Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types

 , ,
, ,  and
and 
{kind=link}
Abstract
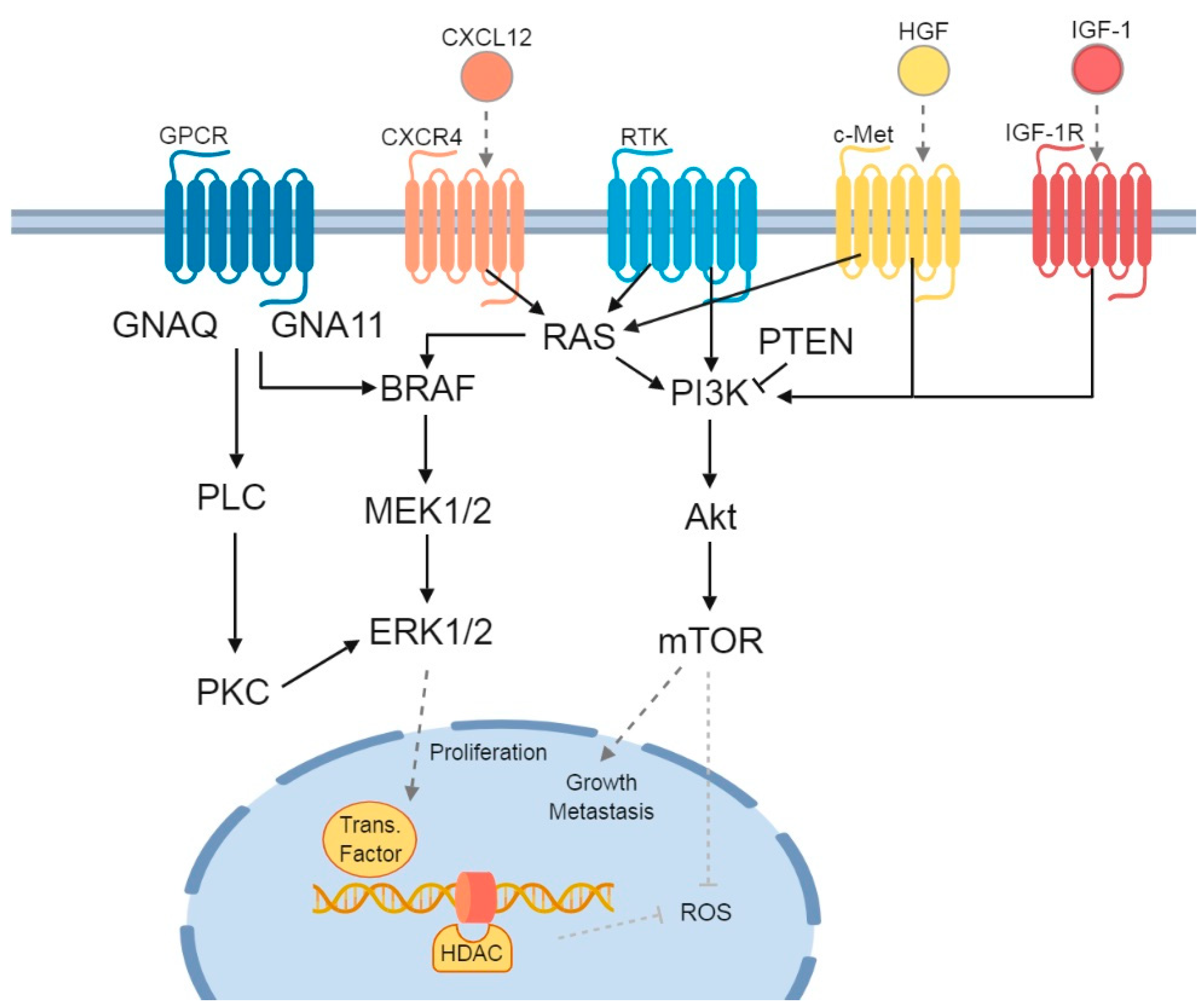
1. Melanocytes and Their Cellular Function
2. Genetic Alterations and Treatment Implications
3. Biological Parameters Underlying Metastasis
4. The Impact of the Immune System
4.1. Primary Tumor
4.2. Metastatic Melanoma
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Westekemper, H.; Murali, R.; Mach, M.; Schilling, B.; Wiesner, T.; Schimming, T.; Livingstone, E.; Sucker, A.; Grabellus, F.; et al. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clin. Cancer Res. 2013, 19, 3143–3152. [Google Scholar] [CrossRef] [PubMed]
- Sarna, T. Properties and function of the ocular melanin—A photobiophysical view. J. Photochem. Photobiol. B 1992, 12, 215–258. [Google Scholar] [CrossRef]
- Weis, E.; Shah, C.P.; Lajous, M.; Shields, J.A.; Shields, C.L. The association between host susceptibility factors and uveal melanoma—A meta-analysis. Arch. Ophthalmol. 2006, 124, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Ny, L.; Nyakas, M.; Hernberg, M.; Koivunen, J.; Oddershede, L.; Yoon, M.-R.; Wang, X.; Guyot, P.; Geisler, J. BRAF mutation as a prognostic marker for survival in malignant melanoma: A systematic review and meta-analysis. J. Clin. Oncol. 2018, 36, e21566. [Google Scholar] [CrossRef]
- Spathis, A.; Katoulis, A.C.; Damaskou, V.; Liakou, A.I.; Kottaridi, C.; Leventakou, D.; Sgouros, D.; Mamantopoulos, A.; Rigopoulos, D.; Karakitsos, P.; et al. BRAF mutation status in primary, recurrent, and metastatic malignant melanoma and its relation to histopathological parameters. Dermatol. Pract. Concept. 2019, 9, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Ihle, M.A.; Fassunke, J.; Konig, K.; Grunewald, I.; Schlaak, M.; Kreuzberg, N.; Tietze, L.; Schildhaus, H.U.; Buttner, R.; Merkelbach-Bruse, S. Comparison of high resolution melting analysis, pyrosequencing, next generation sequencing and immunohistochemistry to conventional Sanger sequencing for the detection of p.V600E and non-p.V600E BRAF mutations. BMC Cancer 2014, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Carvajal, R.D. KIT as an oncogenic driver in melanoma: An update on clinical development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, X.; Liu, J.; Zhao, B.; Cai, W.; Li, Y.; Hu, D. Efficacy and safety of BRAF inhibition alone versus combined BRAF and MEK inhibition in melanoma: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 32258–32269. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Eng. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Eng. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Eng. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Eng. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.-A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Guo, J.; Carvajal, R.D.; Dummer, R.; Hauschild, A.; Daud, A.; Bastian, B.C.; Markovic, S.N.; Queirolo, P.; Arance, A.; Berking, C.; et al. Efficacy and safety of nilotinib in patients with KIT-mutated metastatic or inoperable melanoma: Final results from the global, single-arm, phase II TEAM trial. Ann. Oncol. 2017, 28, 1380–1387. