Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy


Abstract
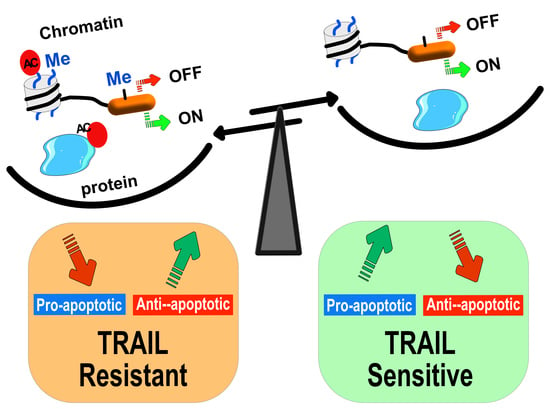
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
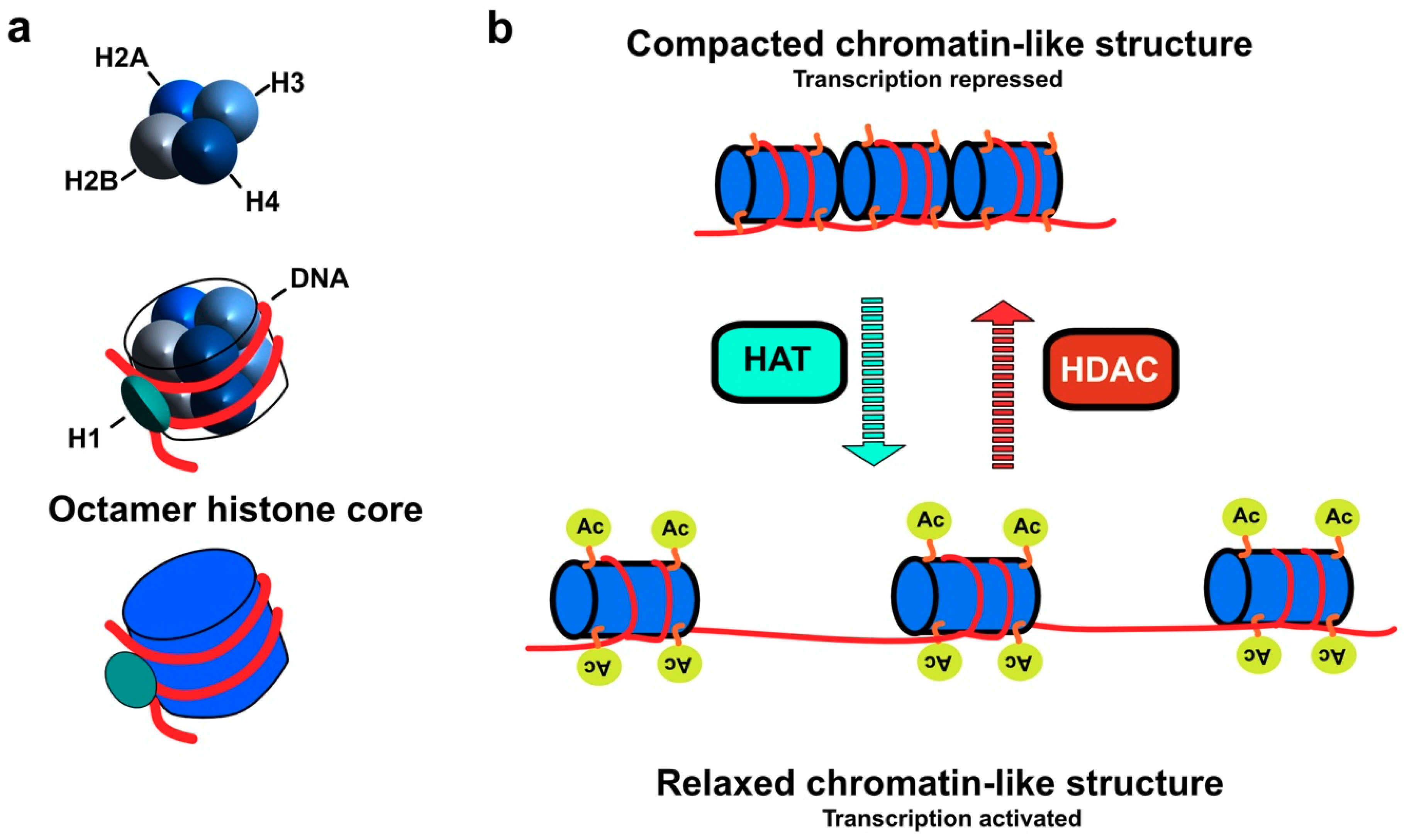
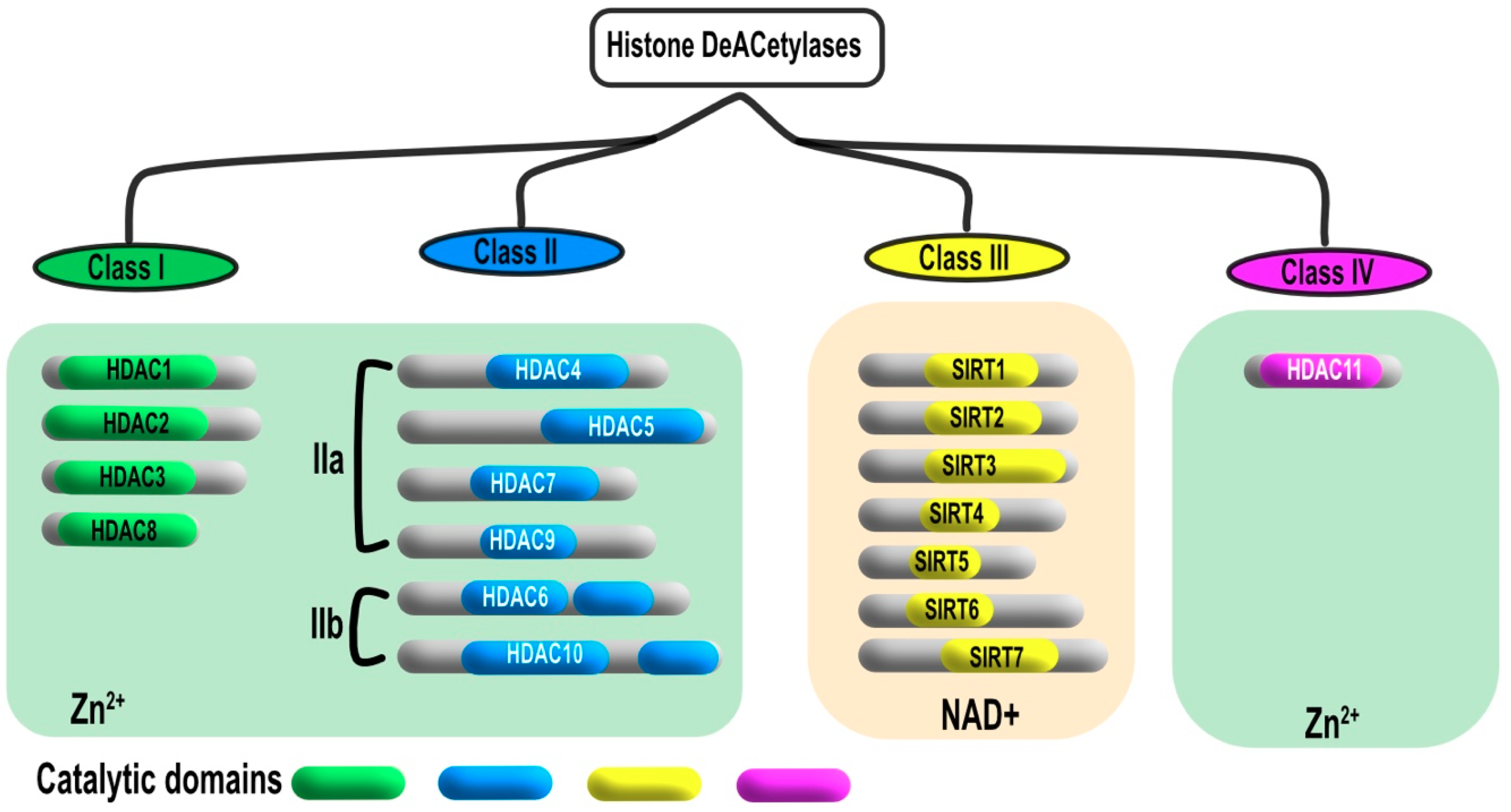
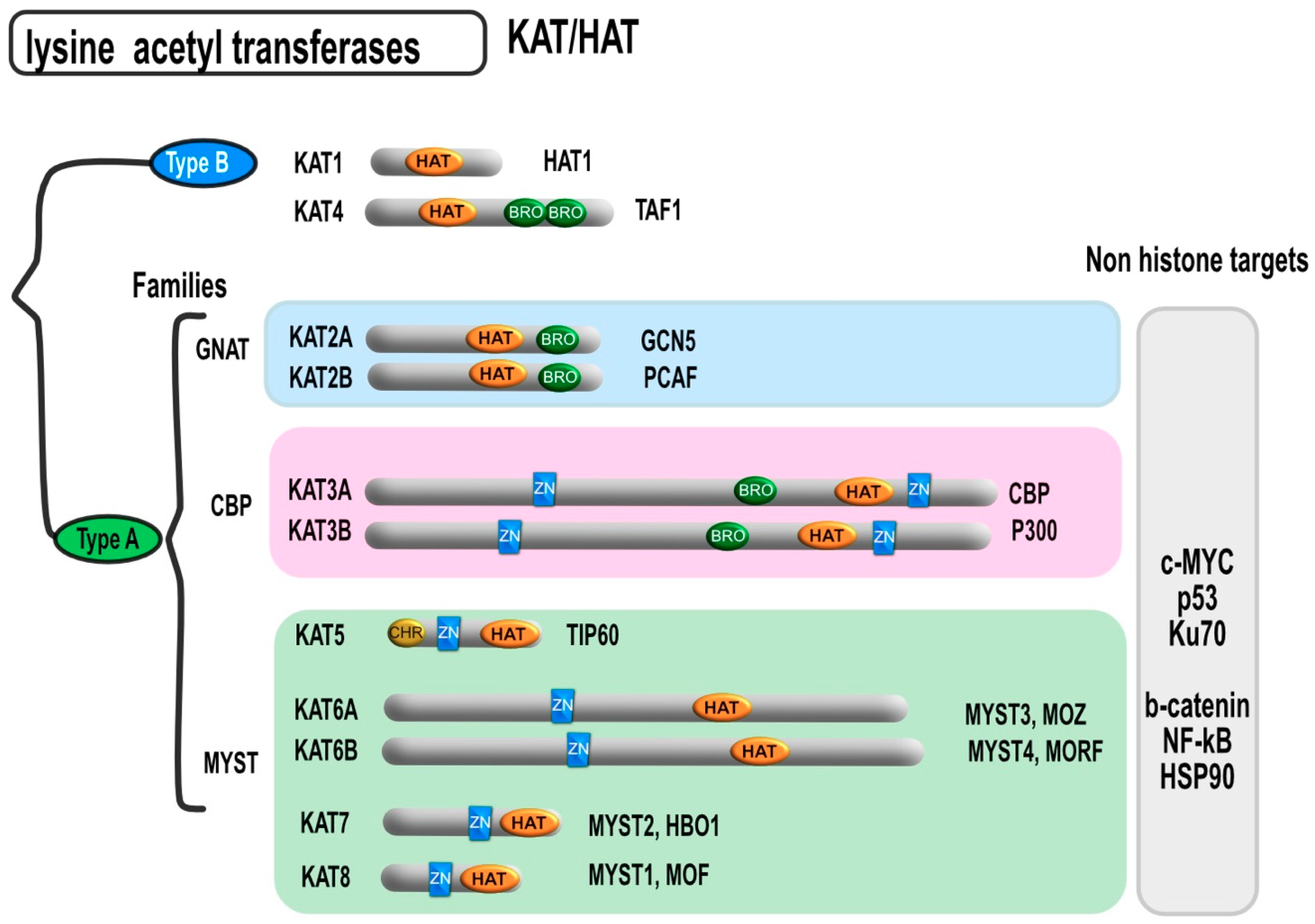
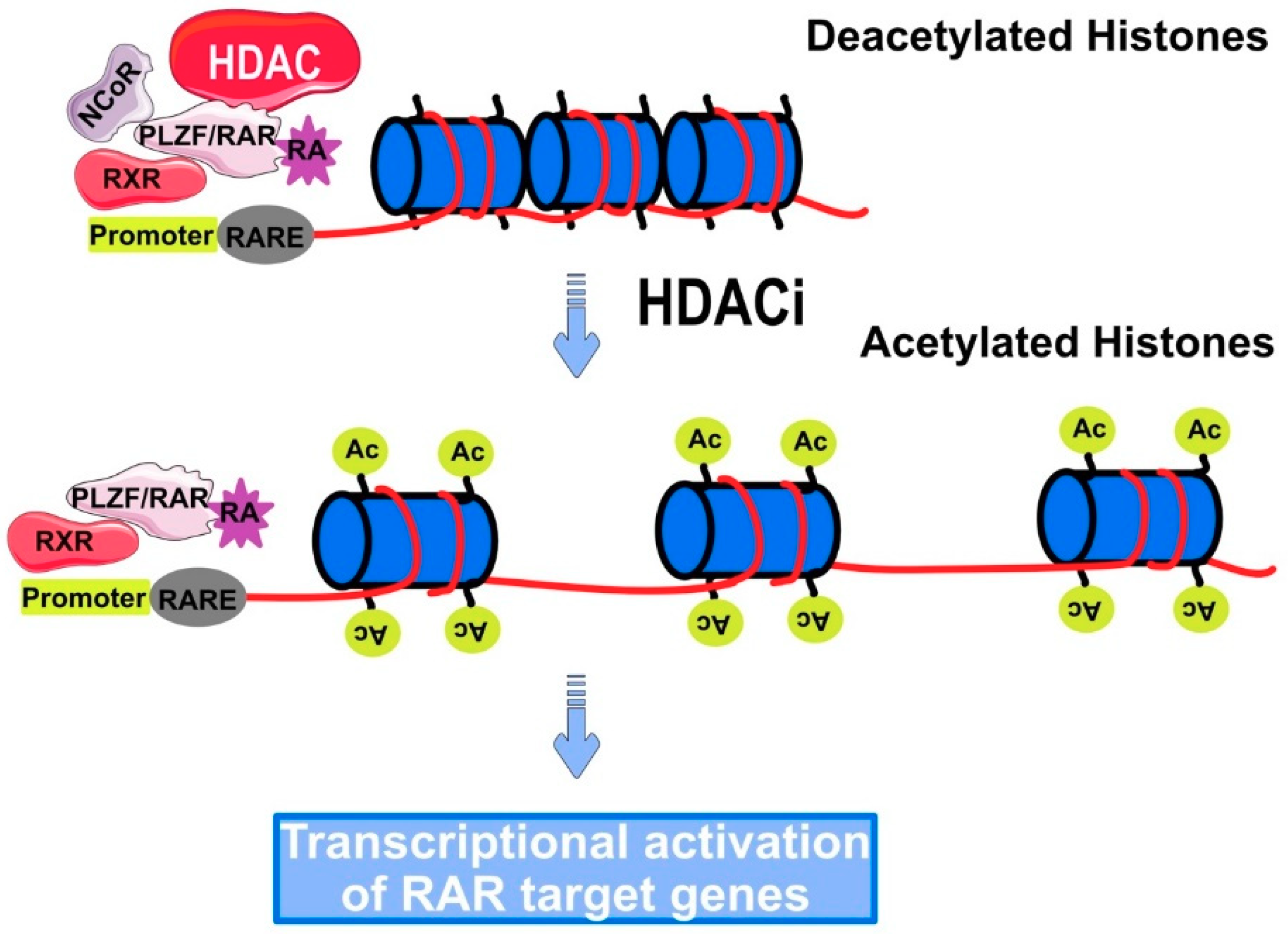
2. Classification and Localization of HDACs
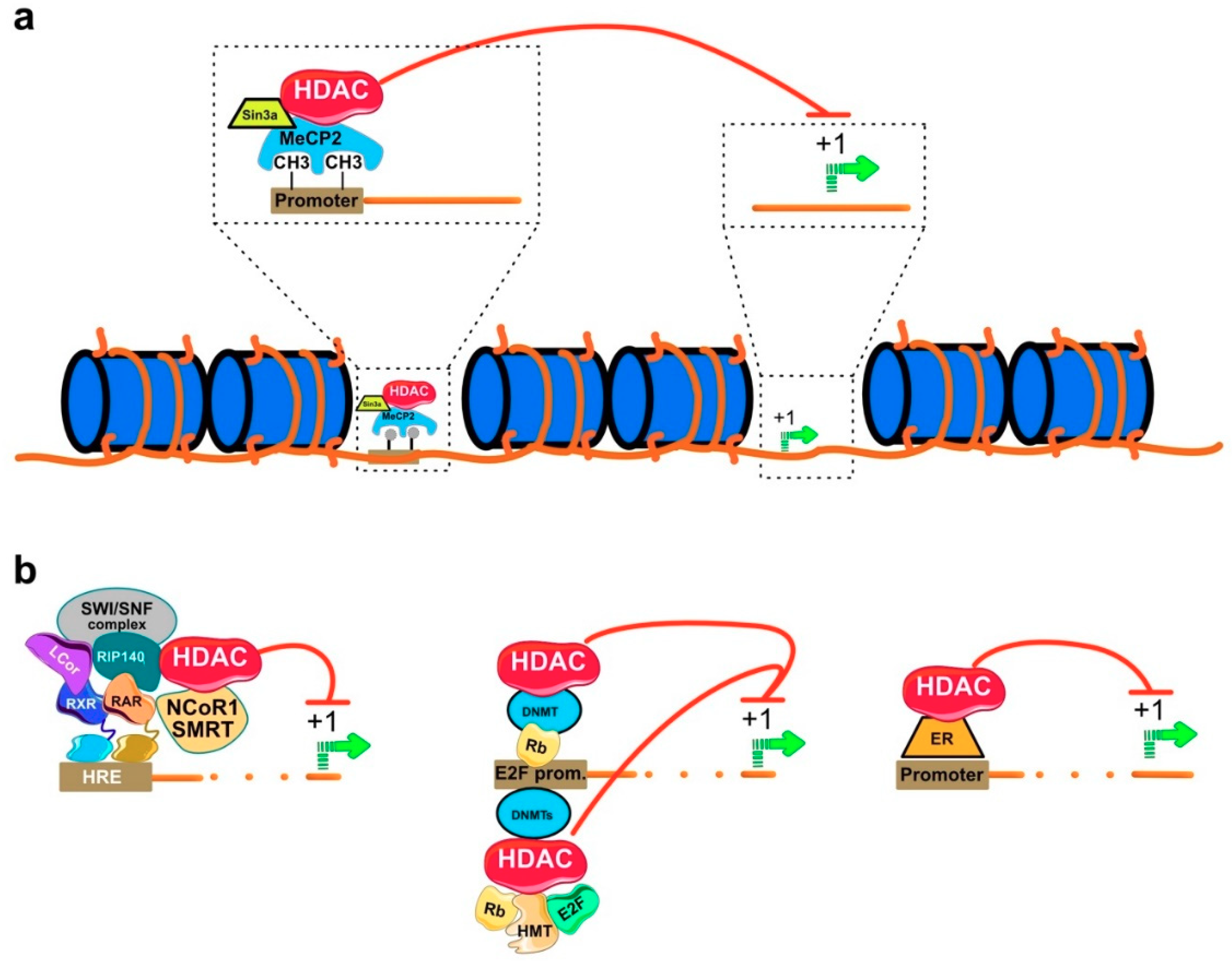
3. Molecular Mechanism of HDAC Action
4. Physiological Function of HDAC
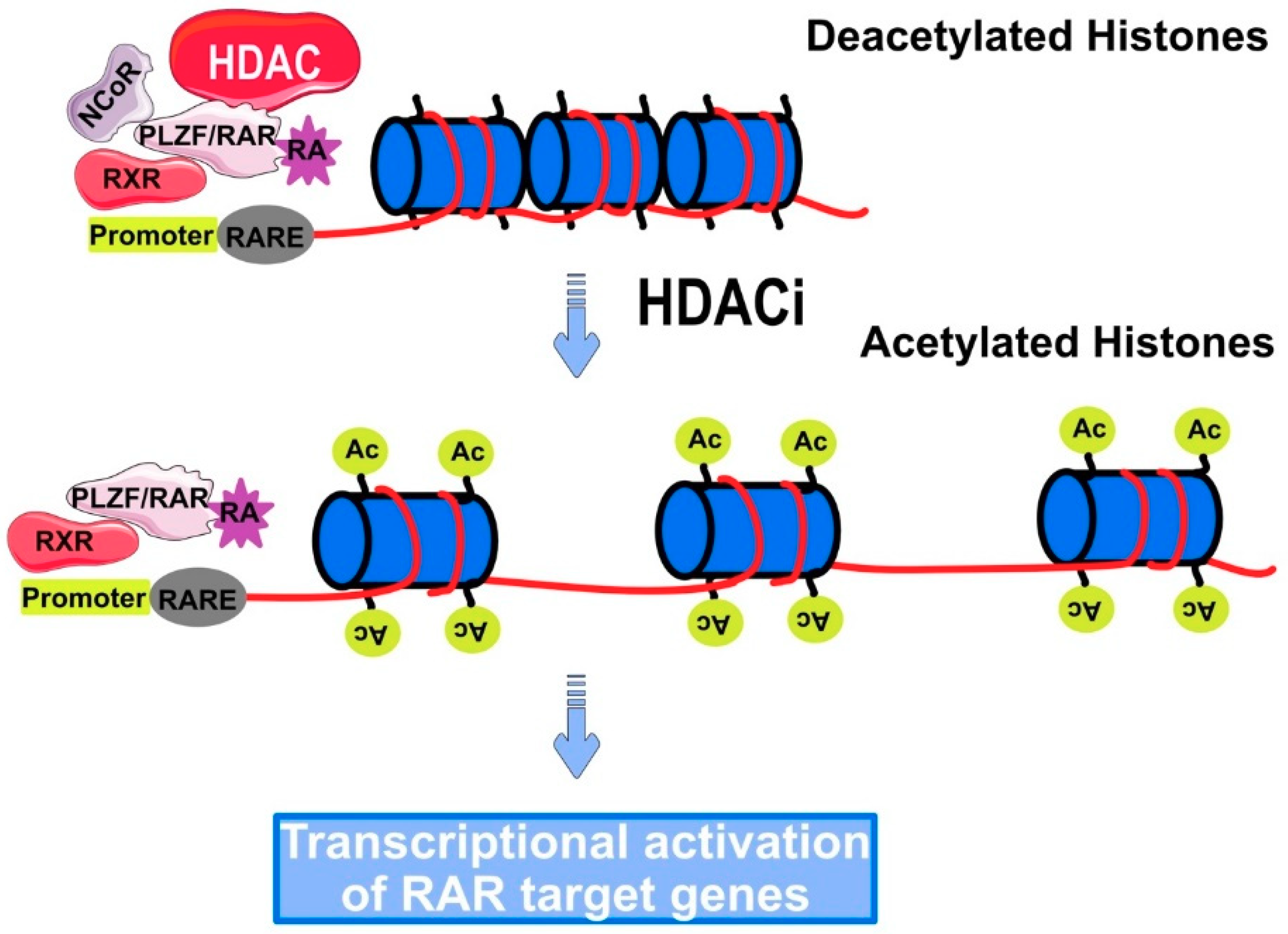
5. Role of HDAC in Cancer
6. DNA Methylation and Cancer
7. Epigenetic Regulation of TRAIL Proapoptotic Signaling
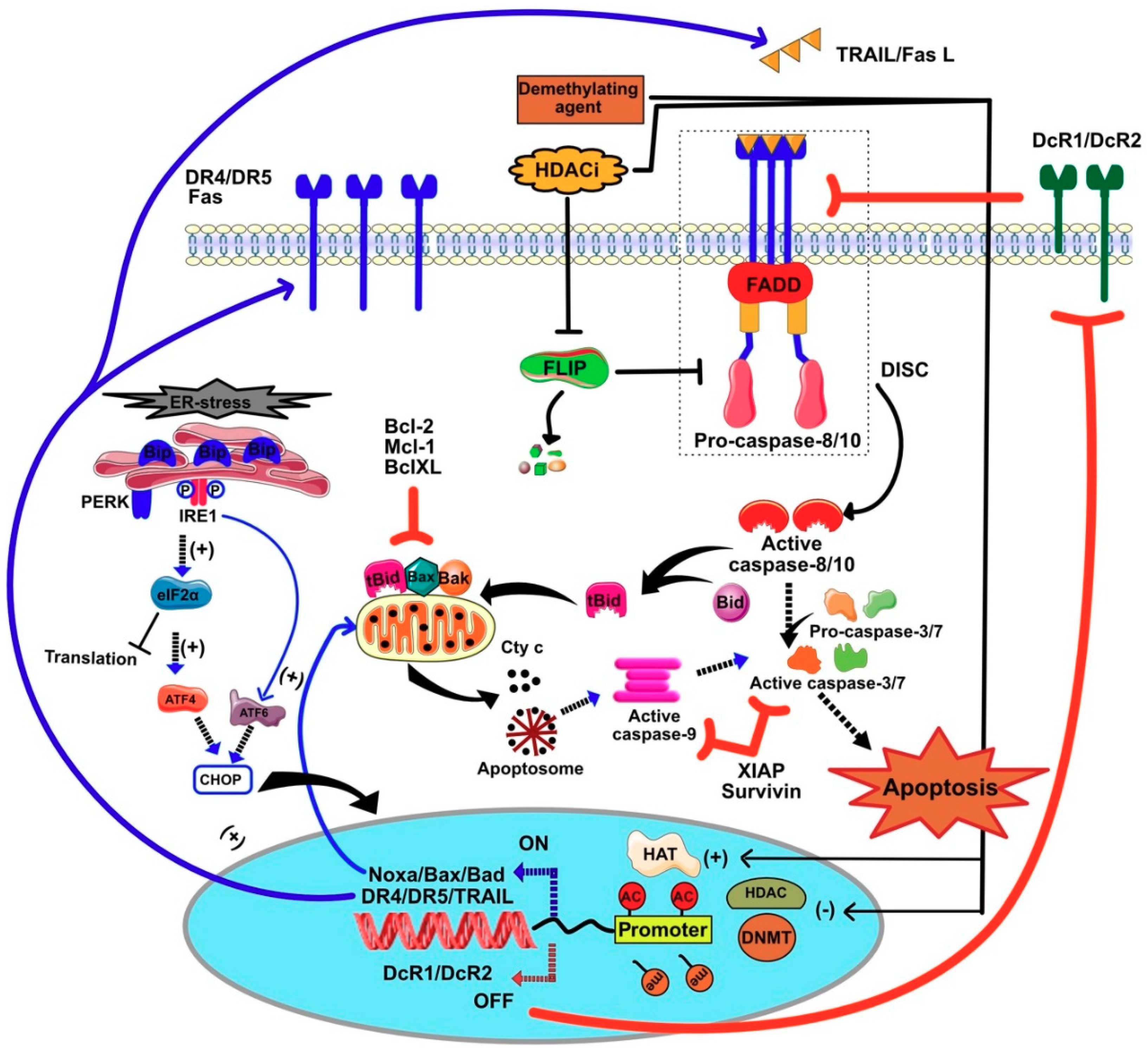
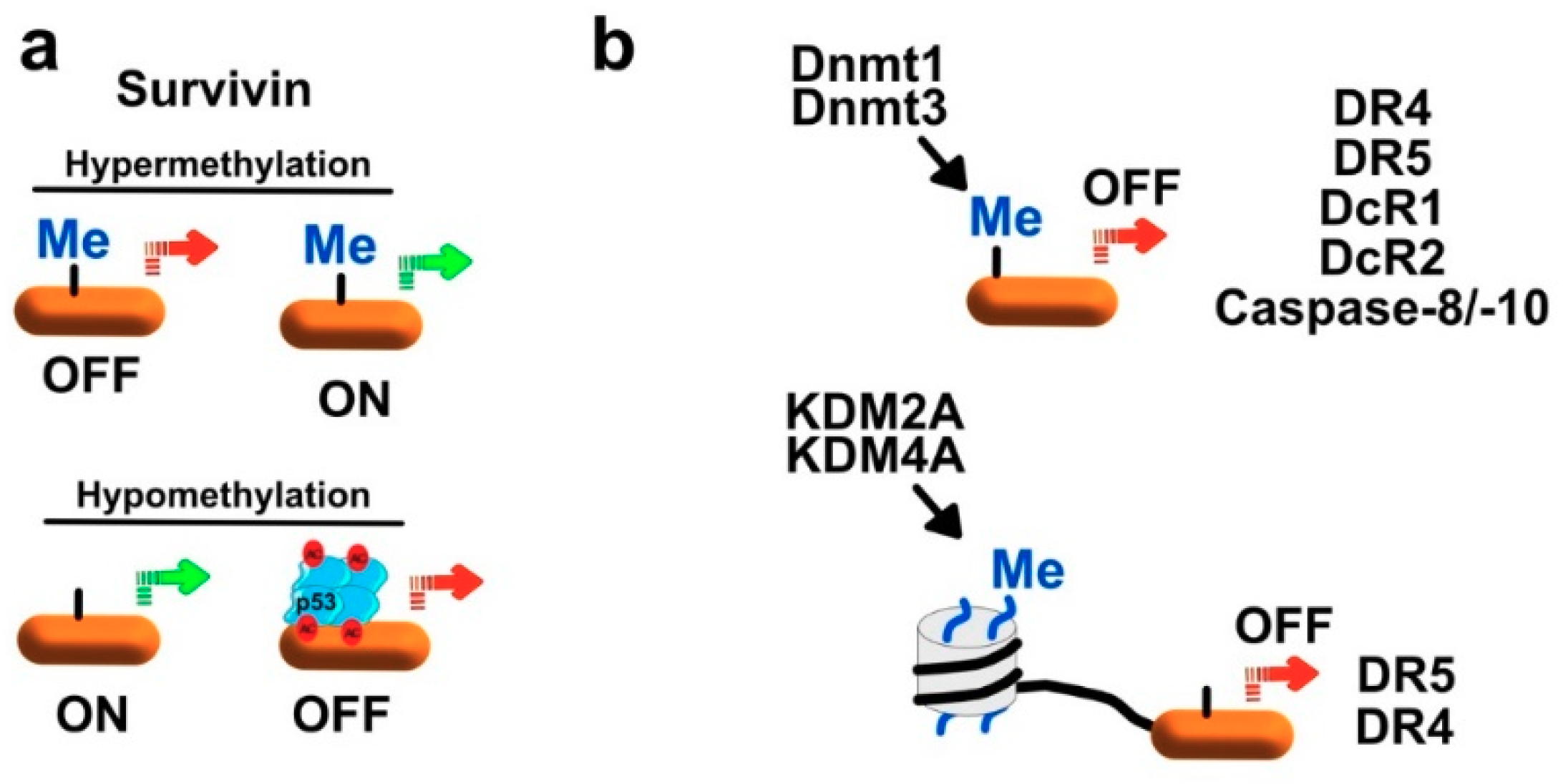
7.1. Restoration of TRAIL-Induced Apoptosis by Demethylating Agents
7.1.1. Regulation of Survivin and XIAP by Methylation
7.1.2. Regulation of Initiator Caspases by Methylation
7.1.3. Regulation of TRAIL Receptors by Methylation
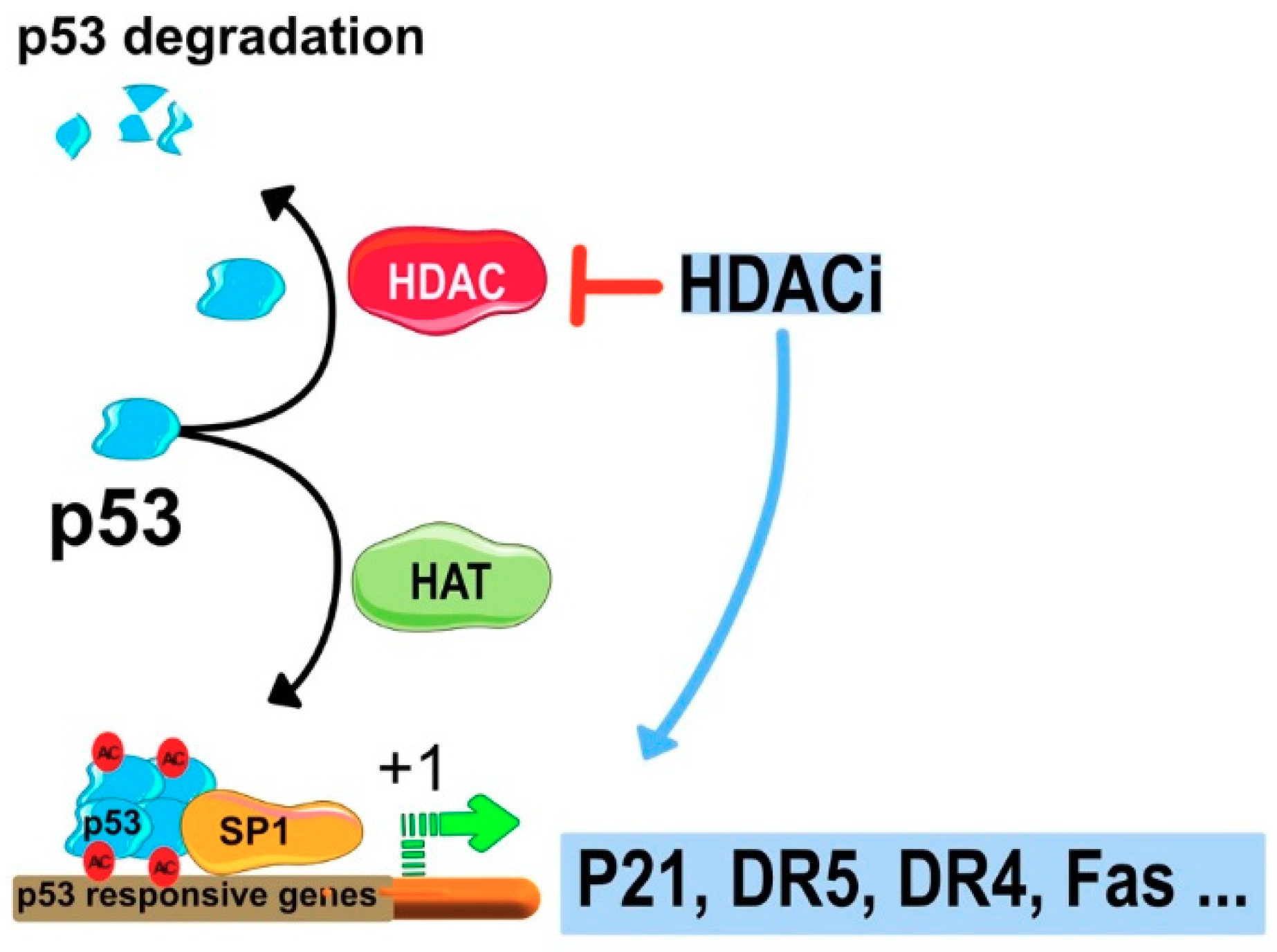
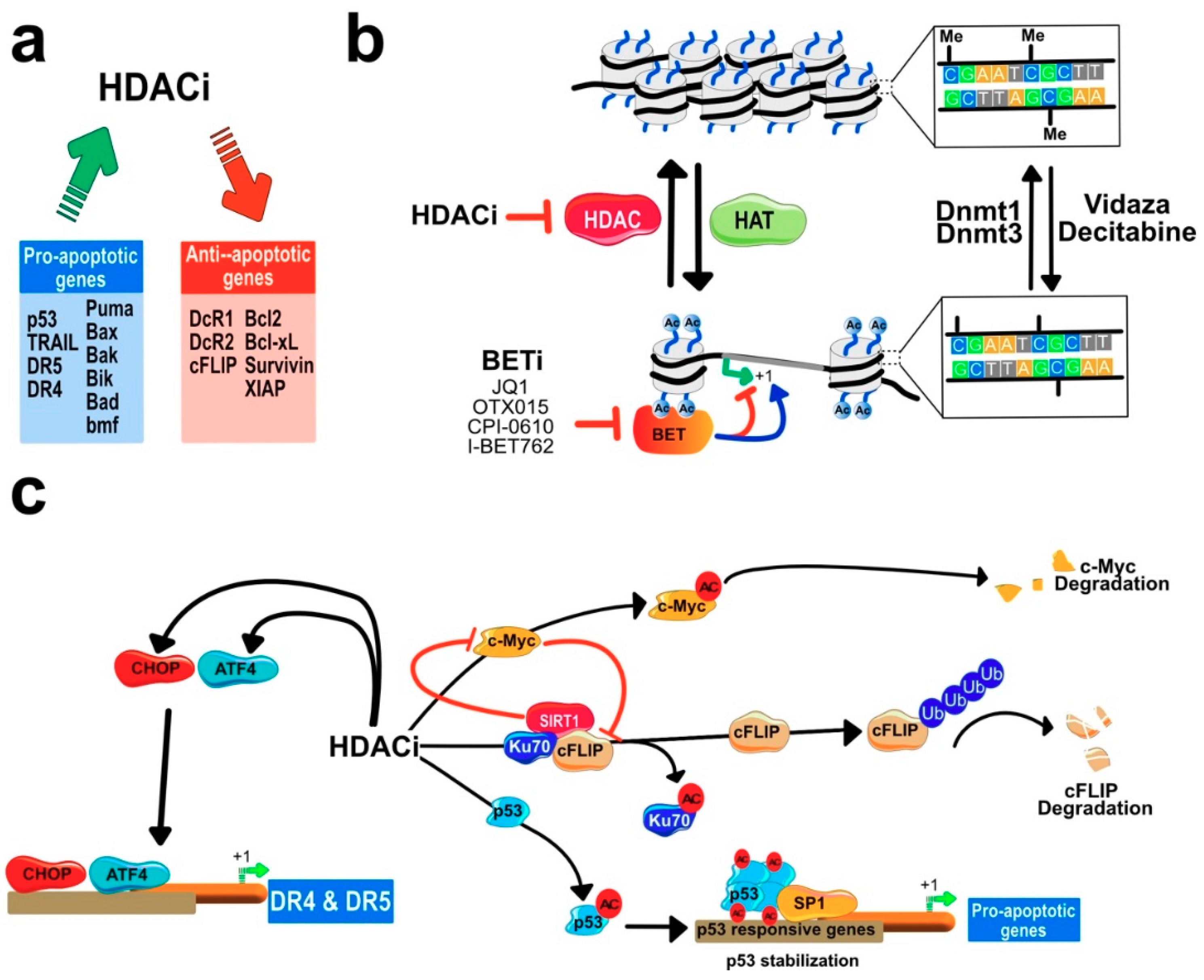
7.2. HDACIs Sensitize Tumor Cells to TRAIL-Mediated Apoptosis
Regulation of Gene Expression by HDACi
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Richmond, T.J.; Davey, C.A. The structure of DNA in the nucleosome core. Nature 2003, 423, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S. The nucleosome: from genomic organization to genomic regulation. Cell 2004, 116, 259–272. [Google Scholar] [CrossRef]
- Brown, R.; Strathdee, G. Epigenomics and epigenetic therapy of cancer. Trends Mol. Med. 2002, 8, S43–S48. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Kramer, O.H.; Gottlicher, M.; Heinzel, T. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab. 2001, 12, 294–300. [Google Scholar] [CrossRef]
- Pandolfi, P.P. Histone deacetylases and transcriptional therapy with their inhibitors. Cancer Chemother. Pharm. 2001, 48 (Suppl. S1), S17–S19. [Google Scholar] [CrossRef]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Kiermer, V.; Dequiedt, F.; Verdin, E. The emerging role of class II histone deacetylases. Biochem. Cell. Biol. 2001, 79, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Brosch, G.; Loidl, P.; Graessle, S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol. Rev. 2008, 32, 409–439. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. The missing linker: an unexpected role for a histone deacetylase. Mol. Cell 2003, 12, 1349–1351. [Google Scholar] [CrossRef]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual regulation of c-Myc by p300 via acetylation-dependent control of Myc protein turnover and coactivation of Myc-induced transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K.; Gabrielli, B.; Johnstone, R.W. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle 2004, 3, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharm. Toxicol 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.R.; Sparreboom, A.; Venitz, J.; Figg, W.D. Rational development of histone deacetylase inhibitors as anticancer agents: a review. Mol. Pharm. 2005, 68, 917–932. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin. Epigenet. 2016, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed]
- Ghizzoni, M.; Haisma, H.J.; Maarsingh, H.; Dekker, F.J. Histone acetyltransferases are crucial regulators in NF-kappaB mediated inflammation. Drug Discov. Today 2011, 16, 504–511. [Google Scholar] [CrossRef]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef]
- Holden, H.M.; Matthews, B.W. The binding of L-valyl-L-tryptophan to crystalline thermolysin illustrates the mode of interaction of a product of peptide hydrolysis. J. Biol. Chem. 1988, 263, 3256–3260. [Google Scholar] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Dowling, D.P.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 2010, 49, 5048–5056. [Google Scholar] [CrossRef]
- Gantt, S.L.; Joseph, C.G.; Fierke, C.A. Activation and inhibition of histone deacetylase 8 by monovalent cations. J. Biol. Chem. 2010, 285, 6036–6043. [Google Scholar] [CrossRef] [PubMed]
- Somoza, J.R.; Skene, R.J.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Vannini, A.; Volpari, C.; Gallinari, P.; Jones, P.; Mattu, M.; Carfi, A.; De Francesco, R.; Steinkuhler, C.; Di Marco, S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007, 8, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Joseph, C.G.; Neidle, S.; Fierke, C.A.; Fuchter, M.J. On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg. Med. Chem. Lett. 2011, 21, 2129–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.N.; Hu, X.; Chandra, D.; Seto, E.; Farooqui, M. Receptor-interacting protein 140 directly recruits histone deacetylases for gene silencing. J. Biol. Chem. 2000, 275, 40782–40787. [Google Scholar] [CrossRef]
- Espada, J.; Ballestar, E.; Fraga, M.F.; Villar-Garea, A.; Juarranz, A.; Stockert, J.C.; Robertson, K.D.; Fuks, F.; Esteller, M. Human DNA methyltransferase 1 is required for maintenance of the histone H3 modification pattern. J. Biol. Chem. 2004, 279, 37175–37184. [Google Scholar] [CrossRef]
