The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
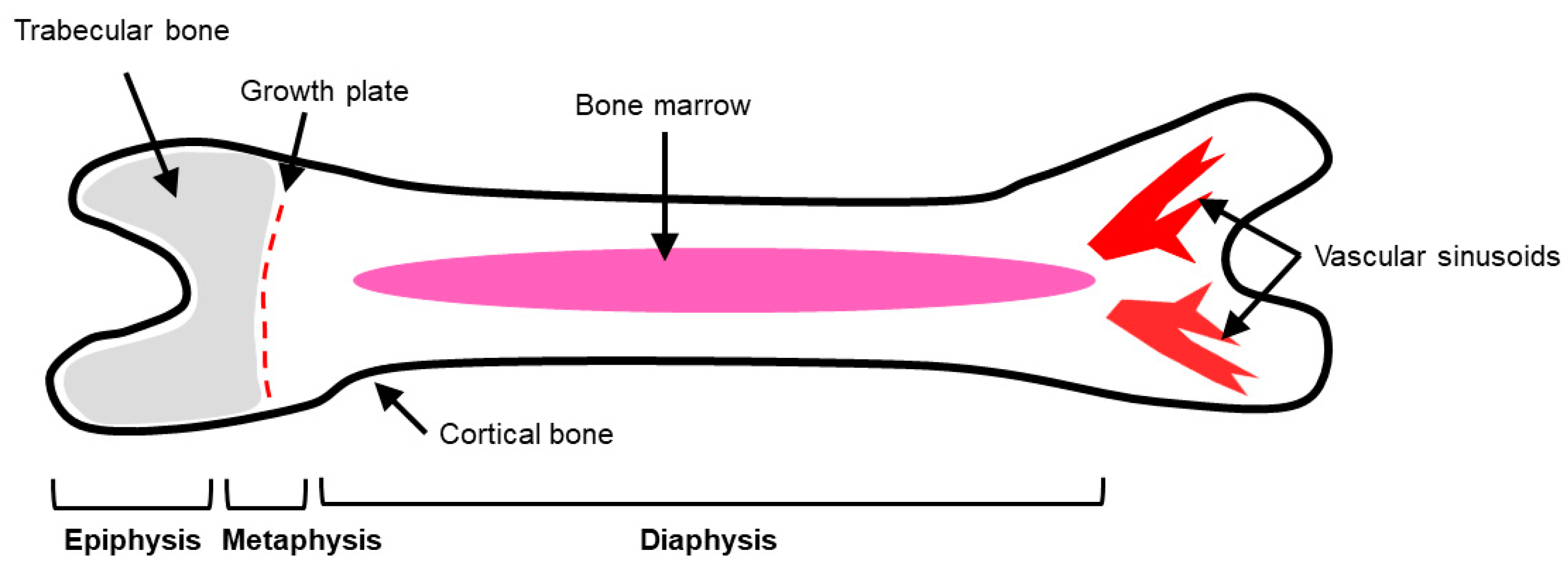
2. Bone Physiology
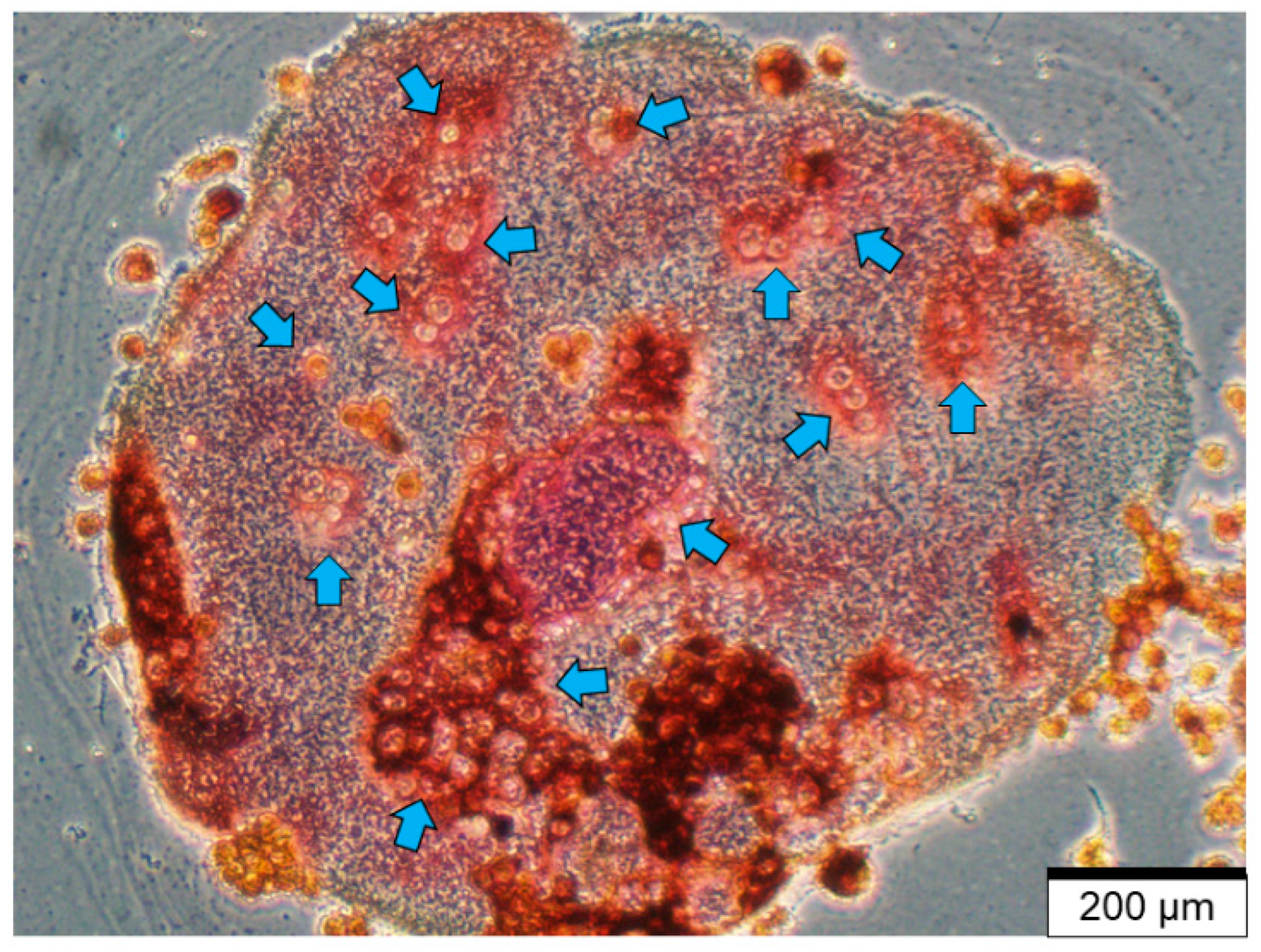
2.1. Bone Cells Modulate the Bone Microenvironment
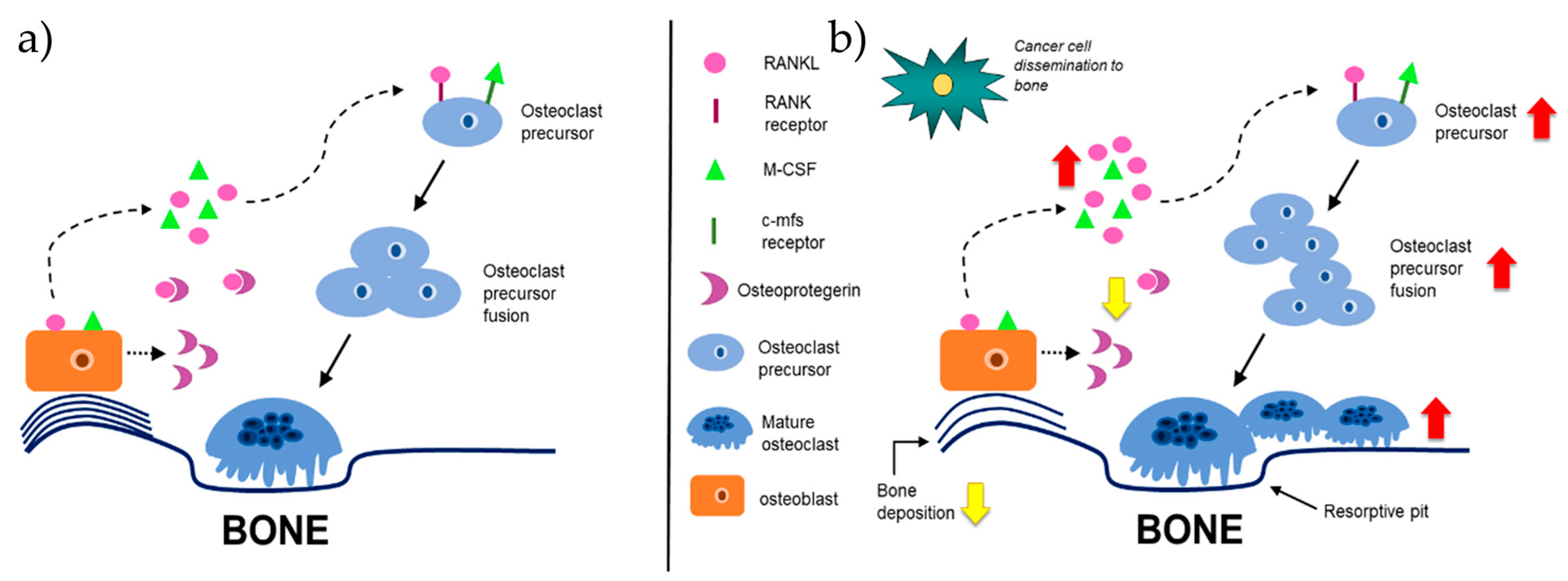
2.2. Bone Remodeling During Homeostasis and Disease
3. The Bone Extracellular Matrix
3.1. Organic Bone Components: Collagenous Proteins
3.2. Organic Bone Components: Non-Collagenous Proteins
3.3. Inorganic Bone Components
4. Bone Metastatic Cancers
4.1. Prostate Cancer
4.2. Breast Cancer
4.3. Multiple Myeloma
4.4. Lung Cancer
5. Current Therapies Targeting Bone Metastases
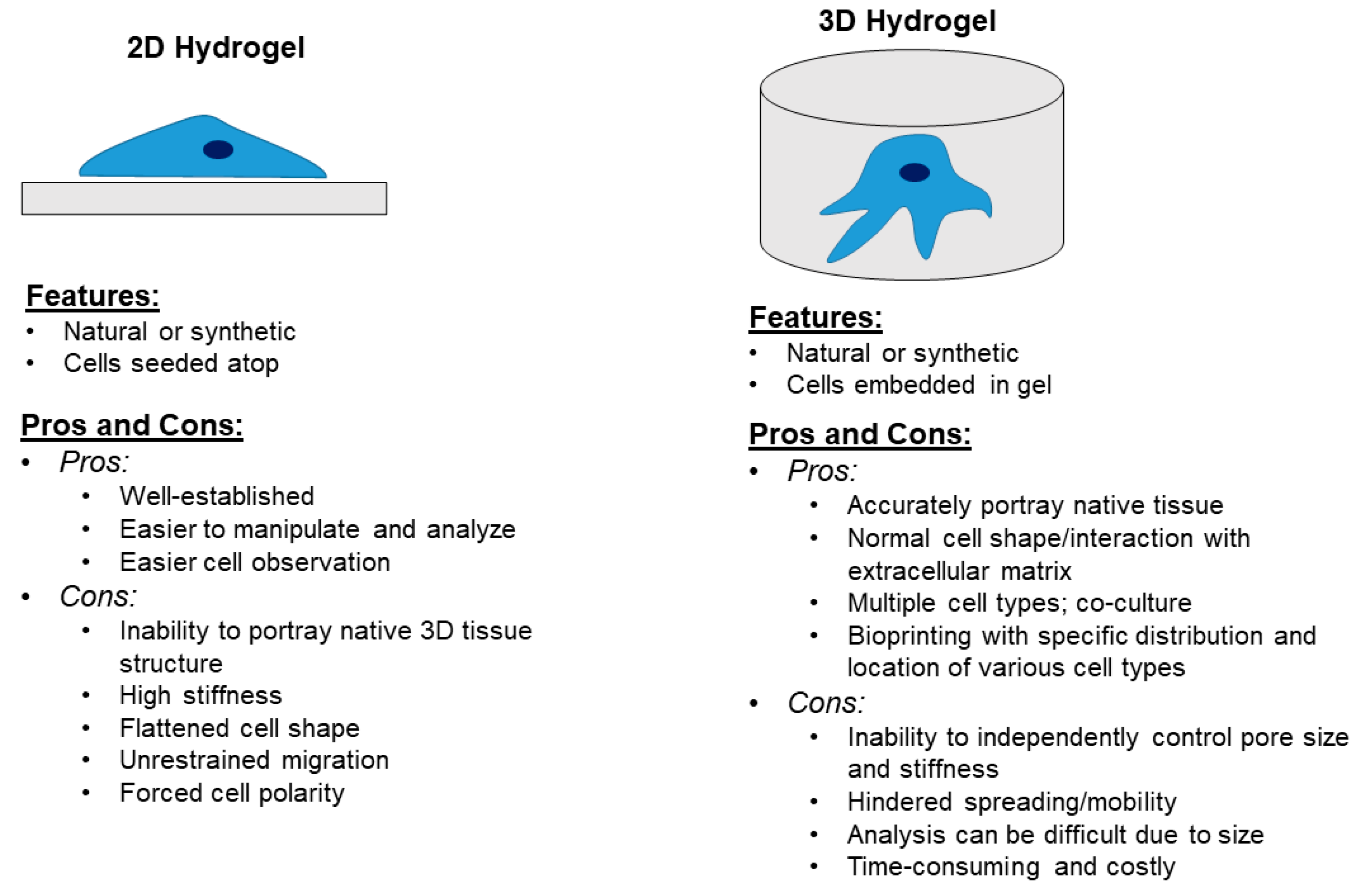
6. Models to Study the Bone and Bone Matrix
7. Concluding Remarks, Challenges, and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, E3028. [Google Scholar] [CrossRef]
- Miller, R.T. Mechanical properties of basement membrane in health and disease. Matrix Biol. 2017, 57–58, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Alford, A.; Kozloff, K.; Hankenson, K. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef]
- Gentili, C.; Cancedda, R. Cartilage and Bone Extracellular Matrix. Curr. Pharm. Des. 2009, 15, 1334–1348. [Google Scholar] [CrossRef]
- Bone Metastasis: Symptoms and Diagnosis. Available online: https://www.breastcancer.org/symptoms/types/recur_metast/metastic/bone (accessed on 11 December 2019).