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X.; et al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, T.M.; Kim, Y.J.; Jang, K.T.; Lee, H.J.; Lee, S.N.; Ahn, M.S.; Hwang, I.G.; Lee, S.; Lee, M.H.; et al. Phase II Trial of nilotinib in patients with metastatic malignant melanoma harboring kit gene aberration: A multicenter trial of korean cancer study group (UN10-06). Oncologist 2015, 20, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.F.; Heijkants, R.C.; Jochemsen, A.G.; Dogrusoz, M.; de Lange, M.J.; van der Velden, P.A.; van der Burg, S.H.; Jager, M.J.; Verdijk, R.M. Targeting of the MAPK and AKT pathways in conjunctival melanoma shows potential synergy. Oncotarget 2017, 8, 58021–58036. [Google Scholar] [CrossRef] [PubMed]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., 3rd; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef]
- Pahlitzsch, M.; Bertelmann, E.; Mai, C. Conjunctival melanoma and BRAF inhibitor therapy. J. Clin. Exp. Ophtalmol. 2014, 5, 322. [Google Scholar] [CrossRef]
- Weber, J.L.; Smalley, K.S.M.; Sondak, V.K.; Gibney, G.T. Conjunctival Melanomas Harbor BRAF and NRAS Mutations-Letter. Clin. Cancer Res. 2013, 19, 6329–6330. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A. Patient survival in uveal melanoma is not affected by oncogenic mutations in GNAQ and GNA11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Staby, K.M.; Gravdal, K.; Mork, S.J.; Heegaard, S.; Vintermyr, O.K.; Krohn, J. Prognostic impact of chromosomal aberrations and GNAQ, GNA11 and BAP1 mutations in uveal melanoma. Acta Ophthalmol. 2018, 96, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Onken, M.D.; Worley, L.A.; Long, M.D.; Duan, S.; Council, M.L.; Bowcock, A.M.; Harbour, J.W. Oncogenic mutations in GNAQ occur early in uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2008, 49, 5230–5234. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Kilic, E.; Vaarwater, J.; Bastian, B.C.; Garbe, C.; de Klein, A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br. J. Cancer 2009, 101, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- De Lange, M.J.; Nell, R.J.; Lalai, R.N.; Versluis, M.; Jordanova, E.S.; Luyten, G.P.M.; Jager, M.J.; van der Burg, S.H.; Zoutman, W.H.; van Hall, T.; et al. Digital PCR-based T-cell quantification-assisted deconvolution of the microenvironment reveals that activated macrophages drive tumor inflammation in uveal melanoma. Mol. Cancer Res. 2018, 16, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- De Lange, M.J.; van Pelt, S.I.; Versluis, M.; Jordanova, E.S.; Kroes, W.G.; Ruivenkamp, C.; van der Burg, S.H.; Luyten, G.P.; van Hall, T.; Jager, M.J.; et al. Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget 2015, 6, 37824–37835. [Google Scholar] [CrossRef]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Kapiteijn, E.; Larkin, J.M.G.; Carvajal, R.D.; Luke, J.J.; Seifert, H.; Roozen, I.; Zoubir, M.; Yang, L.; Choudhury, S.; et al. Phase I dose-escalation study of the protein kinase C (PKC) inhibitor AEB071 in patients with metastatic uveal melanoma. J. Clin. Oncol. 2014, 32, 9030. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Landreville, S.; Agapova, O.A.; Matatall, K.A.; Kneass, Z.T.; Onken, M.D.; Lee, R.S.; Bowcock, A.M.; Harbour, J.W. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin. Cancer Res. 2012, 18, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhou, J.; Jin, B.; Pan, J. Class III-specific HDAC inhibitor Tenovin-6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci. Rep. 2016, 6, 22622. [Google Scholar] [CrossRef]