- Bai, S.; Ghoshal, K.; Datta, J.; Majumder, S.; Yoon, S.O.; Jacob, S.T. DNA methyltransferase 3b regulates nerve growth factor-induced differentiation of PC12 cells by recruiting histone deacetylase 2. Mol. Cell. Biol. 2005, 25, 751–766. [Google Scholar] [CrossRef]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef]
- Murphree, A.L.; Benedict, W.F. Retinoblastoma: clues to human oncogenesis. Science 1984, 223, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Magnaghi-Jaulin, L.; Robin, P.; Harel-Bellan, A.; Trouche, D. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 10493–10498. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, H.; Solomon, D.A.; Gunawardena, R.W.; Wang, Y.; Knudsen, E.S. Histone deacetylation of RB-responsive promoters: requisite for specific gene repression but dispensable for cell cycle inhibition. Mol. Cell. Biol. 2003, 23, 7719–7731. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Macaluso, M.; Cinti, C.; Russo, G.; Russo, A.; Giordano, A. pRb2/p130-E2F4/5-HDAC1-SUV39H1-p300 and pRb2/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 multimolecular complexes mediate the transcription of estrogen receptor-alpha in breast cancer. Oncogene 2003, 22, 3511–3517. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Yasui, W.; Oue, N.; Ono, S.; Mitani, Y.; Ito, R.; Nakayama, H. Histone acetylation and gastrointestinal carcinogenesis. Ann. N. Y. Acad. Sci. 2003, 983, 220–231. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.J.; Sternsdorf, T.; Tini, M.; Evans, R.M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 2001, 20, 7204–7215. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kwon, H.J.; Yoon, B.I.; Kim, J.H.; Han, S.U.; Joo, H.J.; Kim, D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 2001, 92, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Halkidou, K.; Gaughan, L.; Cook, S.; Leung, H.Y.; Neal, D.E.; Robson, C.N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004, 59, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hara, Y.; Kobayashi, S.; Iwase, H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005, 94, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Noh, J.H.; Lee, J.H.; Eun, J.W.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; Park, W.S.; Yoo, N.J.; Lee, J.Y.; et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 2005, 113, 264–268. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Gottlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 2004, 10, 6962–6968. [Google Scholar] [CrossRef]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, T.; Izumi, H.; Wakasugi, T.; Niina, I.; Igarashi, T.; Yoshida, T.; Shibuya, I.; Chijiiwa, K.; Matsuo, K.; Itoh, H.; et al. DNA topoisomerase II poison TAS-103 transactivates GC-box-dependent transcription via acetylation of Sp1. J. Biol. Chem. 2005, 280, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Braun, H.; Koop, R.; Ertmer, A.; Nacht, S.; Suske, G. Transcription factor Sp3 is regulated by acetylation. Nucleic. Acids Res. 2001, 29, 4994–5000. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, D.G.; Das, C.M.; Sinnappah-Kang, N.D.; Joyce, C.; Taylor, P.H.; Wen, S.; Hasselblatt, M.; Paulus, W.; Fuller, G.; Wolff, J.E.; et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J. Neurooncol. 2009, 93, 303–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Sigua, C.; Tao, J.; Bali, P.; George, P.; Li, Y.; Wittmann, S.; Moscinski, L.; Atadja, P.; Bhalla, K. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res. 2004, 64, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Meng, Q.; Zhang, C.; Sun, H.; Lu, R.; Gao, N.; Yang, H.; Li, X.; Aschner, M.; Chen, R. DR4 mediates the progression, invasion, metastasis and survival of colorectal cancer through the Sp1/NF1 switch axis on genomic locus. Int. J. Cancer 2018, 143, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.H.; Chan, S.T.; Chen, C.H.; Yeh, S.L. Quercetin enhances the antitumor activity of trichostatin A through up-regulation of p300 protein expression in p53 null cancer cells. Chem. Biol. Interact. 2019, 306, 54–61. [Google Scholar] [CrossRef]
- Kim, M.J.; Hong, K.S.; Kim, H.B.; Lee, S.H.; Bae, J.H.; Kim, D.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Ku70 acetylation and modulation of c-Myc/ATF4/CHOP signaling axis by SIRT1 inhibition lead to sensitization of HepG2 cells to TRAIL through induction of DR5 and down-regulation of c-FLIP. Int. J. Biochem. Cell Biol. 2013, 45, 711–723. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.W.; Lee, J.Y.; Kwon, T.K. Sodium butyrate sensitizes TRAIL-mediated apoptosis by induction of transcription from the DR5 gene promoter through Sp1 sites in colon cancer cells. Carcinogenesis 2004, 25, 1813–1820. [Google Scholar] [CrossRef]
- Shetty, S.; Graham, B.A.; Brown, J.G.; Hu, X.; Vegh-Yarema, N.; Harding, G.; Paul, J.T.; Gibson, S.B. Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol. Cell. Biol. 2005, 25, 5404–5416. [Google Scholar] [CrossRef]
