. Fas- and FasL-deficient mice are resistant to induction of autoimmune encephalomyelitis. J. Immunol. 1997, 159, 3100–3103. [Google Scholar]
- Sabelko, K.A.; Kelly, K.A.; Nahm, M.H.; Cross, A.H.; Russell, J.H. Fas and Fas ligand enhance the pathogenesis of experimental allergic encephalomyelitis, but are not essential for immune privilege in the central nervous system. J. Immunol. 1997, 159, 3096–3099. [Google Scholar] [PubMed]
- Meyer Zu Horste, G.; Przybylski, D.; Schramm, M.A.; Wang, C.; Schnell, A.; Lee, Y.; Sobel, R.; Regev, A.; Kuchroo, V.K. Fas Promotes T Helper 17 Cell Differentiation and Inhibits T Helper 1 Cell Development by Binding and Sequestering Transcription Factor STAT1. Immunity 2018, 48, 556–569.e557. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, X.; Hsu, H.C.; Tousson, A.; Yang, P.A.; Wu, Q.; Liu, C.; Yu, S.; Zhang, H.G.; Mountz, J.D. CII-DC-AdTRAIL cell gene therapy inhibits infiltration of CII-reactive T cells and CII-induced arthritis. J. Clin. Investig. 2003, 112, 1332–1341. [Google Scholar] [CrossRef]
- Tu-Rapp, H.; Hammermüller, A.; Mix, E.; Kreutzer, H.J.; Goerlich, R.; Köhler, H.; Nizze, H.; Thiesen, H.J.; Ibrahim, S.M. A proinflammatory role for Fas in joints of mice with collagen-induced arthritis. Arthritis Res. Ther. 2004, 6, R404–R414. [Google Scholar] [CrossRef]
- Itoh, N.; Imagawa, A.; Hanafusa, T.; Waguri, M.; Yamamoto, K.; Iwahashi, H.; Moriwaki, M.; Nakajima, H.; Miyagawa, J.; Namba, M.; et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 1997, 186, 613–618. [Google Scholar] [CrossRef]
- Chervonsky, A.V.; Wang, Y.; Wong, F.S.; Visintin, I.; Flavell, R.A.; Janeway, C.A.; Matis, L.A. The role of Fas in autoimmune diabetes. Cell 1997, 89, 17–24. [Google Scholar] [CrossRef]
- Vence, L.; Benoist, C.; Mathis, D. Fas deficiency prevents type 1 diabetes by inducing hyporesponsiveness in islet beta-cell-reactive T-cells. Diabetes 2004, 53, 2797–2803. [Google Scholar] [CrossRef]
- Su, X.; Hu, Q.; Kristan, J.M.; Costa, C.; Shen, Y.; Gero, D.; Matis, L.A.; Wang, Y. Significant role for Fas in the pathogenesis of autoimmune diabetes. J. Immunol. 2000, 164, 2523–2532. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Waiczies, S.; Ehrlich, S.; Wendling, U.; Seeger, B.; Kamradt, T.; Zipp, F. Death ligand TRAIL induces no apoptosis but inhibits activation of human (auto)antigen-specific T cells. J. Immunol. 2002, 168, 4881–4888. [Google Scholar] [CrossRef]
- Rieux-Laucat, F.; Magérus-Chatinet, A.; Neven, B. The Autoimmune Lymphoproliferative Syndrome with Defective FAS or FAS-Ligand Functions. J. Clin. Immunol. 2018, 38, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.H.; Rosenberg, F.J.; Straus, S.E.; Dale, J.K.; Middleton, L.A.; Lin, A.Y.; Strober, W.; Lenardo, M.J.; Puck, J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81, 935–946. [Google Scholar] [CrossRef]
- Watanabe-Fukunaga, R.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Nagata, S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992, 356, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tanaka, M.; Brannan, C.I.; Jenkins, N.A.; Copeland, N.G.; Suda, T.; Nagata, S. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 1994, 76, 969–976. [Google Scholar] [CrossRef]
- Nagata, S.; Suda, T. Fas and Fas ligand: Lpr and gld mutations. Immunol. Today 1995, 16, 39–43. [Google Scholar] [CrossRef]
- Sedger, L.M.; Katewa, A.; Pettersen, A.K.; Osvath, S.R.; Farrell, G.C.; Stewart, G.J.; Bendall, L.J.; Alexander, S.I. Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL and TRAIL double-deficient mice. Blood 2010, 115, 3258–3268. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, H.; Adachi, M.; Suematsu, S.; Miwa, K.; Suda, T.; Yoshida, N.; Nagata, S. Transgenic expression of Fas in T cells blocks lymphoproliferation but not autoimmune disease in MRL-lpr mice. J. Immunol. 1998, 160, 3805–3811. [Google Scholar]