- Hornig, E.; Heppt, M.V.; Graf, S.A.; Ruzicka, T.; Berking, C. Inhibition of histone deacetylases in melanoma-a perspective from bench to bedside. Exp. Dermatol. 2016, 25, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.; Jiang, G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell Mol. Immunol. 2006, 3, 285–290. [Google Scholar] [PubMed]
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The role of histone deacetylase inhibitors in uveal melanoma: Current evidence. Anticancer Res. 2018, 38, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Jin, L.; Gallagher, S.; Mijatov, B.; Zhang, X.D.; Hersey, P. Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv. Pharmacol. 2012, 65, 27–43. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodre, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef]
- Heijkants, R.; Willekens, K.; Schoonderwoerd, M.; Teunisse, A.; Nieveen, M.; Radaelli, E.; Hawinkels, L.; Marine, J.-C.; Jochemsen, A. Combined inhibition of CDK and HDAC as a promising therapeutic strategy for both cutaneous and uveal metastatic melanoma. Oncotarget 2018, 9, 6174–6187. [Google Scholar] [CrossRef]
- Leiter, U.; Meier, F.; Schittek, B.; Garbe, C. The natural course of cutaneous melanoma. J. Surg. Oncol. 2004, 86, 172–178. [Google Scholar] [CrossRef]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative ocular melanoma study group report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A.D.M.H. Paget’s “Seed and Soil” Theory of cancer metastasis: An idea whose time has come. Adv. Anat. Pathol. 2019, 26, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329. Available online: https://www.nature.com/articles/nature15756#supplementary-information (accessed on 24 May 2019). [CrossRef] [PubMed]
- Klein, A.; Sagi-Assif, O.; Meshel, T.; Telerman, A.; Izraely, S.; Ben-Menachem, S.; Bayry, J.; Marzese, D.M.; Ohe, S.; Hoon, D.S.B.; et al. CCR4 is a determinant of melanoma brain metastasis. Oncotarget 2017, 8, 31079–31091. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Wiley, H.E.; Gonzalez, E.B.; Maki, W.; Wu, M.T.; Hwang, S.T. Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J. Natl. Cancer Inst. 2001, 93, 1638–1643. [Google Scholar] [CrossRef] [PubMed]
- Kuhnelt-Leddihn, L.; Muller, H.; Eisendle, K.; Zelger, B.; Weinlich, G. Overexpression of the chemokine receptors CXCR4, CCR7, CCR9, and CCR10 in human primary cutaneous melanoma: A potential prognostic value for CCR7 and CCR10? Arch. Dermatol. Res. 2012, 304, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, T.; Koopmans, A.E.; Vaarwater, J.; van den Berg, M.; de Klein, A.; Verdijk, R.M. Chemokine receptor CCR7 expression predicts poor outcome in uveal melanoma and relates to liver metastasis whereas expression of CXCR4 is not of clinical relevance. Invest. Ophthalmol. Vis. Sci. 2013, 54, 7354–7361. [Google Scholar] [CrossRef]
- Li, H.; Alizadeh, H.; Niederkorn, J.Y. Differential expression of chemokine receptors on uveal melanoma cells and their metastases. Invest. Ophthalmol. Vis. Sci. 2008, 49, 636–643. [Google Scholar] [CrossRef]