- Kavurma, M.M.; Santiago, F.S.; Bonfoco, E.; Khachigian, L.M. Sp1 phosphorylation regulates apoptosis via extracellular FasL-Fas engagement. J. Biol. Chem. 2001, 276, 4964–4971. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Matsui, K.; Fine, A.; Zhu, B.; Marshak-Rothstein, A.; Widom, R.L.; Ju, S.T. FasL promoter activation by IL-2 through SP1 and NFAT but not Egr-2 and Egr-3. Eur J. Immunol. 1999, 29, 3456–3465. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef] [PubMed]
- Alhazzazi, T.Y.; Kamarajan, P.; Verdin, E.; Kapila, Y.L. Sirtuin-3 (SIRT3) and the Hallmarks of Cancer. Genes Cancer 2013, 4, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Zhu, G. Sirtuin-4 (SIRT4), a therapeutic target with oncogenic and tumor-suppressive activity in cancer. Oncotargets 2018, 11, 3395–3400. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Zino, S.; MacDonald, A.I.; Curle, J.; Reilly, J.E.; Mohammed, Z.M.; McMillan, D.C.; Mallon, E.; Payne, A.P.; Edwards, J.; et al. SIRT2: tumour suppressor or tumour promoter in operable breast cancer? Eur J. Cancer 2014, 50, 290–301. [Google Scholar] [CrossRef]
- Miyo, M.; Yamamoto, H.; Konno, M.; Colvin, H.; Nishida, N.; Koseki, J.; Kawamoto, K.; Ogawa, H.; Hamabe, A.; Uemura, M.; et al. Tumour-suppressive function of SIRT4 in human colorectal cancer. Br. J. Cancer 2015, 113, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Paredes, S.; Villanova, L.; Chua, K.F. Molecular pathways: emerging roles of mammalian Sirtuin SIRT7 in cancer. Clin. Cancer Res. 2014, 20, 1741–1746. [Google Scholar] [CrossRef]
- Sebastian, C.; Zwaans, B.M.; Silberman, D.M.; Gymrek, M.; Goren, A.; Zhong, L.; Ram, O.; Truelove, J.; Guimaraes, A.R.; Toiber, D.; et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012, 151, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Shi, L.; Xie, N.; Liu, Z.; Qian, M.; Meng, F.; Xu, Q.; Zhou, M.; Cao, X.; Zhu, W.G.; et al. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat. Commun. 2017, 8, 318. [Google Scholar] [CrossRef] [PubMed]
- Torrens-Mas, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. SIRT3: Oncogene and tumor suppressor in cancer. Cancers 2017, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Che, X.; Wu, Y.; Song, N.; Shi, S.; Wang, S.; Li, C.; Zhang, L.; Zhang, X.; Qu, X.; et al. SIRT5 as a biomarker for response to anthracycline-taxane-based neoadjuvant chemotherapy in triple-negative breast cancer. Oncol. Rep. 2018, 39, 2315–2323. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.; Zinn, R.L.; Ohm, J.E.; McGarvey, K.M.; Kang, S.H.; Watkins, D.N.; Herman, J.G.; Baylin, S.B. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLOS Genet. 2006, 2, e40. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef]
- Ozdag, H.; Teschendorff, A.E.; Ahmed, A.A.; Hyland, S.J.; Blenkiron, C.; Bobrow, L.; Veerakumarasivam, A.; Burtt, G.; Subkhankulova, T.; Arends, M.J.; et al. Differential expression of selected histone modifier genes in human solid cancers. BMC Genom. 2006, 7, 90. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, D.H.; Yen, R.C.; Luo, J.; Gu, W.; Baylin, S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.H.; Min, B.H.; Lee, K.M.; Cho, M.H.; Park, G.H.; Lee, K.H. SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wu, H.; Yang, M.; Ye, S.; Li, L.; Zhang, H.; Hu, J.; Wang, X.; Xu, J.; Liang, A. SIRT1 inhibition impairs non-homologous end joining DNA damage repair by increasing Ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 2016, 7, 13538–13550. [Google Scholar] [CrossRef] [PubMed]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. Biomed. Res. Int 2016, 2016, 8797206. [Google Scholar] [CrossRef] [PubMed]
- Perego, P.; Zuco, V.; Gatti, L.; Zunino, F. Sensitization of tumor cells by targeting histone deacetylases. Biochem. Pharm. 2012, 83, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Seto, E. Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handb. Exp. Pharm. 2011, 206, 39–56. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Ceccacci, E.; Minucci, S. Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. Br. J. Cancer 2016, 114, 605–611. [Google Scholar] [CrossRef] [Green Version]
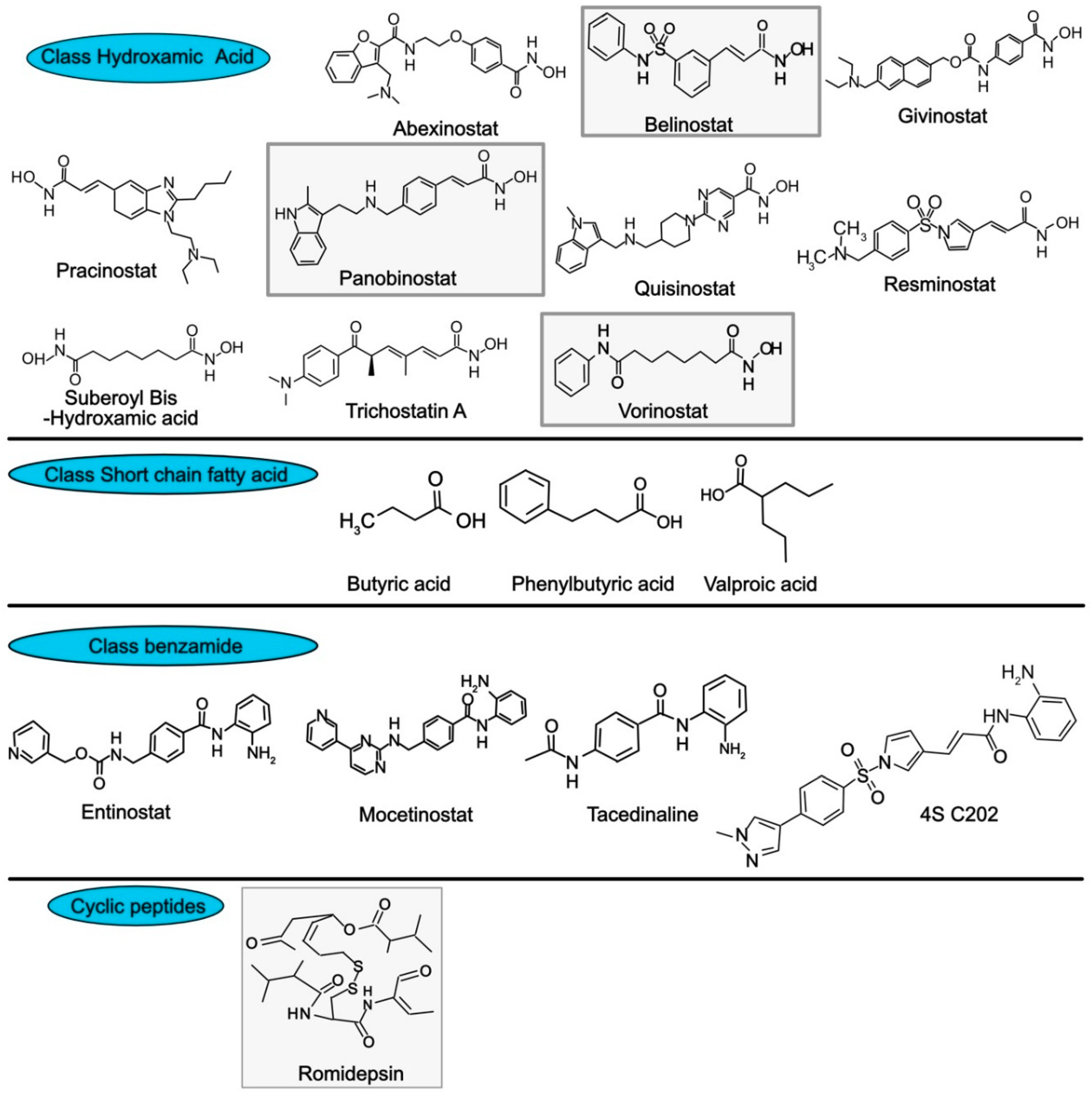
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A structural insight into hydroxamic acid based histone deacetylase inhibitors for the presence of anticancer activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Su, J.M.; Li, X.N.; Thompson, P.; Ou, C.N.; Ingle, A.M.; Russell, H.; Lau, C.C.; Adamson, P.C.; Blaney, S.M. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: a children’s oncology group report. Clin. Cancer Res. 2011, 17, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A. Histone deacetylase inhibitor sulforaphane: The phytochemical with vibrant activity against prostate cancer. Biomed. Pharm. 2016, 81, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell. Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Arima, N.; Honda, S.; Soeda, S. Anticancer agents targeted to sirtuins. Molecules 2014, 19, 20295–20313. [Google Scholar] [CrossRef]