- Komano, H.; Ikegami, Y.; Yokoyama, M.; Suzuki, R.; Yonehara, S.; Yamasaki, Y.; Shinohara, N. Severe impairment of B cell function in lpr/lpr mice expressing transgenic Fas selectively on B cells. Int. Immunol. 1999, 11, 1035–1042. [Google Scholar] [CrossRef][Green Version]
- Stranges, P.B.; Watson, J.; Cooper, C.J.; Choisy-Rossi, C.M.; Stonebraker, A.C.; Beighton, R.A.; Hartig, H.; Sundberg, J.P.; Servick, S.; Kaufmann, G.; et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity 2007, 26, 629–641. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Cooke, M.P.; Ho, W.Y.; Grein, J.; Townsend, S.E.; Davis, M.M.; Goodnow, C.C. CD95 (Fas)-dependent elimination of self-reactive B cells upon interaction with CD4+ T cells. Nature 1995, 376, 181–184. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Seagal, J.; Su, Y.W.; Hong, C.; Haight, J.; Chen, N.J.; Elia, A.; Wakeham, A.; Li, W.Y.; et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 2008, 29, 615–627. [Google Scholar] [CrossRef]
- Bosque, A.; Aguiló, J.I.; Alava, M.A.; Paz-Artal, E.; Naval, J.; Allende, L.M.; Anel, A. The induction of Bim expression in human T-cell blasts is dependent on nonapoptotic Fas/CD95 signaling. Blood 2007, 109, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Aguiló, J.I.; del Rey, M.; Paz-Artal, E.; Allende, L.M.; Naval, J.; Anel, A. Cell cycle regulation by FasL and Apo2L/TRAIL in human T-cell blasts. Implications for autoimmune lymphoproliferative syndromes. J. Leukoc. Biol. 2008, 84, 488–498. [Google Scholar] [CrossRef]
- Cruz, A.C.; Ramaswamy, M.; Ouyang, C.; Klebanoff, C.A.; Sengupta, P.; Yamamoto, T.N.; Meylan, F.; Thomas, S.K.; Richoz, N.; Eil, R.; et al. Fas/CD95 prevents autoimmunity independently of lipid raft localization and efficient apoptosis induction. Nat. Commun. 2016, 7, 13895. [Google Scholar] [CrossRef]
- Daszkiewicz, L.; Vázquez-Mateo, C.; Rackov, G.; Ballesteros-Tato, A.; Weber, K.; Madrigal-Avilés, A.; Di Pilato, M.; Fotedar, A.; Fotedar, R.; Flores, J.M.; et al. Distinct p21 requirements for regulating normal and self-reactive T cells through IFN-γ production. Sci. Rep. 2015, 5, 7691. [Google Scholar] [CrossRef] [PubMed]
- Chakrabandhu, K.; Hérincs, Z.; Huault, S.; Dost, B.; Peng, L.; Conchonaud, F.; Marguet, D.; He, H.T.; Hueber, A.O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 2007, 26, 209–220. [Google Scholar] [CrossRef]
- Feig, C.; Tchikov, V.; Schütze, S.; Peter, M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2007, 26, 221–231. [Google Scholar] [CrossRef]
- Rossin, A.; Durivault, J.; Chakhtoura-Feghali, T.; Lounnas, N.; Gagnoux-Palacios, L.; Hueber, A.O. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates Fas stability. Cell Death Differ. 2015, 22, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Poissonnier, A.; Sanséau, D.; Le Gallo, M.; Malleter, M.; Levoin, N.; Viel, R.; Morere, L.; Penna, A.; Blanco, P.; Dupuy, A.; et al. CD95-Mediated Calcium Signaling Promotes T Helper 17 Trafficking to InflaMed. Organs in Lupus-Prone Mice. Immunity 2016, 45, 209–223. [Google Scholar] [CrossRef]
- Zerafa, N.; Westwood, J.A.; Cretney, E.; Mitchell, S.; Waring, P.; Iezzi, M.; Smyth, M.J. Cutting edge: TRAIL deficiency accelerates hematological malignancies. J. Immunol. 2005, 175, 5586–5590. [Google Scholar] [CrossRef]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- Finnberg, N.; Klein-Szanto, A.J.; El-Deiry, W.S. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J. Clin. Investig. 2008, 118, 111–123. [Google Scholar] [CrossRef]