- Murakami, T.; Cardones, A.R.; Finkelstein, S.E.; Restifo, N.P.; Klaunberg, B.A.; Nestle, F.O.; Castillo, S.S.; Dennis, P.A.; Hwang, S.T. Immune evasion by murine melanoma mediated through CC chemokine receptor-10. J. Exp. Med. 2003, 198, 1337–1347. [Google Scholar] [CrossRef]
- Simonetti, O.; Goteri, G.; Lucarini, G.; Filosa, A.; Pieramici, T.; Rubini, C.; Biagini, G.; Offidani, A. Potential role of CCL27 and CCR10 expression in melanoma progression and immune escape. Eur. J. Cancer 2006, 42, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Dobner, B.C.; Riechardt, A.I.; Joussen, A.M.; Englert, S.; Bechrakis, N.E. Expression of haematogenous and lymphogenous chemokine receptors and their ligands on uveal melanoma in association with liver metastasis. Acta Ophth. 2012, 90, e638–e644. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Hutson, T.E.; Molto, L.; Richmond, A.; Nemec, C.; Mekhail, T.; Elson, P.; Tannenbaum, C.; Olencki, T.; Finke, J.; et al. Clinical and immunologic effects of subcutaneously administered interleukin-12 and interferon alfa-2b: Phase I trial of patients with metastatic renal cell carcinoma or malignant melanoma. J. Clin. Oncol. 2004, 22, 2891–2900. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- O’Boyle, G.; Swidenbank, I.; Marshall, H.; Barker, C.E.; Armstrong, J.; White, S.A.; Fricker, S.P.; Plummer, R.; Wright, M.; Lovat, P.E. Inhibition of CXCR4-CXCL12 chemotaxis in melanoma by AMD11070. Br. J. Cancer 2013, 108, 1634–1640. [Google Scholar] [CrossRef]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Scotton, C.J.; Wilson, J.L.; Scott, K.; Stamp, G.; Wilbanks, G.D.; Fricker, S.; Bridger, G.; Balkwill, F.R. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002, 62, 5930–5938. [Google Scholar]
- Sun, Y.X.; Wang, J.; Shelburne, C.E.; Lopatin, D.E.; Chinnaiyan, A.M.; Rubin, M.A.; Pienta, K.J.; Taichman, R.S. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell Biochem. 2003, 89, 462–473. [Google Scholar] [CrossRef]
- Zhou, Y.; Larsen, P.H.; Hao, C.H.; Yong, V.W. CXCRA is a major chemokine receptor on glioma cells and mediates their survival. J. Biol. Chem. 2002, 277, 49481–49487. [Google Scholar] [CrossRef]
- Cao, H.H.; Cheng, C.Y.; Su, T.; Fu, X.Q.; Guo, H.; Li, T.; Tse, A.K.W.; Kwan, H.Y.; Yu, H.; Yu, Z.L. Quercetin inhibits HGF/c-Met signaling and HGF-stimulated melanoma cell migration and invasion. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Van der Ent, W.; Burrello, C.; de Lange, M.J.; van der Velden, P.A.; Jochemsen, A.G.; Jager, M.J.; Snaar-Jagalska, B.E. Embryonic zebrafish: Different phenotypes after injection of human uveal melanoma cells. Ocul. Oncol. Pathol. 2015, 1, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Bakalian, S.; Marshall, J.C.; Logan, P.; Faingold, D.; Maloney, S.; Di Cesare, S.; Martins, C.; Fernandes, B.F.; Burnier, M.N., Jr. Molecular pathways mediating liver metastasis in patients with uveal melanoma. Clin. Cancer Res. 2008, 14, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.C.; Yu, P.W.; Qian, F.; Chu, F.L.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Griewank, K.; Ding, V.W.; Bastian, B.C. XL184: C-Met inhibition is effective in a mouse xenograft model of metastatic uveal melanoma. Cancer Res. 2011, 71, 3587. [Google Scholar] [CrossRef]