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; American Society for Cell Biology: New York, NY, USA, 2015. [Google Scholar]
- Gong, J.K.; Arnold, J.S.; Cohn, S.H. Composition of trabecular and cortical bone. Anat. Rec. 1964, 149, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Marks, S.C., Jr.; Odgren, P.R. Structure and development of the skeleton. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; Volume 1, pp. 3–16. [Google Scholar]
- Rho, J.-Y.; Kuhn-Spearing, L.; Zioupos, P. Mechanical properties and the hierarchical structure of bone. Med. Eng. Phys. 1998, 20, 92–102. [Google Scholar] [CrossRef]
- Carter, D.R.; Hayes, W. The compressive behavior of bone as a two-phase porous structure. J. Bone Jt. Surg. 1977, 59A, 954–962. [Google Scholar] [CrossRef]
- Gibson, L.J. The mechanical behavior of cancellous bone. J. Biomech. 1985, 18, 317–328. [Google Scholar] [CrossRef]
- Price, J.S.; Oyajobi, B.O.; Russell, R.G. The cell biology of bone growth. Eur. J. Clin. Nutr. 1994, 48, S131–S149. [Google Scholar]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef]
- Yoneda, T.; Hiraga, T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem. Biophys. Res. Comm. 2005, 328, 679–687. [Google Scholar] [CrossRef]
- Guise, T.A.; Mundy, G.R. Cancer and bone. Endocr. Rev. 1998, 19, 18–54. [Google Scholar]
- Erler, J.; Bennewith, K.; Cox, T.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Tomlinson, R.E.; Silva, M.J. Skeletal blood flow in bone repair and maintenance. Bone Res. 2013, 1, 311–322. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Gilbert, S. Osteogenesis: The Development of Bones. In Developmental Biology, 6th ed.; Sinauer Associates: Sutherland, MA, USA, 2000. [Google Scholar]
- Robey, P.G.; Boskey, A. The Composition of Bone. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; American Society of Bone and Mineral Research: Washington, DC, USA, 2008; pp. 32–38. [Google Scholar]
- Yang, C.-M.; Chien, C.-S.; Yao, C.-C.; Hsiao, L.-D.; Huang, Y.-C.; Wu, C.-B. Mechanical Strain Induces Collagenase-3 (MMP-13) Expression in MC3T3-E1 Osteoblastic Cells. J. Biol. Chem. 2004, 279, 22158–22165. [Google Scholar] [CrossRef]
- Krishnan, V.; Vogler, E.A.; Sosnoski, D.; Mastro, A.M. In Vitro Mimics of Bone Remodeling and the Vicious Cycle of Cancer in Bone. J. Cell. Physiol. 2014, 229, 453–462. [Google Scholar] [CrossRef]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.L.; Corey, E.; Padalecki, S.; Suva, L.J.; et al. Basic Mechanisms Responsible for Osteolytic and Osteoblastic Bone Metastases. Clin. Can. Res. 2006, 12. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- Sottnik, J.L.; Dai, J.; Zhang, H.; Campbell, B.; Keller, E.T. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res. 2015, 75, 2151–2158. [Google Scholar] [CrossRef]
- Mullen, C.; Haugh, M.; Schaffler, M.; Majeska, R.; McNamara, L. Osteocyte differentiation is regulated by extracellular matrix stiffness and intercellular separation. J. Mech. Behav. Biomed. Mater. 2013, 28, 183–194. [Google Scholar] [CrossRef]
- Robling, A.G.; Turner, C.H. Mechanical signaling for bone modeling and remodeling. Cirt. Rev. Eukaryot. Expr. 2009, 19, 319–338. [Google Scholar] [CrossRef]
- Vezeridis, P.; Semeins, C.; Chen, Q.; Klein-Nulend, J. Osteocytes subjected to pulsating fluid flow regulate osteoblast proliferation and differentiation. Biochem. Biophys. Res. Commun. 2006, 348, 1082–1088. [Google Scholar] [CrossRef]
- Watanuki, M.; Sakai, A.; Sakata, T.; Tsurukami, H.; Miwa, M.; Uchida, Y.; Watanabe, K.; Ikeda, K.; Nakamura, T. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J. Bone Miner. Res. 2002, 17, 1015–1025. [Google Scholar] [CrossRef]
- Gu, G.; Mulari, M.; Peng, Z.; Hentunen, T.; Vaananen, H.K. Death of osteocytes turns off the inhibition of osteoclasts and triggers local bone resorption. Technol. Health Care 2005, 17, 49–56. [Google Scholar] [CrossRef]
- Wolff, J. Das Gesetz der Transformation der Knochen; Pro Business: Berlin, Germany, 2010. [Google Scholar]
- Klein-Nulend, J.; Bacabac, R.G.; Bakker, A.D. Mechanical loading and how it affects bone cells: The role of the osteocyte cytoskeleton in maintaining our skeleton. Eur. Cell Mater. 2012, 24, 278–291. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; van der Plas, A.; Semeins, C.; Ajubi, N.; Frangos, J.; Nijweide, P.J.; Burger, E.H. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995, 9, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable environment prepared by bone marrow-derived stromal cells. Proc. Nat. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor Necrosis Factor-a Induces Differentiation of and Bone Resorption by Osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastognesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Nat. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.; Kelley, M.J.; Dunstan, C.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Palokangas, H.; Mulari, M.; Vaananen, H.K. Endocytotic pathway from the basal plasma membrane to the ruffled border membrane in bone-resorbing osteoclasts. J. Cell Sci. 1997, 110, 1767–1780. [Google Scholar] [PubMed]
- Boyde, A.; Ali, N.N.; Jones, S.J. Resorption of dentine by isolated osteoclasts in vitro. Br. Dent. J. 1984, 156, 216–220. [Google Scholar] [CrossRef]
- Rumpler, M.; Wurger, T.; Roschger, P.; Zwettler, E.; Sturmlechner, I.; Altmann, P.; Fratzl, P.; Rogers, M.J.; Klaushofer, K. Osteoclasts on Bone and Dentin In Vitro: Mechanism of Trail Formation and Comparision of Resorption Behavior. Calcif Tissue Int. 2013, 93, 526–539. [Google Scholar] [CrossRef]
- Bernhardt, A.; Koperski, K.; Schumacher, M.; Gelinsky, M. Relevance of Osteoclast-specific enzyme activities in cell-based in vitro resorption assays. Eur. Cells Mater. 2017, 33, 28–42. [Google Scholar] [CrossRef]
- Helfrich, M.; Nesbitt, S.A.; Lakkakorpi, P.; Barnes, M.; Bodary, S.; Shankar, G.; Mason, W.; Mendrick, D.; Vaananen, H.K.; Horton, M. Beta 1 integrins and osteoclast function: Involvment in collagen recognition and bone resorption. Bone 1996, 19, 317–328. [Google Scholar] [CrossRef]
- Helfrich, M.; Nesbitt, S.A.; Dorey, E.; Horton, M. Rat osteoclasts adhere to a wide range of RGD (Arg-Gly-Asp) peptide-containing proteins, including the bone sialoproteins and fibronectin, via a beta 3 integrin. J. Bone Miner. Res. 1992, 7, 335–343. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Rodregues da Silva Sasso, G.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015. [Google Scholar] [CrossRef]
- Crane, J.; Cao, X. Bone marrow mesenchymal stem cells and TGF-B signaling in bone remodeling. J. Clin. Investig. 2014, 466–472. [Google Scholar] [CrossRef]
- Crane, J.; Xian, L.; Cao, X. Role of TGF-B signaling in coupling bone remodeling. Methods Mol. Biol. 2016, 287–300. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.; Peng, X.; Hu, J.; et al. TGF-B1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. TRENDS Mol. Med. 2006, 12. [Google Scholar] [CrossRef]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Nat. Acad. Sci. USA 1999, 96, 3540–3545. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakayamada, S.; Okada, Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr. Drug Targets 2005, 4, 325–328. [Google Scholar] [CrossRef]