- Medda, F.; Russell, R.J.; Higgins, M.; McCarthy, A.R.; Campbell, J.; Slawin, A.M.; Lane, D.P.; Lain, S.; Westwood, N.J. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J. Med. Chem. 2009, 52, 2673–2682. [Google Scholar] [CrossRef]
- Figuera-Losada, M.; Stathis, M.; Dorskind, J.M.; Thomas, A.G.; Bandaru, V.V.; Yoo, S.W.; Westwood, N.J.; Rogers, G.W.; McArthur, J.C.; Haughey, N.J.; et al. Cambinol, a novel inhibitor of neutral sphingomyelinase 2 shows neuroprotective properties. PLoS ONE 2015, 10, e0124481. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
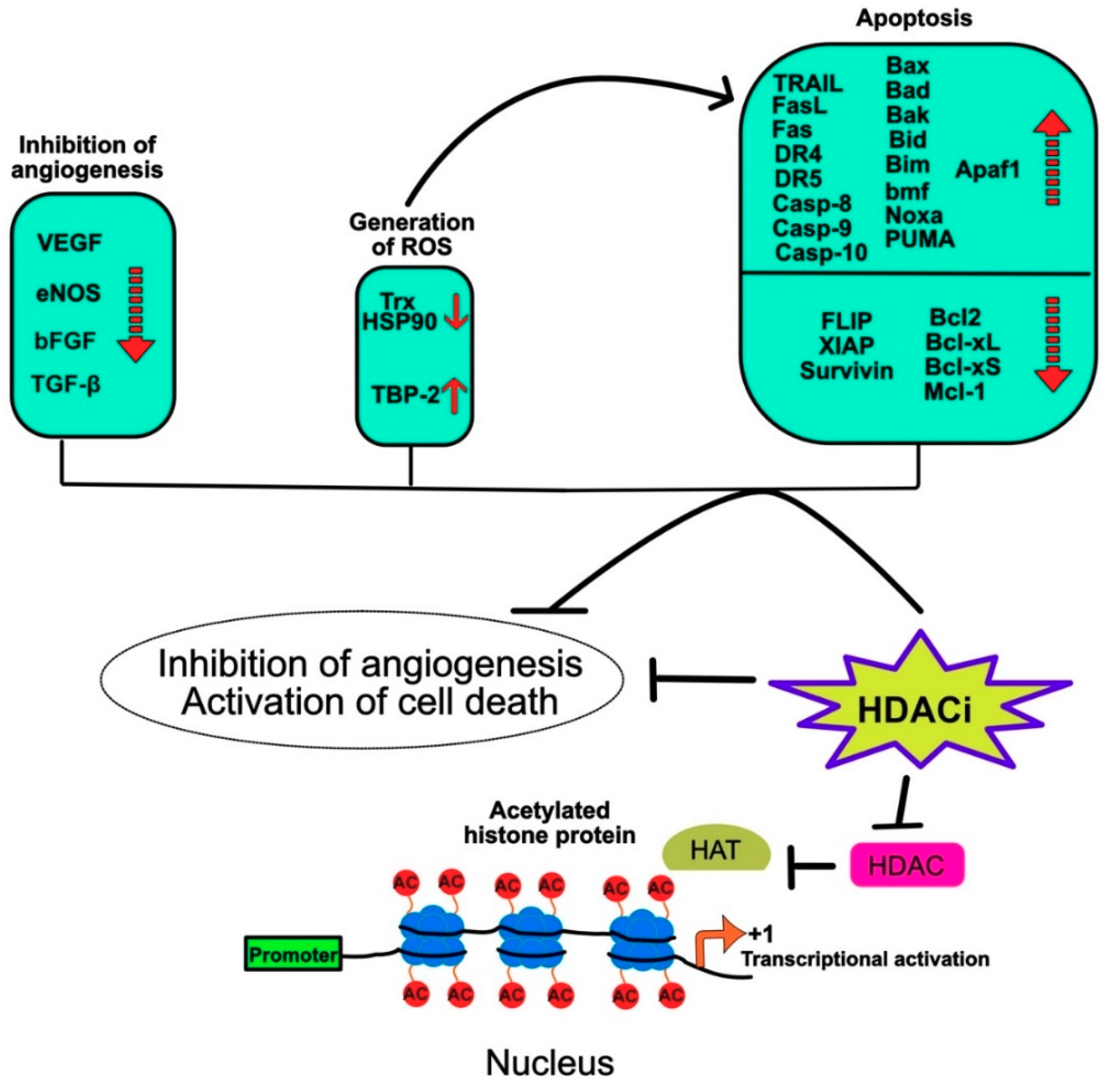
- Zhang, Z.H.; Hao, C.L.; Liu, P.; Tian, X.; Wang, L.H.; Zhao, L.; Zhu, C.M. Valproic acid inhibits tumor angiogenesis in mice transplanted with Kasumi1 leukemia cells. Mol. Med. Rep. 2014, 9, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jiang, S.; Chen, J.; Su, S.B. Suberoylanilide hydroxamic acid suppresses inflammation-induced neovascularization. Can. J. Physiol. Pharm. 2014, 92, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Mobley, A.; Miller, C.; Boklan, J.; Chandra, J. Potentiation of reactive oxygen species is a marker for synergistic cytotoxicity of MS-275 and 5-azacytidine in leukemic cells. Leuk. Res. 2008, 32, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Park, S.; Park, J.A.; Kim, Y.E.; Song, S.; Kwon, H.J.; Lee, Y. Suberoylanilide hydroxamic acid induces ROS-mediated cleavage of HSP90 in leukemia cells. Cell Stress Chaperones 2015, 20, 149–157. [Google Scholar] [CrossRef]
- Bokelmann, I.; Mahlknecht, U. Valproic acid sensitizes chronic lymphocytic leukemia cells to apoptosis and restores the balance between pro- and antiapoptotic proteins. Mol. Med. 2008, 14, 20–27. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, E.J.; Yang, S.H.; Jeong, E.T.; Park, C.; Lee, J.H.; Youn, M.J.; So, H.S.; Park, R. Trichostatin A induces apoptosis in lung cancer cells via simultaneous activation of the death receptor-mediated and mitochondrial pathway? Exp. Mol. Med. 2006, 38, 616–624. [Google Scholar] [CrossRef]
- Morales, J.C.; Ruiz-Magana, M.J.; Carranza, D.; Ortiz-Ferron, G.; Ruiz-Ruiz, C. HDAC inhibitors with different gene regulation activities depend on the mitochondrial pathway for the sensitization of leukemic T cells to TRAIL-induced apoptosis. Cancer Lett. 2010, 297, 91–100. [Google Scholar] [CrossRef]
- Shankar, S.; Singh, T.R.; Fandy, T.E.; Luetrakul, T.; Ross, D.D.; Srivastava, R.K. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: involvement of both death receptor and mitochondrial pathways. Int. J. Mol. Med. 2005, 16, 1125–1138. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Bolden, J.E.; Shi, W.; Jankowski, K.; Kan, C.Y.; Cluse, L.; Martin, B.P.; MacKenzie, K.L.; Smyth, G.K.; Johnstone, R.W. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013, 4, e519. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Imai, K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006, 13, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Fandy, T.E.; Shankar, S.; Ross, D.D.; Sausville, E.; Srivastava, R.K. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia 2005, 7, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, S.; Borrow, J.; Zhang, X.D.; Hersey, P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis 2006, 11, 2251–2265. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef]
- Buurman, R.; Sandbothe, M.; Schlegelberger, B.; Skawran, B. HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma. Eur. J. Med. Res. 2016, 21, 26. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Heckman, C.A.; Boxer, L.M. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 2005, 25, 1608–1619. [Google Scholar] [CrossRef]
- Chueh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 repression of BCL-XL determines apoptotic sensitivity to HDAC inhibitors across tumor types. Clin. Cancer Res. 2017, 23, 5573–5584. [Google Scholar] [CrossRef]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol. Cancer 2004, 3, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, F.; Previdi, S.; Ballarini, M.; Minucci, S.; Perego, P.; La Porta, C.A. Modulation of pro- and anti-apoptotic factors in human melanoma cells exposed to histone deacetylase inhibitors. Apoptosis 2004, 9, 573–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, Y.F.; Huang, S.W.; Hsu, Y.F.; Yu, M.C.; Ou, G.; Huang, W.J.; Hsu, M.J. WMJ-8-B, a novel hydroxamate derivative, induces MDA-MB-231 breast cancer cell death via the SHP-1-STAT3-survivin cascade. Br. J. Pharm. 2017, 174, 2941–2961. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Bruwiere, H.; Verhulst, T.; Steller, U.; Andries, L.; Wouters, W.; Janicot, M.; Arts, J.; Van Heusden, J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J. Pharm. Exp. 2003, 304, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Zimmerman, M.A.; Singh, N.; Martin, P.M.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Shi, H.; Robertson, K.D.; Munn, D.H.; Liu, K. Butyrate suppresses colonic inflammation through HDAC1-dependent Fas upregulation and Fas-mediated apoptosis of T cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1405–G1415. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef]
