Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes

 ,
,
Abstract
:1. Introduction
2. Literature Review
2.1. RhoB Suppressed by Oncogenic Signaling
2.1.1. EGFR Reduces RHOB Promoter Activity Through Ras Signaling
2.1.2. Oncogenic K-Ras Suppresses RhoB and Induces Resistance to 5-Fluorouracil
2.1.3. PI3K Activates Akt through Several Mechanisms
2.1.4. GTP-bound Ras Activates PI3K via MAPK
2.1.5. Differential Regulation between PI3K/AKT and RhoB
2.2. RHOB Epigenetically Regulated by HDAC1/6
2.2.1. HDAC1 Represses RHOB Transcription by Binding Its Promoter
2.2.2. HDAC6 Represses RHOB Transcription through an Unknown Mechanism
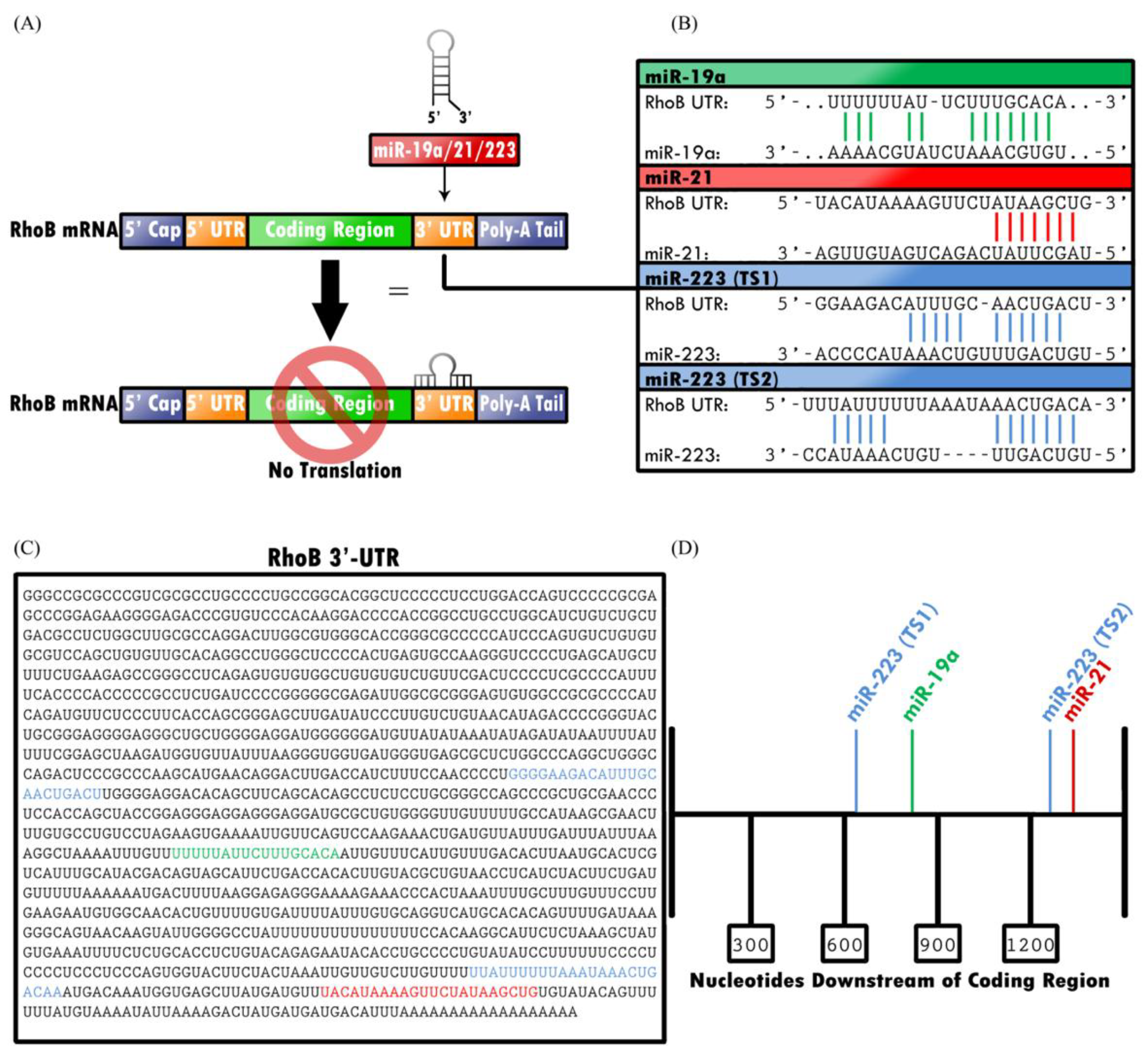
2.3. RHOB Is Targeted by MicroRNAs
2.3.1. miR-19a Downregulates RHOB by Binding to the 3′-UTR with Human Antigen R (HuR)
2.3.2. miR-21 Downregulates RHOB and Other Proteins that Participate in Cell Proliferation
2.3.3. miR-223 Downregulates RhoB Expression, but Can Also Mimic RhoB Expression
2.4. RhoB Loss During Tumorigenesis and Aging in Specific Tissues (Lungs and Muscles)
2.5. Restoration of RhoB for Cancer Prevention and Healthier Aging
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specifcity: Structure, function and local activation. Small GTPases 2014, 5, e968004. [Google Scholar] [CrossRef] [PubMed]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, S.; Mellor, H. Regulation of endocytic traffic by Rho family GTPases. Trends Cell Biol. 2000, 10, 85–88. [Google Scholar] [CrossRef]
- Pruitt, K.; Der, C.J. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001, 171, 1–10. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar]
- Treisman, R.; Alberts, A.S.; Sahai, E. Regulation of SRF Activity by Rho Family GTPases. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 643–652. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Gampel, A.; Mellor, H. Small interfering RNAs as a tool to assign Rho GTPase exchange-factor function in vivo. Biochem. J. 2002, 366, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Ridley, A.J. RhoA, RhoB and RhoC have different roles in cancer cell migration. J. Microsc. 2013, 251, 242–249. [Google Scholar] [CrossRef]
- Ju, J.A.; Gilkes, D.M. RhoB: Team Oncogene or Team Tumor Suppressor? Genes 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Watanabe, S.; Narumiya, S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013, 92, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Nomikou, E.; Livitsanou, M.; Stournaras, C.; Kardassis, D. Transcriptional and post-transcriptional regulation of the genes encoding the small GTPases RhoA, RhoB, and RhoC: Implications for the pathogenesis of human diseases. Cell. Mol. Life Sci. 2018, 75, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.K.; Bravo-Cordero, J.J.; Hodgson, L. Rho GTPase isoforms in cell motility: Don’t fret, we have FRET. Cell Adhes. Migr. 2014, 8, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.P.; Ridley, A.J. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 2004, 301, 43–49. [Google Scholar] [CrossRef]
- Vega, F.; Ridley, A. The RhoB small GTPase in physiology and disease. Small GTPases 2018, 9, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Thomas, M.; Reymond, N.; Ridley, A.J. The Rho GTPase RhoB regulates cadherin expression and epithelial cell-cell interaction. Cell Commun. Signal. 2015, 13, 6. [Google Scholar] [CrossRef]