- Josse, J.; Velard, F.; Gangloff, S.C. Staphylococcus aureus vs. Osteoblast: Relationship and consequences in osteomyelitis. Front. Cell Infect. Microbiol. 2015, 5. [Google Scholar] [CrossRef]
- Mastro, A.M.; Gay, C.V.; Welch, D.R.; Donahue, H.J.; Jewell, J.; Mercer, R.; DiGirolamo, D.; Chislock, E.M.; Guttridge, K. Breast cancer cells induce osteoblast apoptosis: A possible contributor to bone degradation. J. Cell Biochem. 2004, 91, 265–276. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Yoneda, T. Cellular and molecular mechanisms of breast and prostate cancer metastasis to bone. Eur. J. Cancer 1998, 34, 240–245. [Google Scholar] [CrossRef]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef]
- Farbod, K.; Nejadnik, M.R.; Jansen, J.A.; Leeuwenburgh, S.C.G. Interactions Between Inorganic and Organic Phases in Bone Tissue as a Source of Inspiration for Design of Novel Nanocomposites. Tissue Eng. 2014, 20, 173–188. [Google Scholar] [CrossRef]
- Pavalko, F.M.; Norvell, S.M.; Burr, D.B.; Turner, C.H.; Duncan, R.L.; Bidwell, J.P. A Model for mechanotransduction in bone cells: The load-bearing mechanosomes. J. Cell Biochem. 2003, 88, 104–112. [Google Scholar] [CrossRef]
- Paluch, E.K.; Nelson, C.M.; Biais, N.; Fabry, B.; Moeller, J.; Pruitt, B.L.; Wollnik, C.; Kudryasheva, G.; Rehfeldt, F.; Federle, W. Mechanotransduction: Use the force(s). BMC Biol. 2015, 13, 47. [Google Scholar] [CrossRef]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagen/apatite alignment impairs bone mechanical function in osteoblastic metastasis induce by prostate cancer. Bone 2017, 97, 83–93. [Google Scholar] [CrossRef]
- Burstein, A.H.; Zika, J.M.; Heiple, K.G.; Klein, L. Contribution of collagen and mineral to the elastic-plastic properties of bone. J. Bone Jt. Surg. Am. 1975, 57, 956–961. [Google Scholar] [CrossRef]
- Matsugaki, A.; Aramoto, G.; Ninomiya, T.; Sawada, H.; Hata, S.; Nakano, T. Abnormal arrangement of a collagen/apatite extracellular matrix orthogonal to osteoblast alignment is constructed by a nanoscale periodic surface structure. Biomaterials 2015, 37, 134–143. [Google Scholar] [CrossRef]
- Wang, J.; Jia, F.; Gilbert, T.; Woo, S. Cell orientation determines the alignment of cell-produced collagenous matrix. J. Biomech. 2003, 36, 97–102. [Google Scholar] [CrossRef]
- Shi, S.; Kirk, M.; Kahn, A.J. The role of type I collagen in the regulation of the osteoblast phenotype. J. Bone Miner. Res. 1996, 11, 1139–1145. [Google Scholar] [CrossRef]
- Fernandes, H.; Mentink, A.; Bank, R.; Stoop, R.; van Blitterswijk, C.; de Boer, J. Endogenous collagen influences differentiation of human multipotent mesenchymal stromal cells. Tissue Eng. 2010, 16, 1693–1702. [Google Scholar] [CrossRef]
- Ida, T.; Masaru, K.; Kitami, M.; Terajima, M.; Rosales Rocabado, J.; Akiba, Y.; Nagasawa, M.; Yamauchi, M.; Uoshima, K. Extracellular matrix with defective collagen cross-linking affects the differentiation of bone cells. PLoS ONE 2018, 13, e0204306. [Google Scholar] [CrossRef]
- Eyre, D.; Brickley-Parsons, D.; Glimcher, M. Predominance of type I collagen at the surface of avian articular cartilage. FEBS Lett. 1978, 85, 259–263. [Google Scholar] [CrossRef]
- Fonseca, H.; Moreira-Goncalves, D.; Coriolano, H.; Duarte, J. Bone quality: The determinants of bone strength and fragility. Sports Med. 2014, 44, 34–53. [Google Scholar] [CrossRef]
- Sekita, A.; Matsugaki, A.; Nakano, T. Disruption of collagne matrix alignment in osteolytic bone metastasis induce by breast cancer. Mater. Trans. 2016, 57, 2077–2082. [Google Scholar] [CrossRef]
- Liu, X.; Cao, M.; Palomares, M.; Wu, X.; Li, A.; Yan, W.; Fong, M.Y.; Chan, W.-C.; Wang, S.E. Metastatic breast cancer cells overexpress and secrete miR-218 to regulate type I collagen deposition by osteoblasts. Breast Can. Res. 2018, 20. [Google Scholar] [CrossRef]
- Barker, H.; Cox, T.; Erler, J. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.; Rumney, R.; Schoof, E.; Perryman, L.; Hoye, A.; Agrawal, A.; Bird, D.; Latif, N.; Forrest, H.; Evans, H.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Cawston, T.; Young, D. Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 2010, 339, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.W.; Fedarko, N. Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins. Connect. Tissue Res. 2003, 44 (Suppl. 1), 33–40. [Google Scholar] [CrossRef]
- Staines, K.; MacRae, V.; Farquharson, C. The importance of the SIBLING family of proteins on skeletal mineralisation and bone remodeling. J. Endocrinol. 2012, 214, 241–255. [Google Scholar] [CrossRef]
- Gordon, J.A.; Tye, C.; Sampaio, A.; Underhill, T.; Hunter, G.; Goldberg, H. Bone sialoprotein expression enhances osteoblast differentiation and matrix mineralization in vitro. Bone 2007, 41, 462–473. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity, and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Young, M.F.; Kerr, J.M.; Ibaraki, K.; Heegaard, A.M.; Robey, P.G. Structure, expression, and regulation of the major noncollagenous matrix proteins of bone. Clin. Orthop. Relat. Res. 1992, 275–294. [Google Scholar] [CrossRef]
- Gericke, A.; Qin, C.; Spevak, L.; Fujimoto, Y.; Butler, W.T.; Sorensen, E.S.; Boskey, A.L. Importance of Phosphorylation for Osteopontin Regulation of Biomineralization. Calcif Tissue Int. 2005, 77, 45–54. [Google Scholar] [CrossRef]
- Shevde, L.A.; Samant, R.S. Role of Osteopontin in the pathophysiology of cancer. Matrix Biol. 2014, 27, 131–141. [Google Scholar] [CrossRef]
- Hunter, G.K.; Hauschka, P.V.; Poole, A.R.; Rosenberg, L.C.; Goldberg, H. Nucleation and inhibition of hydroxapatite formation by mineralized tissue proteins. Biochem. J. 1996, 317. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Noda, M. Osteopontin Expression and Function: Role in Bone Remodeling. J. Cell. Biochem. Suppl. 1998, 30/31, 92–102. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Roelofsen, J.; Semeins, C.; Bronckers, A.L.; Burger, E.H. Mechanical stimulation of osteopontin mRNA expression and synthesis in bone cell cultures. J. Cell. Physiol. 1997, 170, 174–181. [Google Scholar] [CrossRef]
- Chellaiah, M.A.; Kizer, N.; Biswas, R.; Alvarez, U.; Strauss-Schoenberger, J.; Rifas, L.; Rittling, S.R.; Denhardt, D.T.; Hruska, K.A. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol. Biol. Cell 2003, 14, 173–189. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, G.; Sun, W.; Zhao, C.; Zhang, P.; Song, J.; Zhao, D.; Jin, X.; Li, Q.; Ling, S.; et al. CD44 deficiency inhibits unloading-induced cortical bone loss through downregulation of osteoclast activity. Sci. Rep. 2015, 5, 16124. [Google Scholar] [CrossRef]