- Feltus, F.A.; Lee, E.K.; Costello, J.F.; Plass, C.; Vertino, P.M. Predicting aberrant CpG island methylation. Proc. Natl. Acad. Sci. USA 2003, 100, 12253–12258. [Google Scholar] [CrossRef] [Green Version]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, T.E.; Zaikin, A.; Olhede, S.C.; West, J.; Teschendorff, A.E.; Widschwendter, M. Corruption of the intra-gene DNA methylation architecture is a hallmark of cancer. PLoS ONE 2013, 8, e68285. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Singal, R.; Ginder, G.D. DNA methylation. Blood 1999, 93, 4059–4070. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Bae, M.G.; Kim, J.Y.; Choi, J.K. Frequent hypermethylation of orphan CpG islands with enhancer activity in cancer. BMC Med. Genom. 2016, 9 (Suppl. S1), 38. [Google Scholar] [CrossRef]
- Costello, J.F.; Plass, C. Methylation matters. J. Med. Genet. 2001, 38, 285–303. [Google Scholar] [CrossRef] [Green Version]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes (Basel) 2019, 10, 172. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.; Venkiteswaran, M.; Chen, H.; Xu, G.L.; Plosch, T.; Rots, M.G. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics 2015, 10, 671–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.Y.; Chen, C.C.; Shen, C.K. Active DNA demethylation of the vertebrate genomes by DNA methyltransferases: deaminase, dehydroxymethylase or demethylase? Epigenomics 2014, 6, 353–363. [Google Scholar] [CrossRef]
- Koivunen, P.; Laukka, T. The TET enzymes. Cell. Mol. Life Sci. 2018, 75, 1339–1348. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, N.; Marques, C.J.; Reik, W. DNA demethylases: a new epigenetic frontier in drug discovery. Drug Discov. Today 2011, 16, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013, 2013, 371854. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, C.; Lovato, O.; Bertolaso, A.; Pacelli, L.; Zoratti, E.; Zanolin, E.; Krampera, M.; Zamo, A.; Tecchio, C.; Cassatella, M.A.; et al. The TNF-family cytokine TL1A inhibits proliferation of human activated B cells. PLoS ONE 2013, 8, e60136. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Bang, B.R.; Lu, J.; Eun, S.Y.; Otsuka, M.; Croft, M.; Tobias, P.; Han, J.; Takeuchi, O.; Akira, S.; et al. The TNF family member 4-1BBL sustains inflammation by interacting with TLR signaling components during late-phase activation. Sci. Signal. 2013, 6, ra87. [Google Scholar] [CrossRef]
- Micheau, O. Posttranslational modifications and death receptor signalling. In TRAIL, Fas Ligand, TNF and TLR3 in Cancer; Micheau, O., Ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Micheau, O. Regulation of TNF-related apoptosis-inducing ligand signaling by glycosylation. Int. J. Mol. Sci. 2018, 19, 715. [Google Scholar] [CrossRef] [PubMed]
- Elmallah, M.I.; Micheau, O. Marine drugs regulating apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Mar. Drugs 2015, 13, 6884–6909. [Google Scholar] [CrossRef]
- Pavet, V.; Portal, M.M.; Moulin, J.C.; Herbrecht, R.; Gronemeyer, H. Towards novel paradigms for cancer therapy. Oncogene 2011, 30, 1–20. [Google Scholar] [CrossRef]
- Srivastava, R.K. Intracellular mechanisms of TRAIL and its role in cancer therapy. Mol. Cell. Biol. Res. Commun. 2000, 4, 67–75. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Dufour, F.; Rattier, T.; Constantinescu, A.A.; Zischler, L.; Morle, A.; Ben Mabrouk, H.; Humblin, E.; Jacquemin, G.; Szegezdi, E.; Delacote, F.; et al. TRAIL receptor gene editing unveils TRAIL-R1 as a master player of apoptosis induced by TRAIL and ER stress. Oncotarget 2017, 8, 9974–9985. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Puschel, F.; Leon-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramon, D.; Lucendo, E.; Munoz-Pinedo, C. Glucose deprivation induces ATF4-mediated apoptosis through TRAIL death receptors. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharm. 2013, 169, 1723–1744. [Google Scholar] [CrossRef] [PubMed]
- Hersey, P.; Zhang, X.D. How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer 2001, 1, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Glick, R.D.; Swendeman, S.L.; Coffey, D.C.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999, 59, 4392–4399. [Google Scholar] [PubMed]
- Kwon, S.H.; Ahn, S.H.; Kim, Y.K.; Bae, G.U.; Yoon, J.W.; Hong, S.; Lee, H.Y.; Lee, Y.W.; Lee, H.W.; Han, J.W. Apicidin, a histone deacetylase inhibitor, induces apoptosis and Fas/Fas ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 2002, 277, 2073–2080. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell Death Dis. 2010, 1, e8. [Google Scholar] [CrossRef]
