Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer

 , and
, and
Abstract
:1. Introduction
2. Results
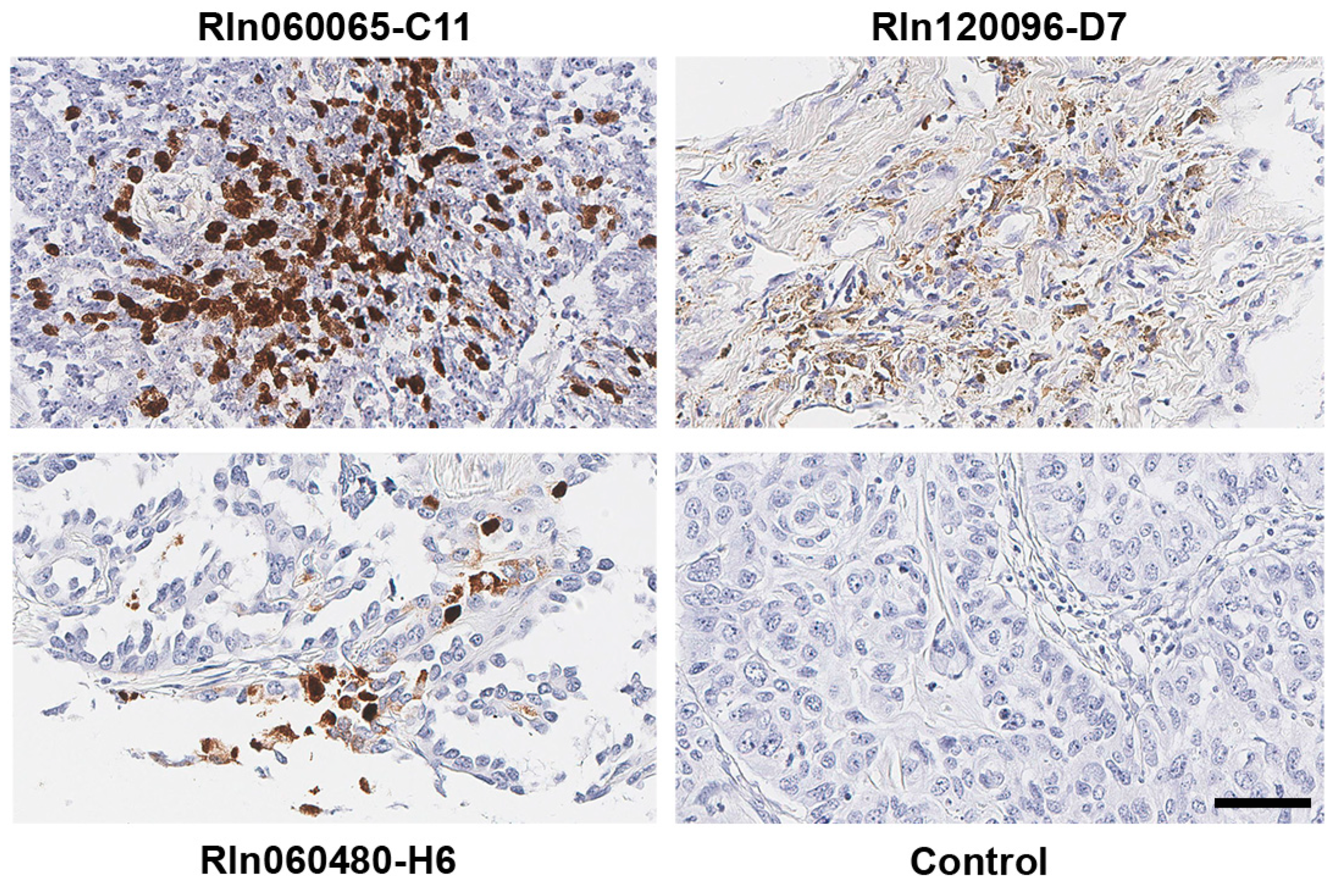
2.1. EBV is Detected in Primary Non-Small Cell Lung Cancer Samples
2.2. Type I EBV is Detected in EBV(+) NSCLC
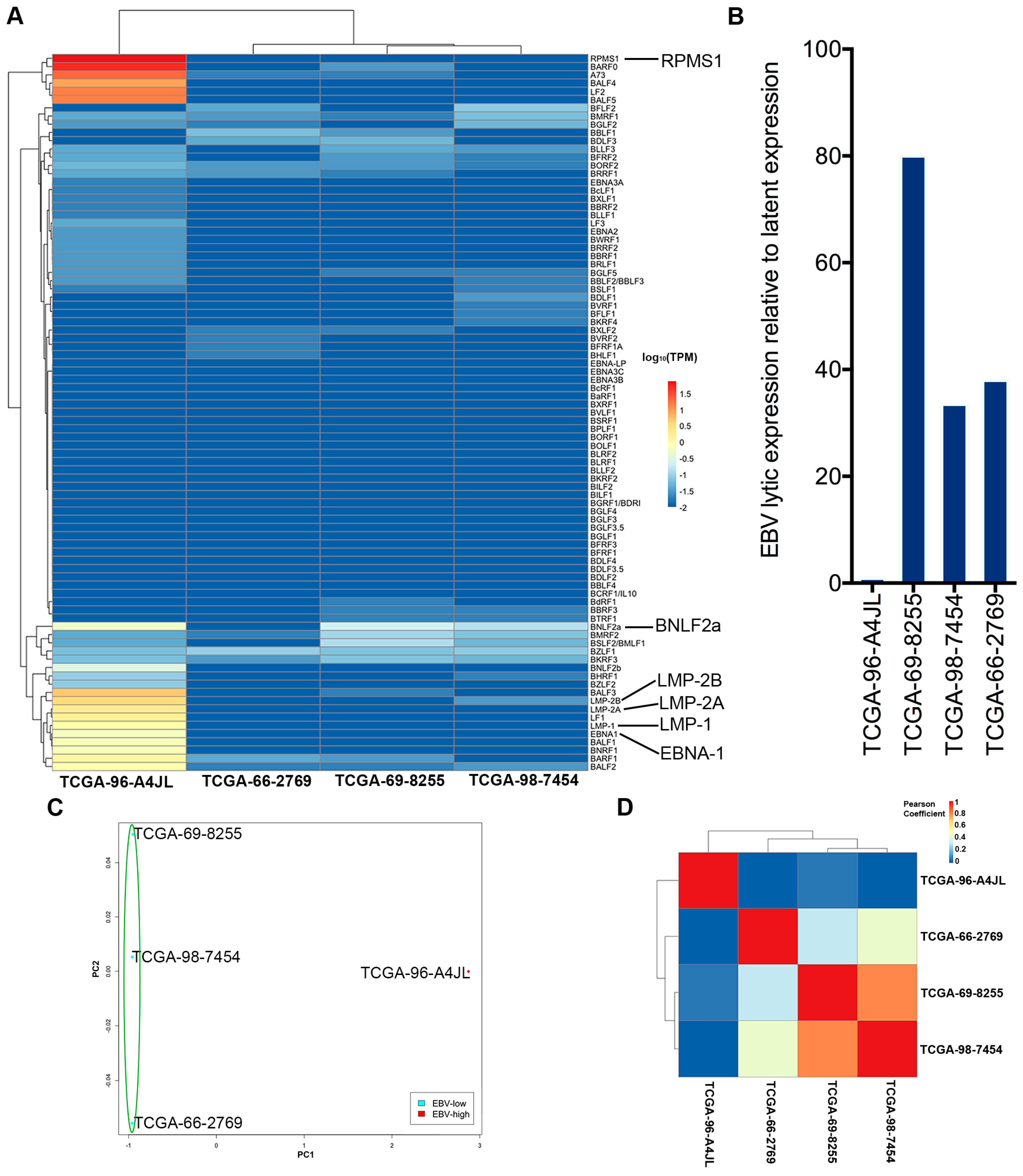
2.3. EBV Transcriptome Analysis
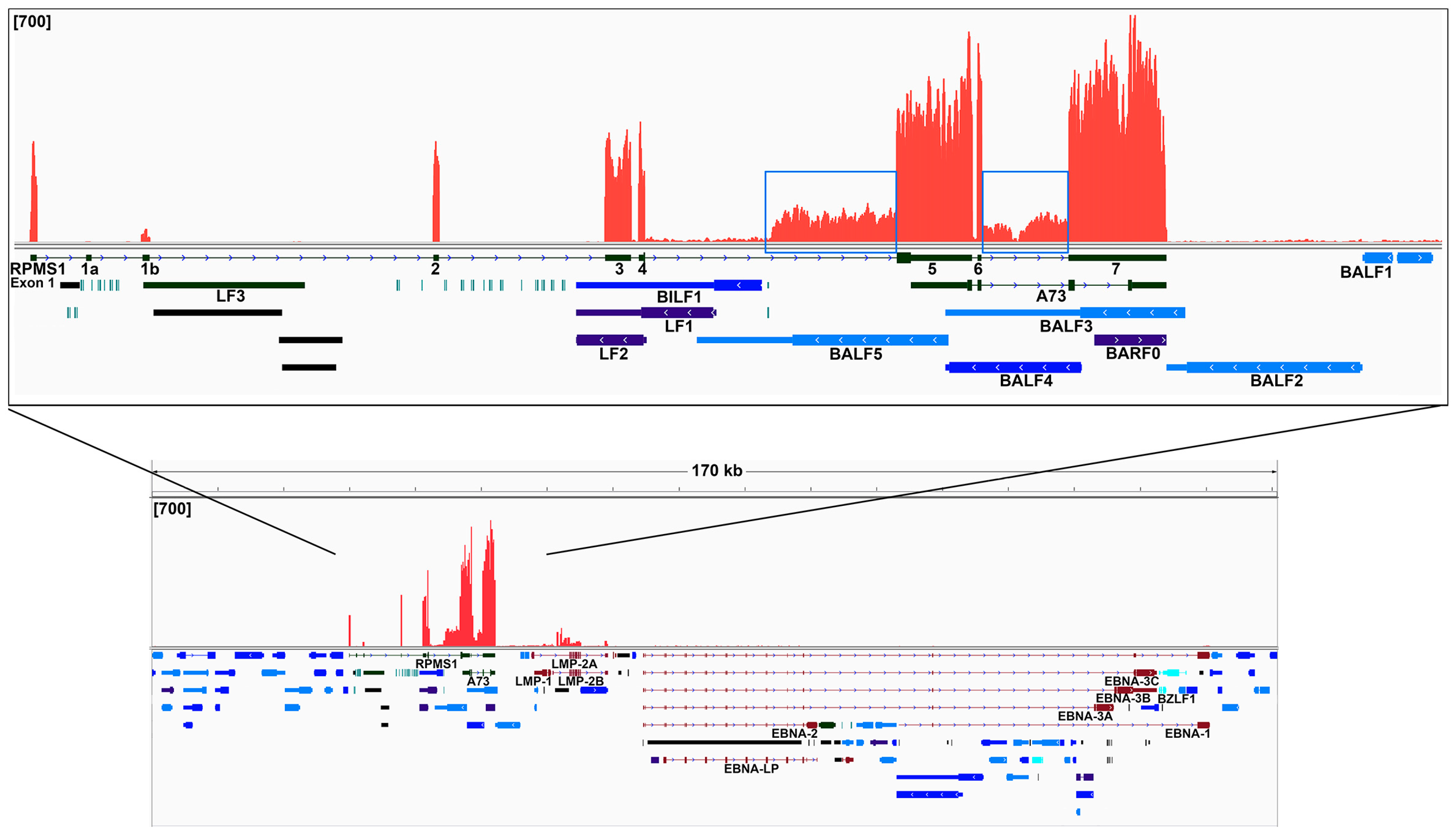
2.4. Analysis of the Highly Transcribed BamHI A Region
2.5. Analysis of the Viral circRNAs
2.6. Differential Cellular Gene Expression Profiles in EBV-High and EBV-Low Lung Squamous Cell Carcinoma
2.7. Elucidation of the Tumor Immune Microenvironment
3. Discussion
4. Materials and Methods
4.1. Sequencing Data Set Acquisition
4.2. RNA CoMPASS Analysis
4.3. Human and EBV Transcriptome Analysis
4.4. Circular RNA (circRNA Backsplice Junction) Analysis
4.5. Dimension Reduction, Correlation and Cluster Analysis of Human and EBV Expression Data
4.6. Deconvolution of Immune Cell Infiltration in the Tumor Tissue
4.7. Ingenuity Pathway Analysis (IPA)
4.8. EBERs In Situ Hybridization
4.9. Histopathology Images of TCGA Lung Cancer Samples
4.10. Cell Culture
4.11. DNA Transfection
4.12. RNA Extraction
4.13. Real-Time RT-PCR Analysis
4.14. Fluorescence Microscopy Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Engels, E.A. Inflammation in the development of lung cancer: Epidemiological evidence. Expert Rev. Anticancer Ther. 2008, 8, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Leroux, C.; Girard, N.; Cottin, V.; Greenland, T.; Mornex, J.F.; Archer, F. Jaagsiekte sheep retrovirus (JSRV): From virus to lung cancer in sheep. Vet. Res. 2007, 38, 211–228. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, C.; de-The, G. Epstein-Barr virus specific iga serum antibodies in nasopharyngeal and other respiratory carcinomas. Int. J. Cancer 1979, 24, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dey, S.; Chatterjee, R. Prevalence of serum IgG and IgM antibodies against Epstein-Barr virus capsid antigen in indian patients with respiratory tract carcinomas. Neoplasma 1994, 41, 29–33. [Google Scholar]
- Lung, M.L.; Lam, W.K.; So, S.Y.; Lam, W.P.; Chan, K.H.; Ng, M.H. Evidence that respiratory tract is major reservoir for Epstein-Barr virus. Lancet 1985, 1, 889–892. [Google Scholar] [CrossRef]