- Kruger, T.E.; Miller, A.H.; Godwin, A.K.; Wang, J. Bone Sialoportein and Osteopontin in Bone Metastasis of Osteotropic Cancers. Crit Rev. Oncol. Hematol. 2014, 89, 330–341. [Google Scholar] [CrossRef]
- Carlinfante, G.; Vassiliou, D.; Svensson, O.; Wendel, M.; Heinegard, D.; Andersson, G. Differential expression of osteopontin and bone sialoprotein in bone metastasis of breast and prostate carcinoma. Clin. Exp. Metastasis 2003, 20, 437–444. [Google Scholar] [CrossRef]
- Zhang, J.H.; Tang, J.; Wang, J.; Ma, W.; Zheng, W.; Yoneda, T.; Chen, J. Over-expression of bone sialoprotein enhances bone metastasis of human breast cancer cells in a mouse model. Int. J. Oncol. 2003, 23, 1043–1046. [Google Scholar] [CrossRef]
- Khan, S.; Cook, A.; Kappil, M.; Gunthert, U.; Chambers, A.F.; Tuck, A.B.; Denhardt, D.T. Enhanced cell surface CD44 variant (v6, v9) expression by osteopontin in breast cacner epithelial cells facilitates tumor cell migration: Novel post-transcriptional, post-translational regulation. Clin. Exp. Metastasis 2005, 22, 663–673. [Google Scholar] [CrossRef]
- Khodavirdi, A.; Song, Z.; Yang, S.; Zhong, C.; Wang, S.; Wu, H.; Pritchard, C.; Nelson, P.; Roy-Burman, P. Increased expression of osteopontin contributes to the progression of prostate cancer. Cancer Res. 2006, 66, 883–888. [Google Scholar] [CrossRef]
- Wai, P.Y.; Kuo, P.C. Osteopontin: Regulation in tumor metastasis. Cancer Metastasis Rev. 2008, 27, 103–118. [Google Scholar] [CrossRef]
- Robey, P.G. Bone matrix proteoglycans and glycoproteins. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: San Diego, CA, USA, 2002; Volume 1, pp. 225–238. [Google Scholar]
- Nikitovic, D.; Aggelidakis, J.; Young, M.F.; Iozzo, R.; Karamanos, N.K.; Tzanakakis, G.N. The Biology of Small Leucine-rich Proteoglycans in Bone Pathophysiology. J. Biol. Chem. 2012, 287, 33926–33933. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R. Decorin: A guardian from the matrix. Am. J. Pathol. 2012, 181, 380–387. [Google Scholar] [CrossRef]
- Lifshitz, V.; Frenkel, D. Chapter 25-TGF-B. In Handbook of Biologically Active Peptides (Second Edition); Kastin, A.J., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 1647–1653. [Google Scholar]
- Massague, J.; Blain, S.W.; Lo, R.S. TGFB signaling in growth control, cancer, and heritable disorders. Cell 2000, 103, 295–309. [Google Scholar] [CrossRef]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.; Heinegard, D.; Twardzik, D.; Border, W.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor B. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef]
- Schonherr, E.; Broszat, M.; Brandan, E.; Bruckner, P.; Kresse, H. Decorin core protein fragment Leu-155-Val-260 interacts with TGF-B but does not compete for decorin binding to type I collagen. Arch. Biochem. Biophys. 1998, 355, 241–248. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Kodama, Y.; Matsumoto, T. Bone Matrix Decorin Binds Transforming Growth Factor-B and Enhances its Bioactivitiy. J. Biol. Chem. 1994, 269, 32634–32638. [Google Scholar]
- Nemani, N.; Santo, L.; Eda, H.; Cirstea, D.; Mishima, Y.; Patel, C.; O’Donnell, E.; Yee, A.; Raje, N. Role of Decorin in Multiple Myeloma (MM) Bone Marrow Microenvironment. J. Bone Miner. Res. 2015, 30, 465–470. [Google Scholar] [CrossRef]
- Zhang, W.; Ge, Y.; Cheng, Q.; Zhang, Q.; Fang, L.; Zheng, J. Decorin is a pivotal effector in the extracellular matrix and tumour microenvironment. Oncotarget 2018, 9, 5480–5491. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef]
- Globus, R.; Doty, S.; Lull, J.; Holmuhamedov, E.; Humphries, M.; Damsky, C. Fibronectin is a survival factor for differentiated osteoblasts. J. Cell Sci. 1998, 111, 1385–1393. [Google Scholar]
- Moursi, A.M.; Damsky, C.H.; Lull, J.; Zimmerman, D.; Doty, S.; Aota, S.; Globus, R. Fibronectin regulates calvarial osteoblast differentiation. J. Cell Sci. 1996, 109, 1369–1380. [Google Scholar]
- Faia-Torres, A.B.; Gorens, T.; Ihalainen, T.O.; Guimond-Lischer, S.; Charnley, M.; Rottmar, M.; Maniura-Weber, K.; Spencer, N.D.; Reis, R.L.; Textor, M.; et al. Regulation of Human Mesenchymal Stem Cell Osteogenesis by Specific Surface Density of Fibronectin: A Gradient Study. ACS Appl. Mater. Interfaces 2015, 7, 2367–2375. [Google Scholar] [CrossRef]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 495–501. [Google Scholar] [CrossRef]
- Kubow, K.E.; Vukmirovic, R.; Zhe, L.; Klotzsch, E.; Smith, M.L.; Gourdon, D.; Luna, S.; Vogel, V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Leikina, E.; Mertts, M.V.; Kuznetsova, N.; Leikin, S. Type I collagen is thermally unstable at body temperature. Proc. Natl. Acad. Sci. USA 2002, 99, 1314–1318. [Google Scholar] [CrossRef]
- Wang, K.; Seo, B.R.; Fischbach, C.; Gourdon, D. Fibronectin Mechanobiology Regulates Tumorigenesis. Cell Mol. Bioeng. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Stenman, S.; Vaheri, A. Fibronectin in human solid tumors. Int. J. Cancer 1981, 27, 427–435. [Google Scholar] [CrossRef]
- Wang, K.; Wu, F.; Seo, B.R.; Fischbach, C.; Chen, W.; Hsu, L.; Gourdon, D. Breast cancer cells alter the dynamics of stromal fibronectin-collagen interactions. Matrix Biology 2017, 60–61, 86–95. [Google Scholar] [CrossRef]
- Radisky, E.; Raeeszadeh-Sarmazdeh, M.; Radisky, D. Therapeutic Potential of Matrix Metalloproteinase Inhibition in Breast Cancer. J. Cell Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef]
- Kusano, K.; Miyaura, C.; Inada, M.; Tamura, T.; Ito, A.; Nagase, H.; Kamoi, K.; Suda, T. Regulation of matrix metalloproteinases (MMP-2, -3, -9, -13) by interleukin-1 and interleukin-6 in mouse calvaria: Association of MMP induction with bone resorption. Endocrinology 1998, 139, 1338–1345. [Google Scholar] [CrossRef]
- MacDougall, J.; Matrisian, L. Contributions of tumor and stromal matrix metalloproteinases to tumor progression, invasion, and metastasis. Cancer Metastasis Rev. 1995, 14, 351–362. [Google Scholar] [CrossRef]
- Meikle, M.; Bord, S.; Hembry, R.; Compston, J.; Croucher, P.I.; Reynolds, J. Human osteoblasts in culture synthesize collagenase and other matrix metalloproteinases in response to osteotropic hormones and cytokines. J. Cell Sci. 1992, 103, 1093–1099. [Google Scholar]
- Tezuka, K.; Nemoto, K.; Tezuka, Y.; Sato, T.; Ikeda, Y.; Kobori, M.; Kawashima, H.; Eguchi, H.; Hakeda, Y.; Kumegawa, M. Identification of matrix metalloproteinase 9 in rabbit osteoclasts. J. Biol. Chem. 1994, 269, 15006–15009. [Google Scholar]
- Fridman, R.; Toth, M.; Pena, D.; Mobashery, S. Activation of progelatinase B (MMP-9) by gelatinase A (MMP-2). Cancer Res. 1995, 55, 2548–2555. [Google Scholar]
- Toth, M.; Chvyrkova, I.; Bernardo, M.; Hernandez-Barrantes, S.; Fridman, R. Pro-MMP-9 activtion by the MT1-MMP/MMP_2 axis and MMP-3: Role of TIMP-2 and plasma membranes. Biochem. Biophys. Res. Commun. 2003, 308, 386–395. [Google Scholar] [CrossRef]
- Knauper, V.; Smith, B.; Lopez-Otin, C.; Murphy, G. Activation of progelatinase B (pro-MMP-9) by active collagenase-3 (MMP-13). Eur. J. Biochem. 1997, 248, 369–373. [Google Scholar] [CrossRef]