- Teodoridis, J.M.; Hall, J.; Marsh, S.; Kannall, H.D.; Smyth, C.; Curto, J.; Siddiqui, N.; Gabra, H.; McLeod, H.L.; Strathdee, G.; et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005, 65, 8961–8967. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ni, M.; Xu, J.; Zhang, H.; Gao, B.; Gu, J.; Chen, J.; Zhang, L.; Wu, M.; Zhen, S.; et al. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer 2002, 2, 29. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, H.; Gu, J.; Lin, S.; Li, J.; Lu, W.; Wang, Y.; Zhu, J. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer 2004, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Chaopatchayakul, P.; Jearanaikoon, P.; Yuenyao, P.; Limpaiboon, T. Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma. Am. J. Obs. Gynecol. 2010, 202, 281.e1–281.e9. [Google Scholar] [CrossRef] [PubMed]
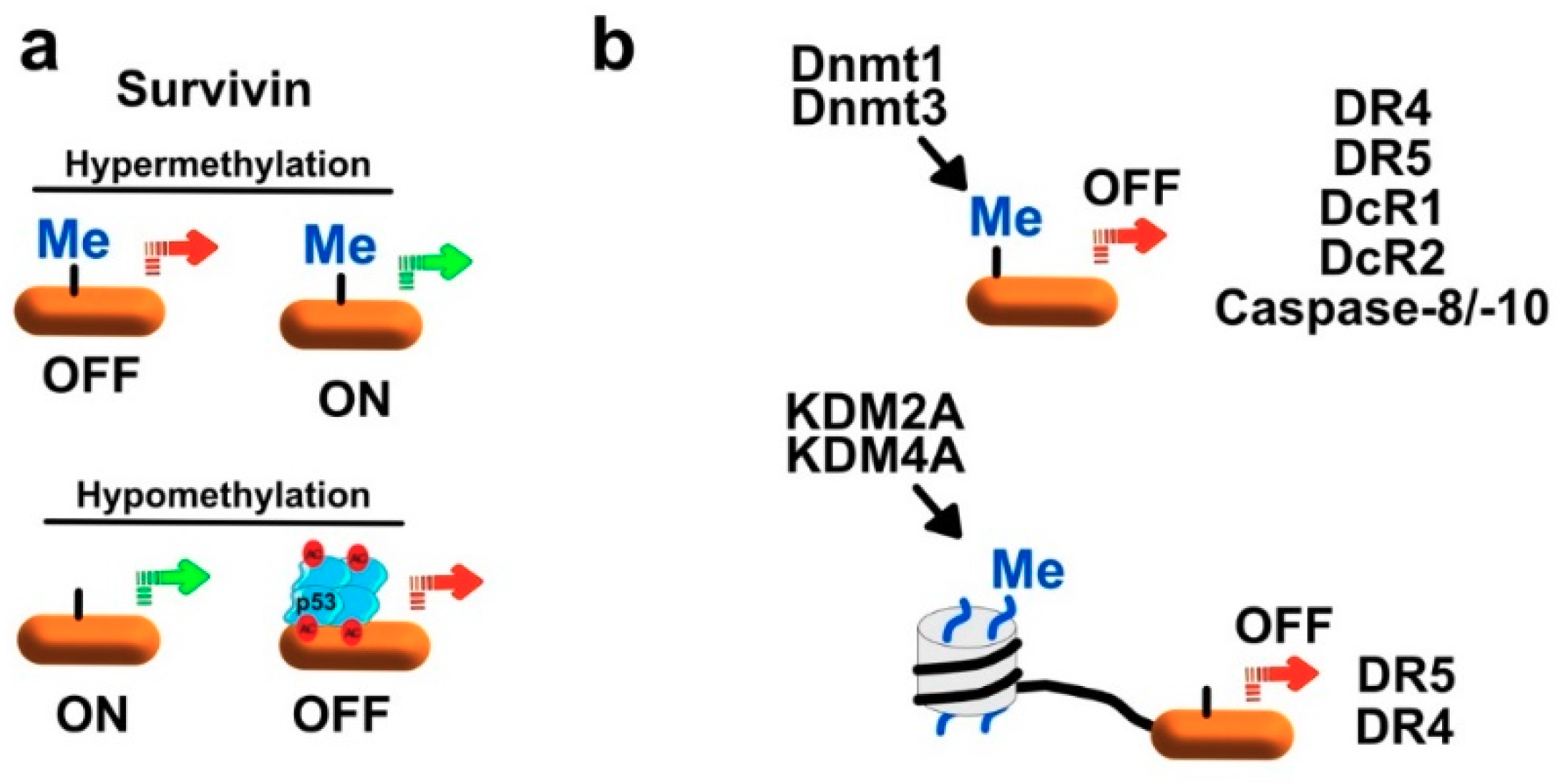
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, D.; Manero-Ruperez, N.; Quesada, R.; Korrodi-Gregorio, L.; Soto-Cerrato, V. Therapeutic strategies involving survivin inhibition in cancer. Med. Res. Rev. 2019, 39, 887–909. [Google Scholar] [CrossRef] [PubMed]
- Nabilsi, N.H.; Broaddus, R.R.; Loose, D.S. DNA methylation inhibits p53-mediated survivin repression. Oncogene 2009, 28, 2046–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminskyy, V.O.; Surova, O.V.; Vaculova, A.; Zhivotovsky, B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis 2011, 32, 1450–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straszewski-Chavez, S.L.; Visintin, I.P.; Karassina, N.; Los, G.; Liston, P.; Halaban, R.; Fadiel, A.; Mor, G. XAF1 mediates tumor necrosis factor-alpha-induced apoptosis and X-linked inhibitor of apoptosis cleavage by acting through the mitochondrial pathway. J. Biol. Chem. 2007, 282, 13059–13072. [Google Scholar] [CrossRef] [PubMed]
- Micali, O.C.; Cheung, H.H.; Plenchette, S.; Hurley, S.L.; Liston, P.; LaCasse, E.C.; Korneluk, R.G. Silencing of the XAF1 gene by promoter hypermethylation in cancer cells and reactivation to TRAIL-sensitization by IFN-beta. BMC Cancer 2007, 7, 52. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Bodmer, J.L.; Bourloud, K.B.; Brognara, C.B.; Tschopp, J.; Gross, N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000, 60, 4315–4319. [Google Scholar]
- Poulaki, V.; Mitsiades, C.S.; McMullan, C.; Fanourakis, G.; Negri, J.; Goudopoulou, A.; Halikias, I.X.; Voutsinas, G.; Tseleni-Balafouta, S.; Miller, J.W.; et al. Human retinoblastoma cells are resistant to apoptosis induced by death receptors: role of caspase-8 gene silencing. Invest. Ophthalmol. Vis. Sci. 2005, 46, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Margetts, C.D.; Astuti, D.; Gentle, D.C.; Cooper, W.N.; Cascon, A.; Catchpoole, D.; Robledo, M.; Neumann, H.P.; Latif, F.; Maher, E.R. Epigenetic analysis of HIC1, CASP8, FLIP, TSP1, DCR1, DCR2, DR4, DR5, KvDMR1, H19 and preferential 11p15.5 maternal-allele loss in von Hippel-Lindau and sporadic phaeochromocytomas. Endocr. Relat. Cancer 2005, 12, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, A.; Grotzer, M.A.; Zuzak, T.J.; Wiewrodt, B.R.; Ho, R.; Ikegaki, N.; Brodeur, G.M. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res. 2001, 61, 1314–1319. [Google Scholar] [PubMed]
- Fulda, S.; Debatin, K.M. 5-Aza-2′-deoxycytidine and IFN-gamma cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene 2006, 25, 5125–5133. [Google Scholar] [CrossRef] [PubMed]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Lee, J.H.; Cho, S.B.; Yoon, K.W.; Park, S.Y.; Lee, W.S.; Park, C.H.; Joo, Y.E.; Kim, H.S.; Choi, S.K.; et al. Epigenetic methylation and expression of caspase 8 and survivin in hepatocellular carcinoma. Pathol. Int. 2010, 60, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Higuchi, H.; Saito, Y.; Nakamoto, N.; Takaishi, H.; Tada, S.; Saito, H.; Gores, G.J.; Hibi, T. DNMT1 and DNMT3b silencing sensitizes human hepatoma cells to TRAIL-mediated apoptosis via up-regulation of TRAIL-R2/DR5 and caspase-8. Cancer Sci. 2010, 101, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Skiriute, D.; Vaitkiene, P.; Saferis, V.; Asmoniene, V.; Skauminas, K.; Deltuva, V.P.; Tamasauskas, A. MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 2012, 12, 218. [Google Scholar] [CrossRef]
- Van Noesel, M.M.; van Bezouw, S.; Salomons, G.S.; Voute, P.A.; Pieters, R.; Baylin, S.B.; Herman, J.G.; Versteeg, R. Tumor-specific down-regulation of the tumor necrosis factor-related apoptosis-inducing ligand decoy receptors DcR1 and DcR2 is associated with dense promoter hypermethylation. Cancer Res. 2002, 62, 2157–2161. [Google Scholar]
- Van Noesel, M.M.; van Bezouw, S.; Voute, P.A.; Herman, J.G.; Pieters, R.; Versteeg, R. Clustering of hypermethylated genes in neuroblastoma. Genes Chromosomes Cancer 2003, 38, 226–233. [Google Scholar] [CrossRef]
- Pal, R.; Srivastava, N.; Chopra, R.; Gochhait, S.; Gupta, P.; Prakash, N.; Agarwal, G.; Bamezai, R.N. Investigation of DNA damage response and apoptotic gene methylation pattern in sporadic breast tumors using high throughput quantitative DNA methylation analysis technology. Mol. Cancer 2010, 9, 303. [Google Scholar] [CrossRef]