- Begin, L.R.; Eskandari, J.; Joncas, J.; Panasci, L. Epstein-Barr virus related lymphoepithelioma-like carcinoma of lung. J. Surg. Oncol. 1987, 36, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.Y.; Ostrowski, M.L.; Barrios, R.; Green, L.K.; Popper, H.H.; Powell, S.; Cagle, P.T.; Ro, J.Y. Relationship between Epstein-Barr virus and lymphoepithelioma-like carcinoma of the lung: A clinicopathologic study of 6 cases and review of the literature. Hum. Pathol. 2001, 32, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Wockel, W.; Hofler, G.; Popper, H.H.; Morresi, A. Lymphoepithelioma-like carcinoma of the lung. Pathol. Res. Pract. 1995, 191, 1170–1174. [Google Scholar] [CrossRef]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Tateishi, R.; Horiuchi, K.; Mishima, K. Lymphoepithelioma-like carcinoma of the lung: Analysis of two cases for Epstein-Barr virus infection. Hum. Pathol. 1995, 26, 1278–1282. [Google Scholar] [CrossRef]
- Ferrara, G.; Nappi, O. Lymphoepithelioma-like carcinoma of the lung. Two cases diagnosed in caucasian patients. Tumori 1995, 81, 144–147. [Google Scholar] [CrossRef]
- Chan, J.K.; Hui, P.K.; Tsang, W.Y.; Law, C.K.; Ma, C.C.; Yip, T.T.; Poon, Y.F. Primary lymphoepithelioma-like carcinoma of the lung. A clinicopathologic study of 11 cases. Cancer 1995, 76, 413–422. [Google Scholar] [CrossRef]
- Han, A.J.; Xiong, M.; Zong, Y.S. Association of epstein-barr virus with lymphoepithelioma-like carcinoma of the lung in southern china. Am. J. Clin. Pathol. 2000, 114, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, J.J.; Martinez, M.N.; Fernandez, S.L.; Val-Bernal, J.F. Epstein-Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod. Pathol. 2009, 22, 530–537. [Google Scholar] [CrossRef]
- Chen, F.F.; Yan, J.J.; Lai, W.W.; Jin, Y.T.; Su, I.J. Epstein-Barr virus-associated nonsmall cell lung carcinoma: Undifferentiated “lymphoepithelioma-like” carcinoma as a distinct entity with better prognosis. Cancer 1998, 82, 2334–2342. [Google Scholar] [CrossRef]
- Li, C.M.; Han, G.L.; Zhang, S.J. [Detection of Epstein-Barr virus in lung carcinoma tissue by in situ hybridization]. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 2007, 21, 288–290. [Google Scholar] [PubMed]
- Kasai, K.; Sato, Y.; Kameya, T.; Inoue, H.; Yoshimura, H.; Kon, S.; Kikuchi, K. Incidence of latent infection of Epstein-Barr virus in lung cancers—An analysis of EBER1 expression in lung cancers by in situ hybridization. J. Pathol. 1994, 174, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Pavlova, B.; Muhlberger, H.; Hollaus, P.; Lintner, F. Detection of the Epstein-Barr virus in primary adenocarcinoma of the lung with signet-ring cells. Virchows Arch. 2002, 441, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiong, H.; Yan, S.; Wu, N.; Lu, Z. Identification and characterization of Epstein-Barr virus genomes in lung carcinoma biopsy samples by next-generation sequencing technology. Sci. Rep. 2016, 6, 26156. [Google Scholar] [CrossRef]