- Huang, M.; Prendergast, G.C. RhoB in cancer suppression. Histol. Histopathol. 2006, 21, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Pedersen, E.D.; Wang, Z.; Brakebusch, C. Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mazières, J.; Tovar, D.; He, B.; Nieto-Acosta, J.; Marty-Detraves, C.; Clanet, C.; Pradines, A.; Jablons, D.; Favre, G. Epigenetic regulation of RhoB loss of expression in lung cancer. BMC Cancer 2007, 7, 220. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Choo, J.H.; Yoo, T.; Kang, K.; Chung, J.H. RhoB is epigenetically regulated in an age- and tissue-specific manner. Biochem. Biophys.Res. Commun. 2007, 362, 164–169. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P. How ageing processes influence cancer. Nat. Rev. Cancer 2013, 13, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Regulation of signal transduction and signal diversity by receptor oligomerization. Trends Biochem. Sci. 1994, 19, 459–463. [Google Scholar] [CrossRef]
- Ono, M.; Kuwano, M. Molecular Mechanisms of Epidermal Growth Factor Receptor (EGFR) Activation and Response to Gefitinib and Other EGFR-Targeting Drugs. Clin. Cancer Res. 2006, 12, 7242–7251. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Der, C.J. Ras family signaling: Therapeutic targeting. Cancer Biol. Ther. 2002, 1, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Brabender, J.; Danenberg, K.D.; Metzger, R.; Schneider, P.M.; Park, J.; Salonga, D.; Hölscher, A.H.; Danenberg, P.V. Epidermal Growth Factor Receptor and HER2-neu mRNA Expression in Non-Small Cell Lung Cancer Is Correlated with Survival. Clin. Cancer Res. 2001, 7, 1850–1855. [Google Scholar] [PubMed]
- Jiang, K.; Delarue, F.L.; Sebti, S.M. EGFR, ErbB2 and Ras but not Src suppress RhoB expression while ectopic expression of RhoB antagonizes oncogene-mediated transformation. Oncogene 2004, 23, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Gampel, A.; Parker, P.J.; Mellor, H. Regulation of epidermal growth factor receptor traffic by the small GTPase RhoB. Curr. Biol. 1999, 9, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Calvayrac, O.; Mazières, J.; Figarol, S.; Marty-Detraves, C.; Raymond-Letron, I.; Bousquet, E.; Farella, M.; Clermont-Taranchon, E.; Milia, J.; Rouquette, I.; et al. The RAS-related GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO Mol. Med. 2017, 9, 238–250. [Google Scholar] [CrossRef]
- Bos, J.L. ras Oncogenes in Human Cancer: A Review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Gysin, S.; Salt, M.; Young, A.; McCormick, F. Therapeutic Strategies for Targeting Ras Proteins. Genes Cancer 2011, 2, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Kjeldgaard, M.; Nyborg, J.; Clark, B.F. The GTP Binding Motif: Variations on A Theme. FASEB J. 1996, 10, 1347–1368. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Immervoll, H.; Hoem, D.; Kugarajh, K.; Steine, S.J.; Molven, A. Molecular analysis of the EGFR-RAS-RAF pathway in pancreatic ductal adenocarcinomas: Lack of mutations in the BRAF and EGFR genes. Virchows Arch. 2006, 448, 788–796. [Google Scholar] [CrossRef]
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.A.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumor-suppressive Pathways in Pancreatic Carcinoma. Cancer Res. 1997, 57, 1731–1734. [Google Scholar]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef] [Green Version]