- Narayan, G.; Xie, D.; Ishdorj, G.; Scotto, L.; Mansukhani, M.; Pothuri, B.; Wright, J.D.; Kaufmann, A.M.; Schneider, A.; Arias-Pulido, H.; et al. Epigenetic inactivation of TRAIL decoy receptors at 8p12-21.3 commonly deleted region confers sensitivity to Apo2L/trail-Cisplatin combination therapy in cervical cancer. Genes Chromosomes Cancer 2016, 55, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; Siegelin, M.D.; Steinmuller, A.; von Deimling, A.; Lass, U.; Korn, B.; Mueller, W. Epigenetic silencing of death receptor 4 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in gliomas. Clin. Cancer Res. 2009, 15, 5457–5465. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Catalano, T.; Fortunato, C.; Oteri, R.; Teti, D.; Venza, I. Impact of DNA methyltransferases on the epigenetic regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in malignant melanoma. Biochem. Biophys. Res. Commun. 2013, 441, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Jeon, Y.K.; Choi, Y.; Kim, C.W. Short-hairpin RNA-induced suppression of adenine nucleotide translocase-2 in breast cancer cells restores their susceptibility to TRAIL-induced apoptosis by activating JNK and modulating TRAIL receptor expression. Mol. Cancer 2010, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Florean, C.; Schnekenburger, M.; Lee, J.Y.; Kim, K.R.; Mazumder, A.; Song, S.; Kim, J.M.; Grandjenette, C.; Kim, J.G.; Yoon, A.Y.; et al. Discovery and characterization of Isofistularin-3, a marine brominated alkaloid, as a new DNA demethylating agent inducing cell cycle arrest and sensitization to TRAIL in cancer cells. Oncotarget 2016, 7, 24027–24049. [Google Scholar] [CrossRef] [PubMed]
- Kurt, I.C.; Sur, I.; Kaya, E.; Cingoz, A.; Kazancioglu, S.; Kahya, Z.; Toparlak, O.D.; Senbabaoglu, F.; Kaya, Z.; Ozyerli, E.; et al. KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis. 2017, 8, e2897. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Wang, L.Y.; Cai, D.; Duan, Z.; Zhang, Y.; Chen, P.; Zou, J.X.; Xu, J.; Chen, X.; et al. Silencing the epigenetic silencer KDM4A for TRAIL and DR5 simultaneous induction and antitumor therapy. Cell Death Differ. 2016, 23, 1886–1896. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: a potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharm. 2003, 66, 1537–1545. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Arle, D. Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: an attractive approach to bypass melanoma immunotherapy resistance. Am. J. Clin. Exp. Immunol. 2013, 2, 55–74. [Google Scholar]
- Jazirehi, A.R.; Kurdistani, S.K.; Economou, J.S. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J. Immunol. 2014, 192, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Harper, N.; Walewska, R.; Dyer, M.J.; Cohen, G.M. Enhanced Fas-associated death domain recruitment by histone deacetylase inhibitors is critical for the sensitization of chronic lymphocytic leukemia cells to TRAIL-induced apoptosis. Mol. Cancer 2009, 8, 3088–3097. [Google Scholar] [CrossRef] [PubMed]
- D’Acunto, C.W.; Carratu, A.; Rodriquez, M.; Taddei, M.; Parente, L.; Petrella, A. LGP1, A histone deacetylase inhibitor analogue of FR235222, sensitizes promyelocytic leukaemia U937 cells to TRAIL-mediated apoptosis. Anticancer Res. 2010, 30, 887–894. [Google Scholar] [PubMed]
- Frew, A.J.; Lindemann, R.K.; Martin, B.P.; Clarke, C.J.; Sharkey, J.; Anthony, D.A.; Banks, K.M.; Haynes, N.M.; Gangatirkar, P.; Stanley, K.; et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc. Natl Acad. Sci. USA 2008, 105, 11317–11322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.H.; Kim, H.S.; Kim, S.U.; Noh, E.J.; Lee, J.S.; Choi, K.S. Sodium butyrate sensitizes human glioma cells to TRAIL-mediated apoptosis through inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene 2005, 24, 6877–6889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhlethaler-Mottet, A.; Flahaut, M.; Bourloud, K.B.; Auderset, K.; Meier, R.; Joseph, J.M.; Gross, N. Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL-induced apoptosis by a caspases-dependent increase of the pro- to anti-apoptotic proteins ratio. BMC Cancer 2006, 6, 214. [Google Scholar] [CrossRef] [PubMed]
- Schuchmann, M.; Schulze-Bergkamen, H.; Fleischer, B.; Schattenberg, J.M.; Siebler, J.; Weinmann, A.; Teufel, A.; Worns, M.; Fischer, T.; Strand, S.; et al. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol Rep. 2006, 15, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar] [CrossRef]
- Earel, J.K., Jr.; VanOosten, R.L.; Griffith, T.S. Histone deacetylase inhibitors modulate the sensitivity of tumor necrosis factor-related apoptosis-inducing ligand-resistant bladder tumor cells. Cancer Res. 2006, 66, 499–507. [Google Scholar] [CrossRef]
- Kim, S.Y.; Hong, M.; Heo, S.H.; Park, S.; Kwon, T.K.; Sung, Y.H.; Oh, Y.; Lee, S.; Yi, G.S.; Kim, I. Inhibition of euchromatin histone-lysine N-methyltransferase 2 sensitizes breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through reactive oxygen species-mediated activating transcription factor 4-C/EBP homologous protein-death receptor 5 pathway activation. Mol. Carcinog. 2018, 57, 1492–1506. [Google Scholar] [CrossRef]