- Jafarian, A.H.; Omidi-Ashrafi, A.; Mohamadian-Roshan, N.; Karimi-Shahri, M.; Ghazvini, K.; Boroumand-Noughabi, S. Association of Epstein Barr virus deoxyribonucleic acid with lung carcinoma. Indian J. Pathol. Microbiol. 2013, 56, 359–364. [Google Scholar]
- Chu, P.G.; Cerilli, L.; Chen, Y.Y.; Mills, S.E.; Weiss, L.M. Epstein-Barr virus plays no role in the tumorigenesis of small-cell carcinoma of the lung. Mod. Pathol. 2004, 17, 158–164. [Google Scholar] [CrossRef]
- Lim, W.T.; Chuah, K.L.; Leong, S.S.; Tan, E.H.; Toh, C.K. Assessment of human papillomavirus and Epstein-Barr virus in lung adenocarcinoma. Oncol. Rep. 2009, 21, 971–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshiol, J.; Gulley, M.L.; Zhao, Y.; Rubagotti, M.; Marincola, F.M.; Rotunno, M.; Tang, W.; Bergen, A.W.; Bertazzi, P.A.; Roy, D.; et al. Epstein-Barr virus micrornas and lung cancer. Br. J. Cancer 2011, 105, 320–326. [Google Scholar] [CrossRef]
- Lin, Z.; Xu, G.; Deng, N.; Taylor, C.; Zhu, D.; Flemington, E.K. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt’s lymphoma cells. J. Virol. 2010, 84, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Puetter, A.; Coco, J.; Xu, G.; Strong, M.J.; Wang, X.; Fewell, C.; Baddoo, M.; Taylor, C.; Flemington, E.K. Detection of murine leukemia virus in the Epstein-Barr virus-positive human B-cell line JY, using a computational RNA-Seq-based exogenous agent detection pipeline, PARSES. J. Virol. 2012, 86, 2970–2977. [Google Scholar] [CrossRef]
- Lin, Z.; Wang, X.; Strong, M.J.; Concha, M.; Baddoo, M.; Xu, G.; Baribault, C.; Fewell, C.; Hulme, W.; Hedges, D.; et al. Whole-genome sequencing of the Akata and Mutu Epstein-Barr virus strains. J. Virol. 2013, 87, 1172–1182. [Google Scholar] [CrossRef]
- Strong, M.J.; O’Grady, T.; Lin, Z.; Xu, G.; Baddoo, M.; Parsons, C.; Zhang, K.; Taylor, C.M.; Flemington, E.K. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin’s diffuse large B-cell lymphoma cohort by using RNA sequencing. J. Virol. 2013, 87, 13059–13062. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Baddoo, M.; Nanbo, A.; Xu, M.; Puetter, A.; Lin, Z. Comprehensive high-throughput RNA sequencing analysis reveals contamination of multiple nasopharyngeal carcinoma cell lines with HeLa cell genomes. J. Virol. 2014, 88, 10696–10704. [Google Scholar] [CrossRef]