- Yue, H.H.; Diehl, G.E.; Winoto, A. Loss of TRAIL-R does not affect thymic or intestinal tumor development in p53 and adenomatous polyposis coli mutant mice. Cell Death Differ. 2005, 12, 94–97. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailey, S.L.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schütz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Investig. 2008, 118, 100–110. [Google Scholar] [CrossRef]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef]
- Peng, S.L.; Robert, M.E.; Hayday, A.C.; Craft, J. A tumor-suppressor function for Fas (CD95) revealed in T cell-deficient mice. J. Exp. Med. 1996, 184, 1149–1154. [Google Scholar] [CrossRef]
- Zörnig, M.; Grzeschiczek, A.; Kowalski, M.B.; Hartmann, K.U.; Möröy, T. Loss of Fas/Apo-1 receptor accelerates lymphomagenesis in E mu L-MYC transgenic mice but not in animals infected with MoMuLV. Oncogene 1995, 10, 2397–2401. [Google Scholar] [PubMed]
- Davidson, W.F.; Giese, T.; Fredrickson, T.N. Spontaneous development of plasmacytoid tumors in mice with defective Fas-Fas ligand interactions. J. Exp. Med. 1998, 187, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rödling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef]
- Gagnoux-Palacios, L.; Awina, H.; Audebert, S.; Rossin, A.; Mondin, M.; Borgese, F.; Planas-Botey, C.; Mettouchi, A.; Borg, J.P.; Hueber, A.O. Cell polarity and adherens junction formation inhibit epithelial Fas cell death receptor signaling. J. Cell Biol. 2018, 217, 3839–3852. [Google Scholar] [CrossRef]
- Straus, S.E.; Jaffe, E.S.; Puck, J.M.; Dale, J.K.; Elkon, K.B.; Rösen-Wolff, A.; Peters, A.M.; Sneller, M.C.; Hallahan, C.W.; Wang, J.; et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 2001, 98, 194–200. [Google Scholar] [CrossRef]
- Dorothée, G.; Vergnon, I.; Menez, J.; Echchakir, H.; Grunenwald, D.; Kubin, M.; Chouaib, S.; Mami-Chouaib, F. Tumor-infiltrating CD4+ T lymphocytes express APO2 ligand (APO2L)/TRAIL upon specific stimulation with autologous lung carcinoma cells: Role of IFN-alpha on APO2L/TRAIL expression and -mediated cytotoxicity. J. Immunol. 2002, 169, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [PubMed]
- Diao, Z.; Shi, J.; Zhu, J.; Yuan, H.; Ru, Q.; Liu, S.; Liu, Y.; Zheng, D. TRAIL suppresses tumor growth in mice by inducing tumor-infiltrating CD4(+)CD25 (+) Treg apoptosis. Cancer Immunol. Immunother. 2013, 62, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Seki, N.; Brooks, A.D.; Carter, C.R.; Back, T.C.; Parsoneault, E.M.; Smyth, M.J.; Wiltrout, R.H.; Sayers, T.J. Tumor-specific CTL kill murine renal cancer cells using both perforin and Fas ligand-mediated lysis in vitro, but cause tumor regression in vivo in the absence of perforin. J. Immunol. 2002, 168, 3484–3492. [Google Scholar] [CrossRef]
- Caldwell, S.A.; Ryan, M.H.; McDuffie, E.; Abrams, S.I. The Fas/Fas ligand pathway is important for optimal tumor regression in a mouse model of CTL adoptive immunotherapy of experimental CMS4 lung metastases. J. Immunol. 2003, 171, 2402–2412. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Catalán, E.; Hervás-Stubbs, S.; Palazón, A.; Azpilikueta, A.; Bolaños, E.; Anel, A.; Pardo, J.; Melero, I. Essential complicity of perforin-granzyme and FAS-L mechanisms to achieve tumor rejection following treatment with anti-CD137 mAb. J. Immunother. Cancer 2013, 1, 3. [Google Scholar] [CrossRef]
- Shanker, A.; Brooks, A.D.; Jacobsen, K.M.; Wine, J.W.; Wiltrout, R.H.; Yagita, H.; Sayers, T.J. Antigen presented by tumors in vivo determines the nature of CD8+ T-cell cytotoxicity. Cancer Res. 2009, 69, 6615–6623. [Google Scholar] [CrossRef]