- Fandy, T.E.; Srivastava, R.K. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother. Pharm. 2006, 58, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacomino, G.; Medici, M.C.; Russo, G.L. Valproic acid sensitizes K562 erythroleukemia cells to TRAIL/Apo2L-induced apoptosis. Anticancer Res. 2008, 28, 855–864. [Google Scholar] [PubMed]
- Frohlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef] [PubMed]
- Pathil, A.; Armeanu, S.; Venturelli, S.; Mascagni, P.; Weiss, T.S.; Gregor, M.; Lauer, U.M.; Bitzer, M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 2006, 43, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Hacker, S.; Dittrich, A.; Mohr, A.; Schweitzer, T.; Rutkowski, S.; Krauss, J.; Debatin, K.M.; Fulda, S. Histone deacetylase inhibitors cooperate with IFN-gamma to restore caspase-8 expression and overcome TRAIL resistance in cancers with silencing of caspase-8. Oncogene 2009, 28, 3097–3110. [Google Scholar] [CrossRef]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Sukhai, M.A.; Hurren, R.; Anyiwe, K.; Mao, X.; Suarez Saiz, F.; Gronda, M.; Eberhard, Y.; et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer 2010, 9, 246–256. [Google Scholar] [CrossRef]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL resistance: Novel targets for TRAIL sensitization in cancer cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Okhovat, J.P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical studies support combined inhibition of BET family proteins and histone deacetylases as epigenetic therapy for cutaneous T-cell lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar] [CrossRef]
- Klingbeil, O.; Lesche, R.; Gelato, K.A.; Haendler, B.; Lejeune, P. Inhibition of BET bromodomain-dependent XIAP and FLIP expression sensitizes KRAS-mutated NSCLC to pro-apoptotic agents. Cell Death Dis. 2016, 7, e2365. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Yue, P.; Khuri, F.R.; Sun, S.Y. The BET bromodomain inhibitor, JQ1, facilitates c-FLIP degradation and enhances TRAIL-induced apoptosis independent of BRD4 and c-Myc inhibition. Oncotarget 2015, 6, 34669–34679. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Toscano, F.; Virard, F.; Jacquemin, G.; Pierrot, A.; Vanbervliet, B.; Bonnin, M.; Lalaoui, N.; Mercier-Gouy, P.; Pacheco, Y.; et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012, 19, 1482–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangert, A.; Cristofanon, S.; Eckhardt, I.; Abhari, B.A.; Kolodziej, S.; Hacker, S.; Vellanki, S.H.; Lausen, J.; Debatin, K.M.; Fulda, S. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene 2012, 31, 4677–4688. [Google Scholar] [CrossRef]
- Lauricella, M.; Ciraolo, A.; Carlisi, D.; Vento, R.; Tesoriere, G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie 2012, 94, 287–299. [Google Scholar] [CrossRef]
- Rao-Bindal, K.; Koshkina, N.V.; Stewart, J.; Kleinerman, E.S. The histone deacetylase inhibitor, MS-275 (entinostat), downregulates c-FLIP, sensitizes osteosarcoma cells to FasL, and induces the regression of osteosarcoma lung metastases. Curr. Cancer Drug Targets 2013, 13, 411–422. [Google Scholar] [CrossRef]
- Sung, E.S.; Kim, A.; Park, J.S.; Chung, J.; Kwon, M.H.; Kim, Y.S. Histone deacetylase inhibitors synergistically potentiate death receptor 4-mediated apoptotic cell death of human T-cell acute lymphoblastic leukemia cells. Apoptosis 2010, 15, 1256–1269. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Beninati, C.; Benfatto, S.; Teti, D.; Venza, I. miR-92a-3p and MYCBP2 are involved in MS-275-induced and c-myc-mediated TRAIL-sensitivity in melanoma cells. Int. Immunopharmacol. 2016, 40, 235–243. [Google Scholar] [CrossRef]
- Zhang, G.; Park, M.A.; Mitchell, C.; Hamed, H.; Rahmani, M.; Martin, A.P.; Curiel, D.T.; Yacoub, A.; Graf, M.; Lee, R.; et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin. Cancer Res. 2008, 14, 5385–5399. [Google Scholar] [CrossRef]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Su, L.; Hao, X.; Zhong, N.; Zhong, D.; Singhal, S.; Liu, X. Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J. Cell. Mol. Med. 2012, 16, 1618–1628. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Cheng, Y.; You, L.; Qian, J.; Qian, W. Homoharringtonine and SAHA synergistically enhance apoptosis in human acute myeloid leukemia cells through upregulation of TRAIL and death receptors. Mol. Med. Rep. 2013, 7, 1838–1844. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmallah, M.I.Y.; Micheau, O. Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 2019, 11, 850. https://doi.org/10.3390/cancers11060850
Elmallah MIY, Micheau O. Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers. 2019; 11(6):850. https://doi.org/10.3390/cancers11060850
Chicago/Turabian StyleElmallah, Mohammed I. Y., and Olivier Micheau. 2019. "Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy" Cancers 11, no. 6: 850. https://doi.org/10.3390/cancers11060850
APA StyleElmallah, M. I. Y., & Micheau, O. (2019). Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers, 11(6), 850. https://doi.org/10.3390/cancers11060850