- Strong, M.J.; Laskow, T.; Nakhoul, H.; Blanchard, E.; Liu, Y.; Wang, X.; Baddoo, M.; Lin, Z.; Yin, Q.; Flemington, E.K. Latent expression of the Epstein-Barr virus (EBV)-encoded major histocompatibility complex class I TAP inhibitor, BNLF2a, in EBV-positive gastric carcinomas. J. Virol. 2015, 89, 10110–10114. [Google Scholar] [CrossRef]
- Strong, M.J.; Blanchard, E.T.; Lin, Z.; Morris, C.A.; Baddoo, M.; Taylor, C.M.; Ware, M.L.; Flemington, E.K. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus—Tumor association. Acta Neuropathol. Commun. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Strong, M.J.; Lacey, M.R.; Baribault, C.; Flemington, E.K.; Taylor, C.M. RNA CoMPASS: A dual approach for pathogen and host transcriptome analysis of RNA-Seq datasets. PLoS ONE 2014, 9, e89445. [Google Scholar] [CrossRef]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Arrand, J.R.; Rymo, L. Characterization of the major Epstein-Barr virus-specific RNA in Burkitt lymphoma-derived cells. J. Virol. 1982, 41, 376–389. [Google Scholar] [PubMed]
- Schwemmle, M.; Clemens, M.J.; Hilse, K.; Pfeifer, K.; Troster, H.; Muller, W.E.; Bachmann, M. Localization of Epstein-Barr virus-encoded RNAs EBER-1 and EBER-2 in interphase and mitotic Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10292–10296. [Google Scholar] [CrossRef]
- Ungerleider, N.; Concha, M.; Lin, Z.; Roberts, C.; Wang, X.; Cao, S.; Baddoo, M.; Moss, W.N.; Yu, Y.; Seddon, M.; et al. The Epstein Barr virus circrnaome. PLoS Pathog. 2018, 14, e1007206. [Google Scholar] [CrossRef]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-Seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. Brca1 regulates ifi16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-beta responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef]
- Liao, G.; Huang, J.; Fixman, E.D.; Hayward, S.D. The Epstein-Barr virus replication protein BBLF2/3 provides an origin-tethering function through interaction with the zinc finger DNA binding protein ZBRK1 and the KAP-1 corepressor. J. Virol. 2005, 79, 245–256. [Google Scholar] [CrossRef]
- Maier, S.; Staffler, G.; Hartmann, A.; Hock, J.; Henning, K.; Grabusic, K.; Mailhammer, R.; Hoffmann, R.; Wilmanns, M.; Lang, R.; et al. Cellular target genes of Epstein-Barr virus nuclear antigen 2. J. Virol. 2006, 80, 9761–9771. [Google Scholar] [CrossRef]
- Yin, Q.; McBride, J.; Fewell, C.; Lacey, M.; Wang, X.; Lin, Z.; Cameron, J.; Flemington, E.K. Microrna-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J. Virol. 2008, 82, 5295–5306. [Google Scholar] [CrossRef]