- Lord, R.V.; O’Grady, R.; Sheehan, C.; Field, A.F.; Ward, R.L. K-ras codon 12 mutations in Barrett’s oesophagus and adenocarcinomas of the oesophagus and oesophagogastric junction. J. Gastroenterol. Hepatol. 2000, 15, 730–736. [Google Scholar] [CrossRef]
- Tajima, Y.; Yamazaki, K.; Makino, R.; Nishino, N.; Aoki, S.; Kato, M.; Morohara, K.; Kaetsu, T.; Kusano, M. Gastric and Intestinal Phenotypic Marker Expression in Early Differentiated-Type Tumors of the Stomach: Clinicopathologic Significance and Genetic Background. Clin. Cancer Res. 2006, 12, 6469–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Lee, J.S.; Suh, C.; Kim, S.W.; Kim, S.B.; Lee, J.H.; Lee, M.S.; Park, M.Y.; Sun, H.S.; Kim, S.H. Clinicopathologic significance of the K-ras gene codon 12 point mutation in stomach cancer. An analysis of 140 cases. Cancer 1995, 75, 2794–2801. [Google Scholar] [CrossRef]
- Yashiro, M.; Nishioka, N.; Hirakawa, K. K-ras mutation influences macroscopic features of gastric carcinoma. J. Surg. Res. 2005, 124, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Ueki, T.; Gao, Y.T.; Houlihan, P.S.; Wallace, C.; Wang, B.S.; Shen, M.C.; Deng, J.; Hsing, A.W. K-ras Mutation, p53 Overexpression, and Microsatellite Instability in Biliary Tract Cancers: A Population-based Study in China. Clin. Cancer Res. 2002, 8, 3156–3163. [Google Scholar] [PubMed]
- Heinemann, V.; Stintzing, S.; Kirchner, T.; Boeck, S.; Jung, A. Clinical relevance of EGFR- and KRAS-status in colorectal cancer patients treated with monoclonal antibodies directed against the EGFR. Cancer Treat. Rev. 2009, 35, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.A.; Wymer, J.; Clements, N. Detection of K-ras gene mutations in non-neoplastic lung tissue and lung cancers. Cancer Lett. 1996, 103, 115–121. [Google Scholar] [CrossRef]
- Suzuki, Y.; Orita, M.; Shiraishi, M.; Hayashi, K.; Sekiya, T. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene 1990, 5, 1037–1043. [Google Scholar]
- Graziano, S.L.; Gamble, G.P.; Newman, N.B.; Abbott, L.Z.; Rooney, M.; Mookherjee, S.; Lamb, M.; Kohman, L.; Poiesz, B.J. Prognostic Significance of K-ras Codon 12 Mutations in Patients with Resected Stage I and II Non–Small-Cell Lung Cancer. J. Clin. Oncol. 1999, 17, 668. [Google Scholar] [CrossRef]
- Meng, D.; Yuan, M.; Li, X.; Chen, L.; Yang, J.; Zhao, X.; Ma, W.; Xin, J. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: A systematic review with meta-analysis. Lung Cancer 2013, 81, 1–10. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Westcott, P.M.K.; To, M.D. The genetics and biology of KRAS in lung cancer. Chin. J. Cancer 2013, 32, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A.; et al. An Integrative Genomic and Proteomic Analysis of PIK3CA, PTEN, and AKT Mutations in Breast Cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS Interaction with PI3K. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Quideau, S.; Rochaix, P.; Caselles, O.; Couderc, B.; Hennebelle, I.; Courbon, F.; Canal, P.; Allal, B.C. Trastuzumab induced in vivo tissue remodelling associated in vitro with inhibition of the active forms of AKT and PTEN and RhoB induction in an ovarian carcinoma model. Br. J. Cancer 2010, 103, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Chang, C.S.; Ho, W.C.; Liao, S.Y.; Lin, W.L.; Wang, C.J. Gallic acid inhibits gastric cancer cells metastasis and invasive growth via increased expression of RhoB, downregulation of AKT/small GTPase signals and inhibition of NF-κB activity. Toxicol. Appl. Pharmacol. 2013, 266, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Chen, J.H.; Chou, F.P.; Wang, C.J. Protocatechuic acid inhibits cancer cell metastasis involving the down-regulation of Ras/Akt/NF-κB pathway and MMP-2 production by targeting RhoB activation. Br. J. Pharmacol. 2011, 162, 237–254. [Google Scholar] [CrossRef]
- Won, K.J.; Kim, B.K.; Han, G.; Lee, K.; Jung, Y.J.; Kim, H.M.; Song, K.B.; Chung, K.S.; Won, M. NSC126188 induces apoptosis of prostate cancer PC-3 cells through inhibition of Akt membrane translocation, FoxO3a activation, and RhoB transcription. Apoptosis Int. J. Program. Cell Death 2014, 19, 179–190. [Google Scholar] [CrossRef]
- Zhang, C.; Elkahloun, A.G.; Liao, H.; Delaney, S.; Saber, B.; Morrow, B.; Prendergast, G.C.; Hollander, M.C.; Gills, J.J.; Dennis, P.A. Expression Signatures of the Lipid-Based Akt Inhibitors Phosphatidylinositol Ether Lipid Analogues in NSCLC Cells. Mol. Cancer Ther. 2011, 10, 1137–1148. [Google Scholar] [CrossRef]
- Bousquet, E.; Calvayrac, O.; Mazières, J.; Lajoie-Mazenc, I.; Boubekeur, N.; Favre, G.; Pradines, A. RhoB loss induces Rac1-dependent mesenchymal cell invasion in lung cells through PP2A inhibition. Oncogene 2016, 35, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, E.; Mazières, J.; Privat, M.; Rizzati, V.; Casanova, A.; Ledoux, A.; Mery, E.; Couderc, B.; Favre, G.; Pradines, A. Loss of RhoB Expression Promotes Migration and Invasion of Human Bronchial Cells Via Activation of AKT1. Cancer Res. 2009, 69, 6092–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Sun, J.; Cheng, J.; Djeu, J.Y.; Wei, S.; Sebti, S. Akt Mediates Ras Downregulation of RhoB, a Suppressor of Transformation, Invasion, and Metastasis. Mol. Cell. Biol. 2004, 24, 5565–5576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazerounian, S.; Gerald, D.; Huang, M.; Chin, Y.R.; Udayakumar, D.; Zheng, N.; O’Donnell, R.K.; Perruzzi, C.; Mangiante, L.; Pourat, J.; et al. RhoB Differentially Controls Akt Function in Tumor Cells and Stromal Endothelial Cells during Breast Tumorigenesis. Cancer Res. 2013, 73, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Adini, I.; Rabinovitz, I.; Sun, J.F.; Prendergast, G.C.; Benjamin, L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003, 17, 2721–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, G.; Brachetti, C.; Bahlmann, F.; Schmidt, M.; Kaina, B. Rho GTPases in human breast tumours: Expression and mutation analyses and correlation with clinical parameters. Br. J. Cancer 2002, 87, 635. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41. [Google Scholar] [CrossRef]
- Wang, S.; Yan-Neale, Y.; Fischer, D.; Zeremski, M.; Cai, R.; Zhu, J.; Asselbergs, F.; Hampton, G.; Cohen, D. Histone deacetylase 1 represses the small GTPase RhoB expression in human nonsmall lung carcinoma cell line. Oncogene 2003, 22, 6204–6213. [Google Scholar] [CrossRef] [Green Version]