- Vu, T.; Shipley, J.; Bergers, G.; Berger, J.; Helms, J.; Hanahan, D.; Shapiro, S.; Senior, R.; Werb, Z. MMP-9/Gelatinase B is a Key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertropic Chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef]
- Manduca, P.; Castagnino, A.; Lombardini, D.; Marchisio, S.; Soldano, S.; Ulivi, V.; Zanotti, S.; Garbi, C.; Ferrari, N.; Palmieri, D. Role of MT1-MMP in the osteogenic differentiation. Bone 2009, 44, 251–265. [Google Scholar] [CrossRef]
- Tauro, M.; Lynch, C.C. Cutting to the Chase: How Matrix Metalloproteinase-2 Activity Controls Breast-Cancer-to-Bone Metastasis. Cancers (Basel) 2018, 10, 185. [Google Scholar] [CrossRef]
- Tauro, M.; Shay, G.; Sansil, S.; Laghezza, A.; Tortorella, P.; Neuger, A.; Soliman, H.; Lynch, C.C. Bone-Seeking Matrix Metalloproteinase-2 Inhibitors Prevent Bone Metastatic Breast Cancer Growth. Mol. Cancer Ther. 2017, 16, 494–505. [Google Scholar] [CrossRef]
- Perentes, J.; Kirkpatrick, N.; Nagano, S.; Smith, E.; Shaver, C.; Sgroi, D.; Garkavtsev, I.; Munn, L.L.; Jain, R.; Boucher, Y. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastatsis in triple-negative mammary tumors. Caner Res. 2011, 71, 4527–4538. [Google Scholar] [CrossRef]
- Bruni-Cardoso, A.; Johnson, L.C.; Vessella, R.L.; Peterson, T.E.; Lynch, C.C. Osteoclast-Derived Matrix Metalloproteinase-9 Directly Affects Angiogenesis in the Prostate Tumor-Bone Microenvironment. Mol. Cancer Res. 2010, 8, 459–470. [Google Scholar] [CrossRef]
- Weiner, S.; Traub, W. Bone structure: From angstroms to microns. FASEB J. 1992, 6, 879–885. [Google Scholar] [CrossRef]
- Nakano, T.; Kaibara, K.; Tabata, Y.; Nagata, N.; Enomoto, S.; Marukawa, E.; Umakoshi, Y. Unique alignment and texture of biological apatite crystallites in typical calcified tissues analyzed by microbeam x-ray diffractometer system. Bone 2002, 31, 479–487. [Google Scholar] [CrossRef]
- Nakano, T.; Kaibara, K.; Ishimoto, T.; Tabata, Y.; Umakoshi, Y. Biological apatite (BAp) cystallographic orientation and texture as a new index for assessing the microstructure and function of bone regnerated by tissue engineering. Bone 2012, 51, 741–747. [Google Scholar] [CrossRef]
- Kimura, Y.; Matsugaki, A.; Sekita, A.; Nakano, T. Alteration of osteoblast arrangment via direct attack by cancer cells: New insights into bone metastasis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef]
- Macedo, F.; Ladeira, K.; Pinho, F.; Saraiva, N.; Bonito, N.; Pinto, L.; Goncalves, F. Bone Metastases: An Overview. Oncol. Rev. 2017, 11, 321. [Google Scholar] [CrossRef]
- Mastro, A.M.; Gay, C.V.; Welch, D.R. The skeleton as a unique environment for breast cancer cells. Clin. Exp. Metastasis 2003, 20, 275–284. [Google Scholar] [CrossRef]
- Kusumbe, A.P. Vascular niches for disseminated tumour cells in bone. J. Bone Oncol. 2016, 5, 112–116. [Google Scholar] [CrossRef][Green Version]
- Hensel, J.; Thalmann, G.N. Biology of bone metastases in prostate cancer. Urology 2016. [Google Scholar] [CrossRef]
- Liede, A.; Jerzak, K.J.; Hernandez, R.K.; Wade, S.W.; Sun, P.; Narod, S.A. The incidence of bone metastasis after early-stage breast cancer in canada. Breast Can. Res. Treat. 2016, 156, 587–595. [Google Scholar] [CrossRef]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef]
- Charhon, S.A.; Chapuy, M.C.; Delvin, E.E.; Valentin-Opran, A.; Edouard, C.M.; Meunier, P.J. Histomorphometric analysis of sclerotic bone metastases from prostatic carcinoma special reference to osteomalacia. Cancer 1983, 51, 918–924. [Google Scholar] [CrossRef]
- Roodman, G.D.; Silbermann, R. Mechanisms of osteolytic and osteoblastic skeletal lesions. BoneKEy Rep. 2015, 4, 753. [Google Scholar]
- Schneider, A.; Kalikin, L.M.; Mattos, A.C.; Keller, E.T.; Allen, M.J.; Pienta, K.J.; McCauley, L.K. Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology 2005, 146, 1727–1736. [Google Scholar] [CrossRef]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-B in bone metastasis: Novel therapeutic perspectives. BoneKEy Rep. 2012, 1. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef]
- Meng, X.; Ark, A.V.; Daft, P.; Woodford, E.; Wang, J.; Madaj, Z.; Li, X. Loss of TGF-B signaling in osteoblasts increases basic-FGF and promotes prostate cancer bone metastasis. Cancer Lett. 2018, 418, 109–118. [Google Scholar] [CrossRef]
- Chen, N.; Ye, X.-C.; Chu, K.; Navone, N.M.; Sage, E.H.; Yu-Lee, L.-Y.; Logothetis, C.J.; Lin, S.-H. A secreted isoform of ErbB3 promotes osteonectin expression in bone and enhances the invasiveness of prostate cancer cells. Cancer Res. 2007, 67, 6544–6548. [Google Scholar] [CrossRef]
- Said, N.; Frierson, H., Jr.; Chernauskas, D.; Motamed, K.; Theodorescu, D. The role of SPARC in the TRAMP model of prostate carciongenesis and progression. Oncogene 2009, 28, 3487–3498. [Google Scholar] [CrossRef]
- Wong, S.; Crowley, D.; Bronson, R.; Hynes, R. Analysis of the role of endogenous SPARC in mouse models of prostate and breast cancer. Clin. Exp. Metastasis 2008, 25, 109–118. [Google Scholar] [CrossRef]
- Kapinas, K.; Lowther, K.; Kessler, C.; Tilbury, K.; Lieberman, J.R.; Tirnauer, J.; Campagnola, P.; Delany, A. Bone Matrix Osteonectin Limits Prostate Cancer Cell Growth and Survival. Matrix Biol. 2012, 31, 299–307. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Phadke, P.A.; Mercer, R.R.; Harms, J.F.; Yujiang, J.; Frost, A.R.; Jewell, J.L.; Bussard, K.M.; Nelson, S.; Moore, C.; Kappes, J.C.; et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin. Cancer Res. 2006, 12, 1431–1440. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef]
- Ubellacker, J.M.; Baryawno, N.; Severe, N.; DeCristo, M.J.; Sceneay, J.; Hutchinson, J.N.; Haider, M.T.; Rhee, C.S.; Qin, Y.; Gregory, W.M.; et al. Modulating Bone Marrow Hematopoietic Lineage Potential to Prevent Bone Metastasis in Breast Cancer. Cancer Res. 2018, 78, 5300–5314. [Google Scholar] [CrossRef]
- Krishnan, V.; Dhurjati, R.; Vogler, E.A.; Mastro, A.M. Osteogenesis in vitro: From pre-osteoblasts to osteocytes: A contribution from the Osteobiology Research Group, The Pennsylvania State University. In Vitro Cell Dev. Biol. Anim. 2010, 46, 28–35. [Google Scholar] [CrossRef]
- Ottewell, P.D. The role of osteblasts in bone metastasis. J. Bone Oncol. 2016, 5, 124–127. [Google Scholar] [CrossRef]
- Hoye, A.; Erler, J. Structural ECM components in the pre-metastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016, 310, C955–C967. [Google Scholar]
- Suarez-Cuervo, C.; Merrell, M.A.; Watson, L.; Harris, K.W.; Rosenthal, E.L.; Vaananen, H.K.; Selander, K.S. Breast cancer cells with inhibition of p38alpha have decreased MMP-9 activity and exhibit decreased bone metastasis in mice. Clin. Exp. Metastasis 2004, 21, 525–533. [Google Scholar] [CrossRef]
- Christensen, J.G.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by Cathepsin K. BMC Res. Notes 2015, 8. [Google Scholar] [CrossRef]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Nat. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar] [CrossRef]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Muntro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and B-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2014, 19, 776–791. [Google Scholar] [CrossRef]