- Cameron, J.E.; Fewell, C.; Yin, Q.; McBride, J.; Wang, X.; Lin, Z.; Flemington, E.K. Epstein-Barr virus growth/latency III program alters cellular microrna expression. Virology 2008, 382, 257–266. [Google Scholar] [CrossRef]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Yue, L.; Yao, R.; Zhou, L.; Yang, Y.; Lu, L.; Gao, W. P53 prevent tumor invasion and metastasis by down-regulating ido in lung cancer. Oncotarget 2017, 8, 54548–54557. [Google Scholar] [CrossRef]
- Yao, H.; Wang, H.; Li, C.; Fang, J.Y.; Xu, J. Cancer cell-intrinsic PD-1 and implications in combinatorial immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef]
- Zhang, H.; Dutta, P.; Liu, J.; Sabri, N.; Song, Y.; Li, W.X.; Li, J. Tumour cell-intrinsic CTLA4 regulates PD-l1 expression in non-small cell lung cancer. J. Cell. Mol. Med. 2019, 23, 535–542. [Google Scholar] [CrossRef]
- Villarroel-Espindola, F.; Yu, X.; Datar, I.; Mani, N.; Sanmamed, M.; Velcheti, V.; Syrigos, K.; Toki, M.; Zhao, H.; Chen, L.; et al. Spatially resolved and quantitative analysis of VISTA/PD-1H as a novel immunotherapy target in human non-small cell lung cancer. Clin. Cancer Res. 2018, 24, 1562–1573. [Google Scholar] [CrossRef]
- Thun, M.J.; Henley, S.J.; Calle, E.E. Tobacco use and cancer: An epidemiologic perspective for geneticists. Oncogene 2002, 21, 7307–7325. [Google Scholar] [CrossRef]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers—A different disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef]
- Ambinder, R.F. Gammaherpesviruses and “Hit-and-Run” oncogenesis. Am. J. Pathol. 2000, 156, 1–3. [Google Scholar] [CrossRef]
- Hu, H.; Luo, M.L.; Desmedt, C.; Nabavi, S.; Yadegarynia, S.; Hong, A.; Konstantinopoulos, P.A.; Gabrielson, E.; Hines-Boykin, R.; Pihan, G.; et al. Epstein-Barr virus infection of mammary epithelial cells promotes malignant transformation. EBioMedicine 2016, 9, 148–160. [Google Scholar] [CrossRef]
- Ho, J.C.; Wong, M.P.; Lam, W.K. Lymphoepithelioma-like carcinoma of the lung. Respirology 2006, 11, 539–545. [Google Scholar] [CrossRef]
- Thun, M.J.; Hannan, L.M.; Adams-Campbell, L.L.; Boffetta, P.; Buring, J.E.; Feskanich, D.; Flanders, W.D.; Jee, S.H.; Katanoda, K.; Kolonel, L.N.; et al. Lung cancer occurrence in never-smokers: An analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 2008, 5, e185. [Google Scholar] [CrossRef]
- Al-Mozaini, M.; Bodelon, G.; Karstegl, C.E.; Jin, B.; Al-Ahdal, M.; Farrell, P.J. Epstein-Barr virus BART gene expression. J. Gen. Virol. 2009, 90, 307–316. [Google Scholar] [CrossRef]