- Kessler, B.; Hudrisier, D.; Schroeter, M.; Tschopp, J.; Cerottini, J.C.; Luescher, I.F. Peptide modification or blocking of CD8, resulting in weak TCR signaling, can activate CTL for Fas- but not perforin-dependent cytotoxicity or cytokine production. J. Immunol. 1998, 161, 6939–6946. [Google Scholar]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Zhu, J.; Powis de Tenbossche, C.G.; Cané, S.; Colau, D.; van Baren, N.; Lurquin, C.; Schmitt-Verhulst, A.M.; Liljeström, P.; Uyttenhove, C.; Van den Eynde, B.J. Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat. Commun. 2017, 8, 1404. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Kim, M.; Rosenwald, A.; von Raden, B.H.; von Rahden, B.; Tsaur, I.; Meier, E.; Heemann, U.; Germer, C.T.; Gasser, M.; et al. Tumour-mediated TRAIL-Receptor expression indicates effective apoptotic depletion of infiltrating CD8+ immune cells in clinical colorectal cancer. Eur. J. Cancer 2010, 46, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Radke, D.I.; Ungefroren, H.; Helm, O.; Voigt, S.; Alp, G.; Braun, H.; Hübner, S.; Dilchert, J.; Sebens, S.; Adam, D.; et al. Negative control of TRAIL-R1 signaling by transforming growth factor β1 in pancreatic tumor cells involves Smad-dependent down regulation of TRAIL-R1. Cell Signal. 2016, 28, 1652–1662. [Google Scholar] [CrossRef]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Stoicov, C.; Liu, J.H.; Zubair, S.; Tsai, E.; Ste Marie, R.; Wang, T.C.; Lyle, S.; Kurt-Jones, E.; et al. Human and mouse colon cancer utilizes CD95 signaling for local growth and metastatic spread to liver. Gastroenterology 2009, 137, 934–944. [Google Scholar] [CrossRef]
- Hoogwater, F.J.; Nijkamp, M.W.; Smakman, N.; Steller, E.J.; Emmink, B.L.; Westendorp, B.F.; Raats, D.A.; Sprick, M.R.; Schaefer, U.; Van Houdt, W.J.; et al. Oncogenic K-Ras turns death receptors into metastasis-promoting receptors in human and mouse colorectal cancer cells. Gastroenterology 2010, 138, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef]
- Azijli, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Würdinger, T.; Dekker, H.; Joore, J.; van Dijk, E.; Quax, W.J.; Peters, G.J.; de Jong, S.; et al. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-STAT3-dependent invasion in resistant non-small cell lung cancer cells. J. Cell Sci. 2012, 125, 4651–4661. [Google Scholar] [CrossRef]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef]
- Chakrabandhu, K.; Huault, S.; Durivault, J.; Lang, K.; Ta Ngoc, L.; Bole, A.; Doma, E.; Dérijard, B.; Gérard, J.P.; Pierres, M.; et al. An Evolution-Guided Analysis Reveals a Multi-Signaling Regulation of Fas by Tyrosine Phosphorylation and its Implication in Human Cancers. PLoS Biol. 2016, 14, e1002401. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Graves, J.D.; Kordich, J.J.; Huang, T.H.; Piasecki, J.; Bush, T.L.; Sullivan, T.; Foltz, I.N.; Chang, W.; Douangpanya, H.; Dang, T.; et al. Apo2L/TRAIL and the death receptor 5 agonist antibody AMG 655 cooperate to promote receptor clustering and antitumor activity. Cancer Cell 2014, 26, 177–189. [Google Scholar] [CrossRef]
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef]
- Xu, Y.; Szalai, A.J.; Zhou, T.; Zinn, K.R.; Chaudhuri, T.R.; Li, X.; Koopman, W.J.; Kimberly, R.P. Fc gamma Rs modulate cytotoxicity of anti-Fas antibodies: Implications for agonistic antibody-based therapeutics. J. Immunol. 2003, 171, 562–568. [Google Scholar] [CrossRef]