- Delarue, F.L.; Adnane, J.; Joshi, B.; Blaskovich, M.A.; Wang, D.A.; Hawker, J.; Bizouarn, F.; Ohkanda, J.; Zhu, K.; Hamilton, A.D.; et al. Farnesyltransferase and geranylgeranyltransferase I inhibitors upregulate RhoB expression by HDAC1 dissociation, HAT association and histone acetylation of the RhoB promoter. Oncogene 2007, 26, 633–640. [Google Scholar] [CrossRef]
- Marlow, L.A.; Reynolds, L.A.; Cleland, A.S.; Cooper, S.J.; Gumz, M.L.; Kurakata, S.; Fujiwara, K.; Zhang, Y.; Sebo, T.; Grant, C.; et al. Reactivation of suppressed RhoB is a critical step for the inhibition of anaplastic thyroid cancer growth. Cancer Res. 2009, 69, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, N.; Ren, K.; Meng, S.; Xie, Y.; Long, Q.; Chen, X.; Zhao, X. Expression loss and revivification of RhoB gene in ovary carcinoma carcinogenesis and development. PLoS ONE 2013, 8, e78417. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Im, J.Y.; Han, G.; Lee, W.J.; Won, K.J.; Chung, K.S.; Lee, K.; Ban, H.S.; Song, K.; Won, M. p300 cooperates with c-Jun and PARP-1 at the p300 binding site to activate RhoB transcription in NSC126188-mediated apoptosis. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Marlow, L.A.; Bok, I.; Smallridge, R.C.; Copland, J.A. RhoB upregulation leads to either apoptosis or cytostasis through differential target selection. Endocr. Relat. Cancer 2015, 22, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Keusch, J.J.; Wang, L.; Saito, M.; Hess, D.; Wang, X.; Melancon, B.J.; Helquist, P.; Gut, H.; Matthias, P. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 2016, 12, 748–754. [Google Scholar] [CrossRef]
- Wang, S.; Yan-Neale, Y.; Zeremski, M.; Cohen, D. Transcription regulation by histone deacetylases. Novartis Found. Symp. 2004, 259, 238–245. [Google Scholar] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Yoda, M.; Kawamata, T.; Paroo, Z.; Ye, X.; Iwasaki, S.; Liu, Q.; Tomari, Y. ATP-dependent human RISC assembly pathways. Nat. Struct. Mol. Biol. 2010, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Glorian, V.; Maillot, G.; Polès, S.; Iacovoni, J.S.; Favre, G.; Vagner, S. HuR-dependent loading of miRNA RISC to the mRNA encoding the Ras-related small GTPase RhoB controls its translation during UV-induced apoptosis. Cell Death Differ. 2011, 18, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Guo, W.; Zhang, Y.; Wu, Y.; Xiang, J. MiR-19a promotes cell proliferation and invasion by targeting RhoB in human glioma cells. Neurosci. Lett. 2016, 628, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Chen, T.; Lin, Q.; Lin, G.; Lin, J.; Chen, G.; Guo, L. Serum miR-19a expression correlates with worse prognosis of patients with non-small cell lung cancer. J. Surg. Oncol. 2013, 107, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Z. MicroRNA-19a functions as an oncogenic microRNA in non-small cell lung cancer by targeting the suppressor of cytokine signaling 1 and mediating STAT3 activation. Int. J. Mol. Med. 2015, 35, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ito, S.; Hanafusa, H.; Shimizu, K.; Ouchida, M. Uncovering Direct Targets of MiR-19a Involved in Lung Cancer Progression. PLoS ONE 2015, 10, e0137887. [Google Scholar] [CrossRef]
- Chen, W.; Niu, S.; Ma, X.; Zhang, P.; Gao, Y.; Fan, Y.; Pang, H.; Gong, H.; Shen, D.; Gu, L.; et al. RhoB Acts as a Tumor Suppressor That Inhibits Malignancy of Clear Cell Renal Cell Carcinoma. PLoS ONE 2016, 11, e0157599. [Google Scholar] [CrossRef]