- Smith, P.R.; de Jesus, O.; Turner, D.; Hollyoake, M.; Karstegl, C.E.; Griffin, B.E.; Karran, L.; Wang, Y.; Hayward, S.D.; Farrell, P.J. Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J. Virol. 2000, 74, 3082–3092. [Google Scholar] [CrossRef]
- Marquitz, A.R.; Mathur, A.; Edwards, R.H.; Raab-Traub, N. Host gene expression is regulated by two types of noncoding rnas transcribed from the Epstein-Barr virus bamhi a rightward transcript region. J. Virol. 2015, 89, 11256–11268. [Google Scholar] [CrossRef]
- Lin, Z.; Flemington, E.K. Mirnas in the pathogenesis of oncogenic human viruses. Cancer Lett. 2011, 305, 186–199. [Google Scholar] [CrossRef]
- Concha, M.; Wang, X.; Cao, S.; Baddoo, M.; Fewell, C.; Lin, Z.; Hulme, W.; Hedges, D.; McBride, J.; Flemington, E.K. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J. Virol. 2012, 86, 1458–1467. [Google Scholar] [CrossRef]
- Fox, C.P.; Haigh, T.A.; Taylor, G.S.; Long, H.M.; Lee, S.P.; Shannon-Lowe, C.; O’Connor, S.; Bollard, C.M.; Iqbal, J.; Chan, W.C.; et al. A novel latent membrane 2 transcript expressed in Epstein-Barr virus-positive NK- and T-cell lymphoproliferative disease encodes a target for cellular immunotherapy. Blood 2010, 116, 3695–3704. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Latent membrane protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar]
- Bell, M.J.; Abbott, R.J.; Croft, N.P.; Hislop, A.D.; Burrows, S.R. An HLA-A2-restricted T-cell epitope mapped to the BNLF2a immune evasion protein of Epstein-Barr virus that inhibits TAP. J. Virol. 2009, 83, 2783–2788. [Google Scholar] [CrossRef]
- Horst, D.; van Leeuwen, D.; Croft, N.P.; Garstka, M.A.; Hislop, A.D.; Kremmer, E.; Rickinson, A.B.; Wiertz, E.J.; Ressing, M.E. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J. Immunol. 2009, 182, 2313–2324. [Google Scholar] [CrossRef]
- Croft, N.P.; Shannon-Lowe, C.; Bell, A.I.; Horst, D.; Kremmer, E.; Ressing, M.E.; Wiertz, E.J.; Middeldorp, J.M.; Rowe, M.; Rickinson, A.B.; et al. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009, 5, e1000490. [Google Scholar] [CrossRef]
- Horst, D.; Favaloro, V.; Vilardi, F.; van Leeuwen, H.C.; Garstka, M.A.; Hislop, A.D.; Rabu, C.; Kremmer, E.; Rickinson, A.B.; High, S.; et al. EBV protein BNLF2a exploits host tail-anchored protein integration machinery to inhibit TAP. J. Immunol. 2011, 186, 3594–3605. [Google Scholar] [CrossRef]