- Daud, A.; Kluger, H.M.; Kurzrock, R.; Schimmoller, F.; Weitzman, A.L.; Samuel, T.A.; Moussa, A.H.; Gordon, M.S.; Shapiro, G.I. Phase II randomised discontinuation trial of the MET/VEGF receptor inhibitor cabozantinib in metastatic melanoma. Br. J. Cancer 2017, 116, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Weroha, S.J.; Haluska, P. The insulin-like growth factor system in cancer. Endocrinol. Metab. Clin. N. Am. 2012, 41, 335. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, S.; Zloto, O.; Pe’er, J.; Barak, V. Insulin-like growth factor-1 as a predictive biomarker for metastatic uveal melanoma in humans. Invest. Ophth. Vis. Sci. 2013, 54, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Kucera, R.; Treskova, I.; Vrzalova, J.; Svobodova, S.; Topolcan, O.; Fuchsova, R.; Rousarova, M.; Treska, V.; Kydlicek, T. Evaluation of IGF1 serum levels in malignant melanoma and healthy subjects. Anticancer Res. 2014, 34, 5217–5220. [Google Scholar]
- Yoshida, M.; Selvan, S.; McCue, P.A.; DeAngelis, T.; Baserga, R.; Fujii, A.; Rui, H.; Mastrangelo, M.J.; Sato, T. Expression of insulin-like growth factor-1 receptor in metastatic uveal melanoma and implications for potential autocrine and paracrine tumor cell growth. Pigment. Cell Melanoma Res. 2014, 27, 297–308. [Google Scholar] [CrossRef]
- All-Ericsson, C.; Girnita, L.; Seregard, S.; Bartolazzi, A.; Jager, M.J.; Larsson, O. Insulin-like growth factor-1 receptor in uveal melanoma: A predictor for metastatic disease and a potential therapeutic target. Invest. Ophth. Vis. Sci. 2002, 43, 1–8. [Google Scholar]
- Economou, M.A.; All-Ericsson, C.; Bykov, V.; Girnita, L.; Bartolazzi, A.; Larsson, O.; Seregard, S. Receptors for the liver synthesized growth factors IGF-1 and HGF/SF in uveal melanoma: Intercorrelation and prognostic implications. Invest. Ophth. Vis. Sci. 2005, 46, 4372–4375. [Google Scholar] [CrossRef] [PubMed]
- Von Manstein, V.; Yang, C.M.; Richter, D.; Delis, N.; Vafaizadeh, V.; Groner, B. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr. Signal Transd. Ther. 2013, 8, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Ramcharan, R.; Aleksic, T.; Kamdoum, W.P.; Gao, S.; Pfister, S.X.; Tanner, J.; Bridges, E.; Asher, R.; Watson, A.J.; Margison, G.P.; et al. IGF-1R inhibition induces schedule-dependent sensitization of human melanoma to temozolomide. Oncotarget 2015, 6, 39877–39890. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Kauffmann, A.; Molle, S.; Schnell, C.; Ferrat, T.; Voshol, H.; Juengert, J.; Erasimus, H.; Marszalek, G.; Kazic-Legueux, M.; et al. Maximizing the efficacy of MAPK-targeted treatment in PTENLOF/BRAF(MUT) melanoma through PI3K and IGF1R inhibition. Cancer Res. 2016, 76, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Flashner-Abramson, E.; Klein, S.; Mullin, G.; Shoshan, E.; Song, R.; Shir, A.; Langut, Y.; Bar-Eli, M.; Reuveni, H.; Levitzki, A. Targeting melanoma with NT157 by blocking Stat3 and IGF1R signaling. Oncogene 2016, 35, 2675–2680. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.; Siim, B.G.; Johnson, R.S. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, H.; Zhao, Y.; Zhang, X.; Liu, J.; Pan, Y.; Bai, J.; Zhang, H. Knockdown of FBXO22 inhibits melanoma cell migration, invasion and angiogenesis via the HIF-1alpha/VEGF pathway. Invest. New Drugs 2019, 1–9. [Google Scholar] [CrossRef]