- Niu, S.; Ma, X.; Zhang, Y.; Liu, Y.N.; Chen, X.; Gong, H.; Yao, Y.; Liu, K.; Zhang, X. MicroRNA-19a and microRNA-19b promote the malignancy of clear cell renal cell carcinoma through targeting the tumor suppressor RhoB. PLoS ONE 2018, 13, e0192790. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Wang, Y. miR-19a promotes invasion and epithelial to mesenchymal transition of bladder cancer cells by targeting RhoB. Off. J. Balk. Union Oncol. 2019, 24, 797–804. [Google Scholar]
- Connolly, E.C.; Doorslaer, K.V.; Rogler, L.E.; Rogler, C.E. Overexpression of miR-21 Promotes an In vitro Metastatic Phenotype by Targeting the Tumor Suppressor RHOB. Mol. Cancer Res. 2010, 8, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Leone, E.; Morelli, E.; Martino, M.T.D.; Amodio, N.; Foresta, U.; Gullà, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 Inhibits In Vitro and In Vivo Multiple Myeloma Cell Growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, Q.; Qiu, M.; Lang, N.; Li, M.; Zheng, Y.; Bi, F. miR-21 targets the tumor suppressor RhoB and regulates proliferation, invasion and apoptosis in colorectal cancer cells. FEBS Lett. 2011, 585, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Schetter, A.J.; Mollerup, S.; Kohno, T.; Skaug, V.; Bowman, E.D.; Mathé, E.A.; Takenoshita, S.; Yokota, J.; Haugen, A.; et al. The Association of MicroRNA Expression with Prognosis and Progression in Early-Stage, Non–Small Cell Lung Adenocarcinoma: A Retrospective Analysis of Three Cohorts. Clin. Cancer Res. 2011, 17, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Song, G.; Roll, G.R.; Frandsen, N.M.; Willenbring, H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Investig. 2012, 122, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Sabatel, C.; Malvaux, L.; Bovy, N.; Deroanne, C.; Lambert, V.; Gonzalez, M.L.A.; Colige, A.; Rakic, J.M.; Noël, A.; Martial, J.A.; et al. MicroRNA-21 Exhibits Antiangiogenic Function by Targeting RhoB Expression in Endothelial Cells. PLoS ONE 2011, 6, e16979. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, Y.; Shi, C.; Chen, H.; Zhang, H.; Chen, N.; Zhang, P.; Wang, F.; Yang, J.; Yang, J.; et al. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB. Biochem. Biophys. Res. Commun. 2013, 434, 746–752. [Google Scholar] [CrossRef]
- Sun, G.; Li, H.; Rossi, J.J. Sequence context outside the target region influences the effectiveness of miR-223 target sites in the RhoB 3′UTR. Nucleic Acids Res. 2010, 38, 239–252. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Kang, K.; Chen, J.; Wu, Z.; Huang, J.; Lu, W.; Chen, Y.; Zhang, J.; Wang, Z.; et al. MicroRNA-223 Attenuates Hypoxia-induced Vascular Remodeling by Targeting RhoB/MLC2 in Pulmonary Arterial Smooth Muscle Cells. Sci. Rep. 2016, 6, 24900. [Google Scholar] [CrossRef]
- Wei, L.J.; Li, J.A.; Bai, D.M.; Song, Y. miR-223-RhoB signaling pathway regulates the proliferation and apoptosis of colon adenocarcinoma. Chem. Biol. Interact. 2018, 289, 9–14. [Google Scholar] [CrossRef]