- Wycisk, A.I.; Lin, J.; Loch, S.; Hobohm, K.; Funke, J.; Wieneke, R.; Koch, J.; Skach, W.R.; Mayerhofer, P.U.; Tampe, R. Epstein-Barr viral BNLF2a protein hijacks the tail-anchored protein insertion machinery to block antigen processing by the transport complex TAP. J. Biol. Chem. 2011, 286, 41402–41412. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef]
- Hwu, P.; Du, M.X.; Lapointe, R.; Do, M.; Taylor, M.W.; Young, H.A. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J. Immunol. 2000, 164, 3596–3599. [Google Scholar] [CrossRef]
- Munn, D.H.; Shafizadeh, E.; Attwood, J.T.; Bondarev, I.; Pashine, A.; Mellor, A.L. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J. Exp. Med. 1999, 189, 1363–1372. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Nix, D.A.; Courdy, S.J.; Boucher, K.M. Empirical methods for controlling false positives and estimating confidence in chip-seq peaks. BMC Bioinform. 2008, 9, 523. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI reference sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using megan4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-Seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gartner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Munz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hilsendegen, T.; Pich, D.; Zeidler, R.; Hammerschmidt, W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 1998, 95, 8245–8250. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Coord 1 | Coord 2 | Gene/Locus | Junction Counts |
|---|---|---|---|---|
| EBV Akata Strain | 47242 | 47383 | RPMS1 | 1 |
| EBV Akata Strain | 47516 | 47631 | RPMS1 | 2 |
| EBV Akata Strain | 51610 | 51683 | RPMS1 | 1 |
| Canonical Pathways | Z-Score |
|---|---|
| Role of BRCA1 in DNA Damage Response | 2.6 |
| HIPPO signaling | 2.3 |
| TNFR1 Signaling | 1.8 |
| TNFR2 Signaling | 1.7 |
| CDK5 Signaling | 1.5 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 1.5 |
| Sirtuin Signaling Pathway | 1.5 |
| Sonic Hedgehog Signaling | 1.4 |
| Glutamate Receptor Signaling | 1.3 |
| Death Receptor Signaling | 1.0 |
| Calcium Transport I | 1.0 |
| p53 Signaling | 0.7 |
| Fatty Acid alpha-oxidation | 0.7 |
| Th2 Pathway | 0.7 |
| Cell Cycle: G1/S Checkpoint Regulation | 0.6 |
| Canonical Pathways | Z-Score |
|---|---|
| Oxidative Phosphorylation | −3.3 |
| Leukocyte Extravasation Signaling | −3.2 |
| ILK Signaling | −3.2 |
| TGF-beta Signaling | −3.1 |
| IL-8 Signaling | −2.9 |
| HMGB1 Signaling | −2.7 |
| fMLP Signaling in Neutrophils | −2.5 |
| Neuregulin Signaling | −2.5 |
| BMP signaling pathway | −2.5 |