- Martinez-Garcia, M.A.; Riveiro-Falkenbach, E.; Rodriguez-Peralto, J.L.; Nagore, E.; Martorell-Calatayud, A.; Campos-Rodriguez, F.; Farre, R.; Hernandez Blasco, L.; Banuls Roca, J.; Chiner Vives, E.; et al. A prospective multicenter cohort study of cutaneous melanoma: Clinical staging and potential associations with HIF-1alpha and VEGF expressions. Melanoma Res. 2017, 27, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; You, S.; Zhang, Q.; Osuka, S.; Devi, N.S.; Kaluz, S.; Ferguson, J.H.; Yang, H.; Chen, G.; Wang, B.; et al. Arylsulfonamide 64B inhibits hypoxia/HIF-induced expression of c-met and CXCR4 and reduces primary tumor growth and metastasis of uveal melanoma. Clin. Cancer Res. 2019, 25, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Gartrell, R.D.; Marks, D.K.; Hart, T.D.; Li, G.; Davari, D.R.; Wu, A.; Blake, Z.; Lu, Y.; Askin, K.N.; Monod, A.; et al. Quantitative analysis of immune infiltrates in primary melanoma. Cancer Immunol. Res. 2018, 6, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, A.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejos, Z.; Gandi, I.; Timar, J. FOXP3(+) Cell density in primary tumor has no prognostic impact in patients with cutaneous malignant melanoma. Pathol. Oncol. Res. 2010, 16, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hillen, F.; Baeten, C.I.M.; van de Winkel, A.; Creytens, D.; van der Schaft, D.W.J.; Winnepenninckx, V.; Griffioen, A.W. Leukocyte infiltration and tumor cell plasticity are parameters of aggressiveness in primary cutaneous melanoma. Cancer Immunol. Immun. 2008, 57, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Miracco, C.; Mourmouras, V.; Biagioli, M.; Rubegni, P.; Mannucci, S.; Monciatti, I.; Cosci, E.; Tosi, P.; Luzi, P. Utility of tumour-infiltrating CD25(+)FOXP3(+) regulatory T cell evaluation in predicting local recurrence in vertical growth phase cutaneous melanoma. Oncol. Rep. 2007, 18, 1115–1122. [Google Scholar] [PubMed]
- Santegoets, S.J.A.M.; Dijkgraaf, E.M.; Battaglia, A.; Beckhove, P.; Britten, C.M.; Gallimore, A.; Godkin, A.; Gouttefangeas, C.; de Gruijl, T.D.; Koenen, H.J.P.M.; et al. Monitoring regulatory T cells in clinical samples: Consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol. Immun. 2015, 64, 1271–1286. [Google Scholar] [CrossRef] [PubMed]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Whelchel, J.C.; Farah, S.E.; Mclean, I.W.; Burnier, M.N. Immunohistochemistry of infiltrating lymphocytes in uveal malignant-melanoma. Invest. Ophth. Vis. Sci. 1993, 34, 2603–2606. [Google Scholar]