- Eisele, G.; Roth, P.; Hasenbach, K.; Aulwurm, S.; Wolpert, F.; Tabatabai, G.; Wick, W.; Weller, M. APO010, a synthetic hexameric CD95 ligand, induces human glioma cell death in vitro and in vivo. Neuro-Oncology 2011, 13, 155–164. [Google Scholar] [CrossRef]
- Blaes, J.; Thomé, C.M.; Pfenning, P.N.; Rübmann, P.; Sahm, F.; Wick, A.; Bunse, T.; Schmenger, T.; Sykora, J.; von Deimling, A.; et al. Inhibition of CD95/CD95L (FAS/FASLG) Signaling with APG101 Prevents Invasion and Enhances Radiation Therapy for Glioblastoma. Mol. Cancer Res. 2018, 16, 767–776. [Google Scholar] [CrossRef]
- Wick, W.; Fricke, H.; Junge, K.; Kobyakov, G.; Martens, T.; Heese, O.; Wiestler, B.; Schliesser, M.G.; von Deimling, A.; Pichler, J.; et al. A phase II, randomized, study of weekly APG101+reirradiation versus reirradiation in progressive glioblastoma. Clin. Cancer Res. 2014, 20, 6304–6313. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Autoimmune Mouse Model | Ligand /Receptor Status | Outcome | References |
|---|---|---|---|
| Experimental autoimmune encephalomyelitis (EAE) | TRAIL neutralization (sDR5) | Exacerbation of symptoms | [44] |
| TRAIL neutralization (TRAIL Abs) | [38] | ||
| TRAIL −/− mice | [38,39] | ||
| TRAIL-R −/− mice | [40] | ||
| TRAIL injection | Attenuation of symptoms | [38] | |
| TRAIL expressing DC | [47,48] | ||
| lpr mice | [49,50,51] | ||
| gld mice | [49,50] | ||
| Experimental autoimmune thyroiditis (EAT) | TRAIL injection | Attenuation of symptoms | [42,43] |
| Collagen-induced rheumatoid arthritis (RA) | TRAIL neutralization (sDR5) | Exacerbation of symptoms | [45] |
| TRAIL −/− mice | [41] | ||
| TRAIL expressing DC | Attenuation of symptoms | [52] | |
| lpr mice | [53] | ||
| Type 1 diabetes | TRAIL neutralization (sDR5) | Exacerbation of symptoms | [46] |
| TRAIL −/− mice | [41] | ||
| NOD lpr mice | Attenuation of symptoms | [54,55,56] | |
| NOD gld mice | [57] |
| Ligand/Receptor Status | Cancer Induction | Outcome | References |
|---|---|---|---|
| TRAIL −/− mice | Spontaneous | Late-age lymphoma | [79] |
| p53 +/− mice | Sarcoma, lymphoma | [79] | |
| Her2/neu mice | No symptoms | [79] | |
| A20 cell line transfer | Lymphoma | [36] | |
| Renca cell line transfer | Liver metastasis | [80] | |
| 4T1 cell line transfer | Mammary carcinoma Lung and liver metastasis | [80] | |
| MCA induction | Fibrosarcoma | [80] | |
| TRAIL neutralization by Abs | p53 +/− mice | Sarcoma, lymphoma | [84] |
| L929 cell line transfer | Liver metastasis | [85] | |
| Renca cell line transfer | Liver metastasis | [86] | |
| MCA induction | Fibrosarcoma | [84] | |
| TRAIL-R −/− mice | Eu-myc mice | Lymphoma | [81] |
| p53 −/− mice | No symptoms | [82] | |
| APC min/+ mice | No symptoms | [82] | |
| DMBA/TPA | Lymph node metastasis | [83] | |
| DEN treatment | Hepato carcinoma | [81] | |
| lpr mice | T cell deficient | Lymphoma | [87] |
| Eu-myc mice | Lymphoma | [88] | |
| gld mice | Spontaneous | Lymphoma | [89] |
| FasL−/− T cells (CD8+) | Lymphoma cells transfer in rag1−/− mice | Lymphoma | [90] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossin, A.; Miloro, G.; Hueber, A.-O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639. https://doi.org/10.3390/cancers11050639
Rossin A, Miloro G, Hueber A-O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers. 2019; 11(5):639. https://doi.org/10.3390/cancers11050639
Chicago/Turabian StyleRossin, Aurélie, Giorgia Miloro, and Anne-Odile Hueber. 2019. "TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity" Cancers 11, no. 5: 639. https://doi.org/10.3390/cancers11050639
APA StyleRossin, A., Miloro, G., & Hueber, A.-O. (2019). TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers, 11(5), 639. https://doi.org/10.3390/cancers11050639