- Zhang, C.; Elkahloun, A.G.; Robertson, M.; Gills, J.J.; Tsurutani, J.; Shih, J.H.; Fukuoka, J.; Hollander, M.C.; Harris, C.C.; Travis, W.D.; et al. Loss of Cytoplasmic CDK1 Predicts Poor Survival in Human Lung Cancer and Confers Chemotherapeutic Resistance. PLoS ONE 2011, 6, e23849. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Nuevo, A.; O’Donnell, R.; Rosendahl, A.; Chung, J.H.; Benjamin, L.E.; Odaka, C. RhoB deficiency in thymic medullary epithelium leads to early thymic atrophy. Int. Immunol. 2011, 23, 593–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Cerniglia, G.J.; Bernhard, E.J.; Prendergast, G.C. RhoB is required to mediate apoptosis in neoplasticallytransformed cells after DNA damage. Proc. Natl. Acad. Sci. USA 2001, 98, 6192–6197. [Google Scholar] [CrossRef] [PubMed]
- Mamouni, K.; Cristini, A.; Guirouilh-Barbat, J.; Monferran, S.; Lemarié, A.; Faye, J.C.; Lopez, B.S.; Favre, G.; Sordet, O. RhoB Promotes γH2AX Dephosphorylation and DNA Double-Strand Break Repair. Mol. Cell. Biol. 2014, 34, 3144–3155. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Guo, L.; Wu, Q.; Zeng, T.; Lin, Q.; Qiao, Y.; Wang, Q.; Liu, M.; Zhang, X.; Ren, L.; et al. ATR/Chk1/Smurf1 pathway determines cell fate after DNA damage by controlling RhoB abundance. Nat. Commun. 2014, 5, 4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, G.C. Actin’ up: RhoB in cancer and apoptosis. Nat. Rev. Cancer 2001, 1, 162–168. [Google Scholar] [CrossRef]
- Calvayrac, O.; Nowosad, A.; Cabantous, S.; Lin-Po, L.; Figarol, S.; Jeannot, P.; Serres, M.; Callot, C.; Perchey, R.; Creff, J.; et al. Cytoplasmic p27Kip1 promotes tumorigenesis via suppression of RhoB activity. J. Pathol. 2018, 247, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Luis-Ravelo, D.; Antón, I.; Zandueta, C.; Valencia, K.; Pajares, M.; Agorreta, J.; Montuenga, L.; Vicent, S.; Wistuba, I.; Rivas, J.; et al. RHOB influences lung adenocarcinoma metastasis and resistance in a host-sensitive manner. Mol. Oncol. 2014, 8, 192–206. [Google Scholar] [CrossRef]
- Calvayrac, O.; Pradines, A.; Raymond-Letron, I.; Rouquette, I.; Bousquet, E.; Lauwers-Cances, V.; Filleron, T.; Cadranel, J.; Beau-Faller, M.; Casanova, A.; et al. RhoB Determines Tumor Aggressiveness in a Murine EGFRL858R-Induced Adenocarcinoma Model and Is a Potential Prognostic Biomarker for Lepidic Lung Cancer. Clin. Cancer Res. 2014, 20, 6541–6550. [Google Scholar] [CrossRef]
- Couderc, B.; Pradines, A.; Rafii, A.; Golzio, M.; Deviers, A.; Allal, C.; Berg, D.; Penary, M.; Tessie, J.; Favre, G. In vivorestoration of RhoB expression leads to ovarian tumor regression. Cancer Gene Ther. 2008, 15, 456–464. [Google Scholar] [CrossRef]
- Tan, Y.; Yin, H.; Zhang, H.; Fang, J.; Zheng, W.; Li, D.; Li, Y.; Cao, W.; Sun, C.; Liang, Y.; et al. Sp1-driven up-regulation of miR-19a decreases RHOB and promotes pancreatic cancer. Oncotarget 2015, 6, 17391–17403. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Alic, N.; Foley, A.; Cabecinha, M.; Hoddinott, M.P.; Partridge, L. The Ras-Erk-ETS-Signaling Pathway Is a Drug Target for Longevity. Cell 2015, 162, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, A.; Yamashita, M.; Yoshida, Y.; Shimizu, I.; Ichimiya, H.; Kamimura, N.; Kobayashi, Y.; Ohta, S.; Ishii, N.; Minamino, T. Haploinsufficiency of Akt1 Prolongs the Lifespan of Mice. PLoS ONE 2013, 8, e69178. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging following genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Miller, R.A. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Molina, A.; Efeyan, A.; Lopez-Guadamillas, E.; Muñoz-Martin, M.; Gómez-López, G.; Cañamero, M.; Mulero, F.; Pastor, J.; Martinez, S.; Romanos, E.; et al. Pten Positively Regulates Brown Adipose Function, Energy Expenditure, and Longevity. Cell Metab. 2012, 15, 382–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Kamasani, U.; Prendergast, G.C. RhoB facilitates c-Myc turnover by supporting efficient nuclear accumulation of GSK-3. Oncogene 2006, 25, 1281–1289. [Google Scholar] [CrossRef]
- Kümper, S.; Mardakheh, F.K.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jørgensen, C.; Guichard, S.; et al. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 2016, 15, e12203. [Google Scholar] [CrossRef]
- Grassi, E.S.; Vezzoli, V.; Negri, I.; Lábadi, Á.; Fugazzola, L.; Vitale, G.; Persani, L. SP600125 has a remarkable anticancer potential against undifferentiated thyroid cancer through selective action on ROCK and p53 pathways. Oncotarget 2015, 6, 36383–36399. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.S.; Hinds, P.W. Phosphorylation of ezrin by cyclin-dependent kinase 5 induces the release of Rho GDP dissociation inhibitor to inhibit Rac1 activity in senescent cells. Cancer Res. 2006, 66, 2708–2715. [Google Scholar] [CrossRef]
- Lawson, C.D.; Fan, C.; Mitin, N.; Baker, N.M.; George, S.D.; Graham, D.M.; Perou, C.M.; Burridge, K.; Der, C.J.; Rossman, K.L. Rho GTPase Transcriptome Analysis Reveals Oncogenic Roles for Rho GTPase-Activating Proteins in Basal-like Breast Cancers. Cancer Res. 2016, 76, 3826–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Suppression of RhoB through oncogenic pathways | Reference |
| [30,109] |
| Regulation of RhoB activity through the PI3K/Akt pathway | |
| [56,58,61,62,63,64] |
| Epigenetic regulation of RhoB during aging and cancer | |
| [23,24,69,70,74,75] |
| Regulation of RHOB expression by miRNA | |
| [83,91,92,93,99] |
| Impact of RhoB in the control of genome stability and response to stress | |
| [23,24,101,103,104,105] |
| Potential therapeutic benefits using RhoB targeting | |
| [57,58,83,84,110,111] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez, E.; Cahatol, I.; Bailey, C.A.R.; Lafargue, A.; Zhang, N.; Song, Y.; Tian, H.; Zhang, Y.; Chan, R.; Gu, K.; et al. Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes. Cancers 2019, 11, 818. https://doi.org/10.3390/cancers11060818
Gutierrez E, Cahatol I, Bailey CAR, Lafargue A, Zhang N, Song Y, Tian H, Zhang Y, Chan R, Gu K, et al. Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes. Cancers. 2019; 11(6):818. https://doi.org/10.3390/cancers11060818
Chicago/Turabian StyleGutierrez, Eutiquio, Ian Cahatol, Cedric A.R. Bailey, Audrey Lafargue, Naming Zhang, Ying Song, Hongwei Tian, Yizhi Zhang, Ryan Chan, Kevin Gu, and et al. 2019. "Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes" Cancers 11, no. 6: 818. https://doi.org/10.3390/cancers11060818
APA StyleGutierrez, E., Cahatol, I., Bailey, C. A. R., Lafargue, A., Zhang, N., Song, Y., Tian, H., Zhang, Y., Chan, R., Gu, K., Zhang, A. C. C., Tang, J., Liu, C., Connis, N., Dennis, P., & Zhang, C. (2019). Regulation of RhoB Gene Expression during Tumorigenesis and Aging Process and Its Potential Applications in These Processes. Cancers, 11(6), 818. https://doi.org/10.3390/cancers11060818