- Page, J.M.; Merkel, A.R.; Ruppender, N.S.; Guo, R.; Dadwal, U.C.; Cannonier, S.A.; Basu, S.; Guelcher, S.A.; Sterling, J.A. Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin B3 and TGF-B receptor type II. Biomaterials 2015, 64, 33–44. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in Multiple Myeloma Progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Kyle, R.A.; Rajkumar, S.V. Plasma cell disorders. In Cecil Textbook of Medicine, 22nd ed.; Goldman, L., Ausiello, D.A., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2004; pp. 1184–1195. [Google Scholar]
- Kyle, R.A.; Rajkumar, S.V. Multiple Myeloma. NEJM 2004, 351, 1860–1873. [Google Scholar] [CrossRef]
- Wu, D.; Guo, X.; Su, J.; Chen, R.; Berenzon, D.; Guthold, M.; Bonin, K.; Zhao, W.; Zhou, X. CD138-negative myeloma cells regulate mechanical properties of bone marrow stromal cells through SDF-1/CXCR4/AKT signaling pathway. Biochem. Biophys. Acta 2014, 1853, 338–347. [Google Scholar] [CrossRef][Green Version]
- Matsui, W.; Huff, C.; Wang, Q.; Malehorn, M.; Barber, J.; Tanhehco, Y.; Smith, B.; Civin, C.; Jones, R. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar] [CrossRef]
- Reghunathan, R.; Bi, C.; Liu, S.; Loong, K.; Chung, T.; Huang, G.; Chng, W. Clonogenic multiple myeloma cells have shared stemness signature associated with patient survival. Oncotarget 2013, 4, 1230–1240. [Google Scholar] [CrossRef]
- Alsayed, Y.; Ngo, H.; Runnels, J.; Leleu, X.; Singha, U.; Pitsillides, C.; Spencer, J.; Kimlinger, T.; Ghobrial, J.; Jia, X.; et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood 2007, 109, 2708–2717. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Sun, Y.-X.; Song, W.; Nor, J.E.; Wang, C.Y.; Taichman, R.S. Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cell Signal. 2005, 17, 1578–1592. [Google Scholar] [CrossRef]
- Vallet, S.; Pozzi, S.; Patel, K.; Vaghela, N.; Fulciniti, M.T.; Veiby, P.; Hideshima, T.; Santo, L.; Cirstea, D.; Scadden, D.T.; et al. A Novel Role for CCL3 (MIP-1 alpha) in Myeloma-induced Bone Disease via Osteocalcin Downregulation and Inhibition of Osteoblast Function. Leukemia 2011, 25, 1174–1181. [Google Scholar] [CrossRef]
- Lentzsch, S.; Gries, M.; Janz, M.; Bargou, R.; Dorken, B.; Mapara, M. Macrophage inflammatory protein 1-alpha (MIP-1-alpha) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood 2003, 101, 3568–3573. [Google Scholar] [CrossRef]
- Popper, H.H. Progression and metastasis of lung cancer. Cancer Metastasis Rev. 2016, 35, 75–91. [Google Scholar] [CrossRef]
- Catena, R.; Luis-Ravelo, D.; Antón, I.; Zandueta, C.; Salazar-Colocho, P.; Larzábal, L.; Calvo, A.; Lecanda, F. PDGFR Signaling Blockade in Marrow Stroma Impairs Lung Cancer Bone Metastasis. Cancer Res. 2011, 71, 164. [Google Scholar] [CrossRef]
- Baker, J.; Falconer, A.M.D.; Wilkinson, D.J.; Europe-Finner, G.N.; Litherland, G.J.; Rowan, A.D. Protein kinase D3 modulates MMP1 and MMP13 expression in human chondrocytes. PLoS ONE 2018, 13, e0195864. [Google Scholar] [CrossRef]
- Vicent, S.; Luis-Ravelo, D.; Antón, I.; García-Tuñón, I.; Borrás-Cuesta, F.; Dotor, J.; De Las Rivas, J.; Lecanda, F. A Novel Lung Cancer Signature Mediates Metastatic Bone Colonization by a Dual Mechanism. Cancer Res. 2008, 68, 2275. [Google Scholar] [CrossRef]
- Tang, C.-H.; Tan, T.-W.; Fu, W.-M.; Yang, R.-S. Involvement of matrix metalloproteinase-9 in stromal cell-derived factor-1/CXCR4 pathway of lung cancer metastasis. Carcinogenesis 2007, 29, 35–43. [Google Scholar] [CrossRef]
- Sugiura, H.; Yamada, K.; Sugiura, T.; Hida, T.; Mitsudomi, T. Predictors of surivival in patients with bone metastasis of lung cancer. Clin. Ortho. Relat. Res. 2008, 466, 729–736. [Google Scholar] [CrossRef]
- Coleman, R. Metastatic bone disease: Clinical features, pathopysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Hill, C.A. Bronchioloalveolar carcinoma: A review. Radiology 1984, 150, 15–20. [Google Scholar] [CrossRef]
- Kang, E.J.; Lee, S.Y.; Kim, H.J.; Min, K.H.; Hur, G.Y.; Shim, J.J.; Kang, K.H.; Oh, S.C.; Seo, J.H.; Lee, S.Y.; et al. Prognostic Factors and Skeletal-related Events in Patients with Small Cell Lung Cancer with Bone Metastases at the Time of Diagnosis. Oncology 2016, 90, 103–111. [Google Scholar] [CrossRef]
- Lang, J.; Zhao, Q.; He, Y.; Yu, X. Bone turnover markers and novel biomarkers in lung cancer bone metastases. Biomarkers 2018, 23, 518–526. [Google Scholar] [CrossRef]
- Papotti, M.; Kalebic, T.; Volante, M.; Chiusa, L.; Bacillo, E.; Cappia, S.; Lausi, P.; Novello, S.; Borasio, P.; Scagliotti, G.V. Bone sialoprotein is predictive of bone metastases in resectable non-small-cell lung cancer: A retrospective case-control study. J. Clin. Oncol. 2006, 24, 4814–4824. [Google Scholar] [CrossRef]
- He, J.J.; Zhi, K.; Liu, G.F. Predicitive value of serum bone sialoprotein in patients with bone metastasis of non-small cell lung cancer. Onkologie 2011, 34, 584–588. [Google Scholar] [CrossRef]
- Brown, J.E.; Cook, R.J.; Major, P.; Lipton, A.; Saad, F.; Smith, M.; Lee, K.A.; Zheng, M.; Hei, Y.J.; Coleman, R.E. Bone turnover markers as predictors of skeletal complications in prostate cancer, lung cancer, and other solid tumors. J. Natl. Cancer Inst. 2005, 97, 59–69. [Google Scholar] [CrossRef]
- Costa, L.; Demers, L.M.; Gouveia-Oliveira, A.; Schaller, J.; Costa, E.B.; de Moura, M.C.; Lipton, A. Prospective evaluation of the peptide-bound collagen type I cross-links N-telopeptide and C-telopeptide in predicting bone metastases status. J. Clin. Oncol. 2002, 20, 850–856. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, S.; Su, Y.; Chen, H.; Wang, S.; Sun, B.; Zhang, L. Serum cross-linked N-telopeptide of type I collagen as a biomarker of bone metastases for patients with lung cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2018, 11, 12864–12869. [Google Scholar]
- Liu, B.; Zhao, Y.; Yuan, J.; Zeng, L.; Sun, R.; Meng, X.; Yang, S. Elevated N-telopeptide as a potential diagnostic marker for bone metastasis in lung cancer: A meta-analysis. PLoS ONE 2017, 12, e0187860. [Google Scholar] [CrossRef]
- Valencia, K.; Ormazabal, C.; Zandueta, C.; Luis-Ravelo, D.; Anton, I.; Pajares, M.J.; Agorreta, J.; Montuenga, L.M.; Martinez-Canarias, S.; Leitinger, B.; et al. Inhibition of Collagen Receptor Discoidin Domain Receptor-1 (DDR1) Reduces Cell Survival, Homing, and Colonization in Lung Cancer Bone Metastasis. Clin. Cancer Res. 2012, 18, 969–980. [Google Scholar] [CrossRef]
- Hu, Z.; Lin, D.; Yuan, J.; Xiao, T.; Zhang, H.; Sun, W.; Han, N.; Ma, Y.; Di, X.; Gao, M.; et al. Overexpression of osteopontin is associated with more aggressive phenotypes in human non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 4646–4652. [Google Scholar] [CrossRef]