- Gezgin, G.; Dogrusoz, M.; van Essen, T.H.; Kroes, W.G.M.; Luyten, G.P.M.; van der Velden, P.A.; Walter, V.; Verdijk, R.M.; van Hall, T.; van der Burg, S.H.; et al. Genetic evolution of uveal melanoma guides the development of an inflammatory microenvironment. Cancer Immunol. Immunother. 2017, 66, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, T.H.; van Pelt, S.I.; Bronkhorst, I.H.G.; Versluis, M.; Nemati, F.; Laurent, C.; Luyten, G.P.M.; van Hall, T.; van den Elsen, P.J.; Decaudin, D.; et al. Upregulation of HLA expression in primary uveal melanoma by infiltrating leukocytes. PLoS ONE 2016, 11, e0164292. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, I.H.G.; Ly, L.V.; Jordanova, E.S.; Vrolijk, J.; Versluis, M.; Luyten, G.P.M.; Jager, M.J. Detection of M2-macrophages in uveal melanoma and relation with survival. Invest. Ophth. Vis. Sci. 2011, 52, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, I.H.G.; Vu, T.H.K.; Jordanova, E.S.; Luyten, G.P.M.; van der Burg, S.H.; Jager, M.J. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Invest. Ophth. Vis. Sci. 2012, 53, 5370–5378. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Johansson, C.C.; Trocme, E.; All-Ericsson, C.; Economou, M.A.; Larsson, O.; Seregard, S.; Kiessling, R. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2 positive uveal melanoma. Cancer 2010, 116, 2224–2233. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Fu, Y.X. Tumor-infiltrating T lymphocytes: Friends or foes? Lab. Invest. 2006, 86, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Melief, S.M.; Visconti, V.V.; Visser, M.; van Diepen, M.; Kapiteijn, E.H.W.; van den Berg, J.H.; Haanen, J.B.A.G.; Smit, V.T.H.B.M.; Oosting, J.; van der Burg, S.H.; et al. Long-term survival and clinical benefit from adoptive T-cell transfer in stage IV melanoma patients is determined by a four-parameter tumor immune signature. Cancer Immunol. Res. 2017, 5, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.P.; Salama, A.K. Updates in therapy for advanced melanoma. Cancers Basel 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Eng. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef]
- Luke, J.J.; Callahan, M.K.; Postow, M.A.; Romano, E.; Ramaiya, N.; Bluth, M.; Giobbie-Hurder, A.; Lawrence, D.P.; Ibrahim, N.; Ott, P.A.; et al. Clinical activity of ipilimumab for metastatic uveal melanoma: A retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial Sloan-Kettering Cancer Center, and University Hospital of Lausanne experience. Cancer 2013, 119, 3687–3695. [Google Scholar] [CrossRef] [PubMed]
- Kelderman, S.; van der Kooij, M.K.; van den Eertwegh, A.J.; Soetekouw, P.M.; Jansen, R.L.; van den Brom, R.R.; Hospers, G.A.; Haanen, J.B.; Kapiteijn, E.; Blank, C.U. Ipilimumab in pretreated metastastic uveal melanoma patients. Results of the Dutch working group on immunotherapy of oncology (WIN-O). Acta Oncol. 2013, 52, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Vaubel, J.; Mohr, P.; Hauschild, A.; Utikal, J.; Simon, J.; Garbe, C.; Herbst, R.; Enk, A.; Kampgen, E.; et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS ONE 2015, 10, e0118564. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Danielli, R.; Chiarion-Sileni, V.; Pigozzo, J.; Parmiani, G.; Ridolfi, R.; De, R.F.; Del, V.M.; Di, G.L.; Queirolo, P.; et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann. Oncol. 