| EIF2 Signaling | −2.4 |
| NRF2-mediated Oxidative Stress Response | −2.4 |
| B Cell Receptor Signaling | −2.2 |
| Regulation of eIF4 and p70S6K Signaling | −2.1 |
| CXCR4 Signaling | −2.1 |
| Regulation of Cellular Mechanics by Calpain Protease | −2.1 |
| Upstream Regulator | Z-Score |
|---|---|
| IFNL1 | 3.6 |
| miR-21-5p (and other miRNAs w/seed AGCUUAU) | 3.5 |
| miR-155-5p (miRNAs w/seed UAAUGCU) | 3.3 |
| NANOG | 2.9 |
| PML | 2.9 |
| NEUROG1 | 2.8 |
| HSF1 | 2.7 |
| GSK3B | 2.7 |
| KDM5B | 2.5 |
| miR-17-5p (and other miRNAs w/seed AAAGUGC) | 2.4 |
| miR-30c-5p (and other miRNAs w/seed GUAAACA) | 2.4 |
| ZNF281 | 2.4 |
| MSC | 2.3 |
| SIN3A | 2.2 |
| NR5A1 | 2.2 |
| Upstream Regulator | Z-Score |
|---|---|
| TGFB3 | −4.3 |
| IL1A | −3.9 |
| TGFB1 | −3.8 |
| CSF1 | −3.6 |
| IL6 | −3.5 |
| CSF2 | −3.3 |
| IGFBP2 | −3.3 |
| IL4 | −3.1 |
| SMAD3 | −2.9 |
| YAP1 | −2.8 |
| SMARCA4 | −2.7 |
| KLF6 | −2.7 |
| ELF4 | −2.6 |
| TGFB2 | −2.6 |
| ATF4 | −2.6 |
| Patient ID | Diagnosis | Gender | Race | TNM Stage | Tumor Stage | Smoking History | Age | Source |
|---|---|---|---|---|---|---|---|---|
| Rln060065-C11 | LUSC | FEMALE | N/A | T2-N2-M0 | Stage IIIA | N/A | 51 | Biomax |
| Rln120096-D7 | Adenosquamous | FEMALE | N/A | T2-N0-M0 | Stage IB | N/A | 64 | Biomax |
| Rln060480-H6 | LUAD | MALE | N/A | T2-N0-M0 | Stage IB | N/A | 49 | Biomax |
| TCGA-96-A4JL | LUSC | FEMALE | ASIAN | T2a-N1-M0 | Stage IIA | Lifelong Non-smoker | 78 | TCGA |
| TCGA-69-8255 | LUAD | MALE | WHITE | T1a-N0-M0 | Stage IA | smoker | 71 | TCGA |
| TCGA-98-7454 | LUSC | MALE | WHITE | T2a-N0-M0 | Stage IB | smoker | 73 | TCGA |
| TCGA-66-2769 | LUSC | MALE | N/A | T4-N0-M0 | Stage IIIB | smoker | 75 | TCGA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. https://doi.org/10.3390/cancers11060759
Kheir F, Zhao M, Strong MJ, Yu Y, Nanbo A, Flemington EK, Morris GF, Reiss K, Li L, Lin Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers. 2019; 11(6):759. https://doi.org/10.3390/cancers11060759
Chicago/Turabian StyleKheir, Fayez, Mengmeng Zhao, Michael J. Strong, Yi Yu, Asuka Nanbo, Erik K. Flemington, Gilbert F. Morris, Krzysztof Reiss, Li Li, and Zhen Lin. 2019. "Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer" Cancers 11, no. 6: 759. https://doi.org/10.3390/cancers11060759
APA StyleKheir, F., Zhao, M., Strong, M. J., Yu, Y., Nanbo, A., Flemington, E. K., Morris, G. F., Reiss, K., Li, L., & Lin, Z. (2019). Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers, 11(6), 759. https://doi.org/10.3390/cancers11060759