- Kothari, A.N.; Arffa, M.L.; Chang, V.; Blackwell, R.H.; Syn, W.K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin-A Master Regulator of Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 39. [Google Scholar] [CrossRef]
- Shojaei, F.; Scott, N.; Kang, X.; Lappin, P.B.; Fitzgerald, A.A.; Karlicek, S.; Simmons, B.H.; Wu, A.; Lee, J.H.; Berggvist, S.; et al. Osteopontin induces growth of metastatic tumors in a preclinical model of non-small lung cancer. J. Exp. Clin. Cancer Res. 2012, 31. [Google Scholar] [CrossRef]
- Roman, J.; Ritzenthaler, J.D.; Roser-Page, S.; Sun, X.J.; Han, S. a5B1-integrin Expression is Essential for Tumor Progression in Experimental Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2010, 43, 684–691. [Google Scholar] [CrossRef]
- Russell, R.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef]
- Body, J.J. Rationale for the use of bisphosphonates in osteoblastic and osteolytic bone lesions. Breast 2003, 12 (Suppl. 2), S37–44. [Google Scholar] [CrossRef]
- Holen, I.; Coleman, R.E. Bisphosphonates as treatment of bone metastases. Curr. Pharm. Des. 2010, 16, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Selvaggi, G.; Scagliotti, G.V. Management of bone metastases in cancer: A review. Clin. Rev. Oncol. Hematol. 2005, 56, 365–378. [Google Scholar] [CrossRef] [PubMed]
- von Moos, R.; Costa, L.; Gonzalez-Suarez, E.; Terpos, E.; Niepel, D.; Body, J.J. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat. Rev. 2019, 76, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Gralow, J.; Tripathy, D. Managing Metastatic Bone Pain: The Role of Bisphosphonates. J. Pain Sym. Man. 2007, 33, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Gdowski, A.S.; Ranjan, A.; Vishwanatha, J.K. Current concepts in bone metastasis, contemporary therapeutic strategies, and ongoing clinical trials. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Benford, H.L.; McGowan, N.W.; Helfrich, M.H.; Nuttall, M.E.; Rogers, M.J. Visualization of bisphosphonate-induced caspase-3 activity in apoptotic osteoclasts in vitro. Bone 2001, 28, 465–473. [Google Scholar] [CrossRef]
- Rogers, M.J.; Gordon, S.; Benford, H.L.; Coxon, F.P.; Luckman, S.P.; Monkkonen, J.; Frith, J.C. Cellular and molecular mechanisms of action of bisphosphonates. Cancer 2000, 88, 2961–2978. [Google Scholar] [CrossRef]
- Lipton, A.; Theriault, R.L.; Hortobagyi, G.N.; Simeone, J.; Knight, R.D.; Mellars, K.; Reitsma, D.J.; Heffernan, M.; Seaman, J.J. Pamidronate prevents skeletal complications and is effective palliative treatment in women with breast carcinoma and osteolytic bone metastases. Cancer 2000, 88, 1082–1090. [Google Scholar] [CrossRef]
- Rouach, V.; Goldshtein, I.; Wolf, I.; Catane, R.; Chodick, G.; Iton, A.; Stern, N.; Cohen, D. Exposure to alendronate is associated with a lower risk of bone metastases in osteoporotic women with early breast cancer. J. Bone Oncol. 2018, 12, 91–95. [Google Scholar] [CrossRef]
- Bock, O.; Felsenberg, D. Bisphosphonates in the management of postmenopausal osteoporosis-optimizing efficacy in clinical practice. Clin. Interv. Aging 2008, 3, 279–297. [Google Scholar] [CrossRef]
- Rosen, L.S.; Gordon, D.H.; Dugan Jr., W.; Major, P.; Eisenberg, P.D.; Provencher, L.; Kaminski, M.; Simeone, J.; Seaman, J.; Chen, B.L.; et al. Zoledronic acid is superior to pamidronate for the treatment of bone metastases in breast carcinoma patients with at least one osteolytic lesion. Cancer 2004, 100, 36–43. [Google Scholar] [CrossRef]
- Barrett-Lee, P.; Casbard, A.; Abraham, J.; Hood, K.; Coleman, R.; Simmonds, P.; Timmins, H.; Wheatley, D.; Grieve, R.; Griffiths, G.; et al. Oral ibandronic acid versus intravenous zoledronic acid in treatment of bone metastases from breast cancer: A randomized, open lable, non-inferiority phase 3 trial. Lancet Oncol. 2014, 15, 114–122. [Google Scholar] [CrossRef]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Zheng, M.; et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J. Natl. Cancer Inst. 2004, 96, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Biskup, E.; Cai, F.; Vetter, M. Bone targeted therapies in advanced breast cancer. Swiss Med. Wkly. 2017, 147, w14440. [Google Scholar]
- Body, J.J. Denosumab for the management of bone disease in patients with solid tumors. Expert Rev. Anticancer Ther. 2012, 12, 307–322. [Google Scholar] [CrossRef]
- Lewiecki, E.M. Role of sclerostin in bone and cartilage and its potential as a therapeutic target in bone diseases. Ther. Adv. Musculoskelet Dis. 2014, 6, 48–57. [Google Scholar] [CrossRef]
- Ominsky, M.S.; Boyd, S.K.; Varela, A.; Jolette, J.; Felx, M.; Doyle, N.; Mellal, N.; Smith, S.Y.; Locher, K.; Buntich, S.; et al. Romosozumab Improves Bone Mass and Strength While Maintaining Bone Quality in Ovariectomized Cynomolgus Monkeys. J. Bone Miner. Res. 2017, 32, 788–801. [Google Scholar] [CrossRef]
- Mendoza-Villanueva, L.; Zeef, L.; Shore, P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFbeta-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. 2011, 13, R106. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef]
- Hesse, E.; Schroder, S.; Brandt, D.; Pamperin, J.; Saito, H.; Taipaleenmaki, H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight 2019, 4, e125543. [Google Scholar] [CrossRef]
- Zhu, M.; Liu, C.; Li, S.; Zhang, S.; Yao, Q.; Song, Q. Sclerostin induced tumor growth, bone metastasis and osteolysis in breast cancer. Sci. Rep. 2017, 7, 11399. [Google Scholar] [CrossRef]
- Paszek, M.; Zahir, N.; Johnson, K.; Lakins, J.; Rozenberg, G.; Gefen, A.; Reinhart-King, C.; Margulies, S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Caliari, S.R.; Burdick, J.A. A Practical Guide to Hydrogels for Cell Culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef]
- Cassereau, L.; Miroshnikova, Y.; Ou, G.; Lakins, J.; Weaver, V.M. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J. Biotechnol. 2015, 193, 66–69. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef]
- Seib, F.P.; Berry, J.E.; Shiozawa, Y.; Taichamn, R.S.; Kaplan, D.L. Tissue engineering a surrogate niche for metastatic cancer cells. Biomaterials 2015, 51, 313–319. [Google Scholar] [CrossRef]
- Carpenter, R.A.; Kwak, J.-G.; Peyton, S.R.; Lee, J. Implantable pre-metastatic niches for the study of the microenvironmental regulation of disseminated human tumour cells. Nat. Biomed. Eng. 2018. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolb, A.D.; Bussard, K.M. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers 2019, 11, 1020. https://doi.org/10.3390/cancers11071020
Kolb AD, Bussard KM. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers. 2019; 11(7):1020. https://doi.org/10.3390/cancers11071020
Chicago/Turabian StyleKolb, Alexus D., and Karen M. Bussard. 2019. "The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases" Cancers 11, no. 7: 1020. https://doi.org/10.3390/cancers11071020
APA StyleKolb, A. D., & Bussard, K. M. (2019). The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers, 11(7), 1020. https://doi.org/10.3390/cancers11071020