2013, 24, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Kottschade, L.A.; McWilliams, R.R.; Markovic, S.N.; Block, M.S.; Villasboas Bisneto, J.; Pham, A.Q.; Esplin, B.L.; Dronca, R.S. The use of pembrolizumab for the treatment of metastatic uveal melanoma. Melanoma Res. 2016, 26, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Bender, C.; Enk, A.; Gutzmer, R.; Hassel, J.C. Anti-PD-1 antibodies in metastatic uveal melanoma: A treatment option? Cancer Med. 2017, 6, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Heinzerling, L.; Kahler, K.C.; Forschner, A.; Kirchberger, M.C.; Loquai, C.; Meissner, M.; Meier, F.; Terheyden, P.; Schell, B.; et al. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur. J. Cancer 2017, 82, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Van der Kooij, M.K.; Joosse, A.; Speetjens, F.M.; Hospers, G.A.; Bisschop, C.; de Groot, J.W.; Koornstra, R.; Blank, C.U.; Kapiteijn, E. Anti-PD1 treatment in metastatic uveal melanoma in the Netherlands. Acta Oncol. 2017, 56, 101–103. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.P.; Kim, I.K.; Esmaeli, B.; Amin-Mansour, A.; Treacy, D.J.; Carter, S.L.; Hodis, E.; Wagle, N.; Seepo, S.; Yu, X.; et al. Systematic genomic and translational efficiency studies of uveal melanoma. PLoS ONE 2017, 12, e0178189. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Wolf, Y.; Feferman, T.; Barnea, E.; Greenstein, E.; Reshef, D.; Tirosh, I.; Reuben, A.; Patkar, S.; Levy, R.; et al. Combined analysis of antigen presentation and t-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 2018, 8, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Petaccia de Macedo, M.; Reuben, A.; Forget, M.A.; Haymaker, C.; Bernatchez, C.; Spencer, C.N.; Gopalakrishnan, V.; Reddy, S.; Cooper, Z.A.; et al. Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: A pilot study. Oncoimmunology 2017, 6, e1321187. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T-regs in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Komatsubara, K.M.; Gartrell, R.D.; Bayan, C.-A.; Pradhan, J.S.; Hasan, S.S.; Hart, T.D.; Borgardus, M.; Lu, Y.; Marks, D.K.; Yang, J.; et al. Characterization and spatial localization of the tumor immune microenvironment in metastatic uveal melanoma. J. Clin. Oncol. 2018, 36, 9570. [Google Scholar] [CrossRef]
- Chandran, S.S.; Somerville, R.P.T.; Yang, J.C.; Sherry, R.M.; Klebanoff, C.A.; Goff, S.L.; Wunderlich, J.R.; Danforth, D.N.; Zlott, D.; Paria, B.C.; et al. Treatment of metastatic uveal melanoma with adoptive transfer of tumour-infiltrating lymphocytes: A single-centre, two-stage, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 792–802. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Kooij, M.K.; Speetjens, F.M.; van der Burg, S.H.; Kapiteijn, E. Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types. Cancers 2019, 11, 845. https://doi.org/10.3390/cancers11060845
van der Kooij MK, Speetjens FM, van der Burg SH, Kapiteijn E. Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types. Cancers. 2019; 11(6):845. https://doi.org/10.3390/cancers11060845
Chicago/Turabian Stylevan der Kooij, Monique K., Frank M. Speetjens, Sjoerd H. van der Burg, and Ellen Kapiteijn. 2019. "Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types" Cancers 11, no. 6: 845. https://doi.org/10.3390/cancers11060845
APA Stylevan der Kooij, M. K., Speetjens, F. M., van der Burg, S. H., & Kapiteijn, E. (2019). Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types. Cancers, 11(6), 845. https://doi.org/10.3390/cancers11060845