The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer


Abstract
:1. Introduction
2. Breast Cancer Stem Cells
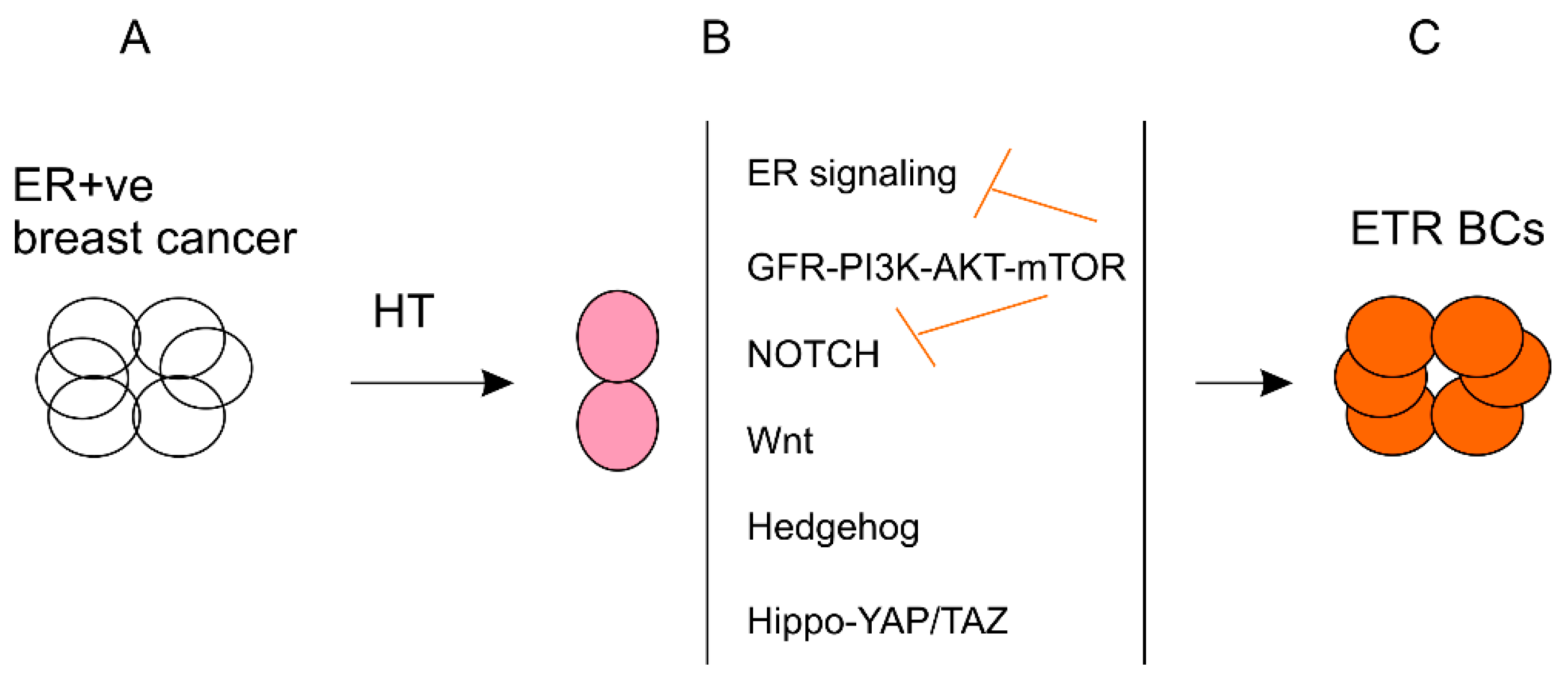
3. Association of Endocrine Therapy Resistance with BCSC Enrichment
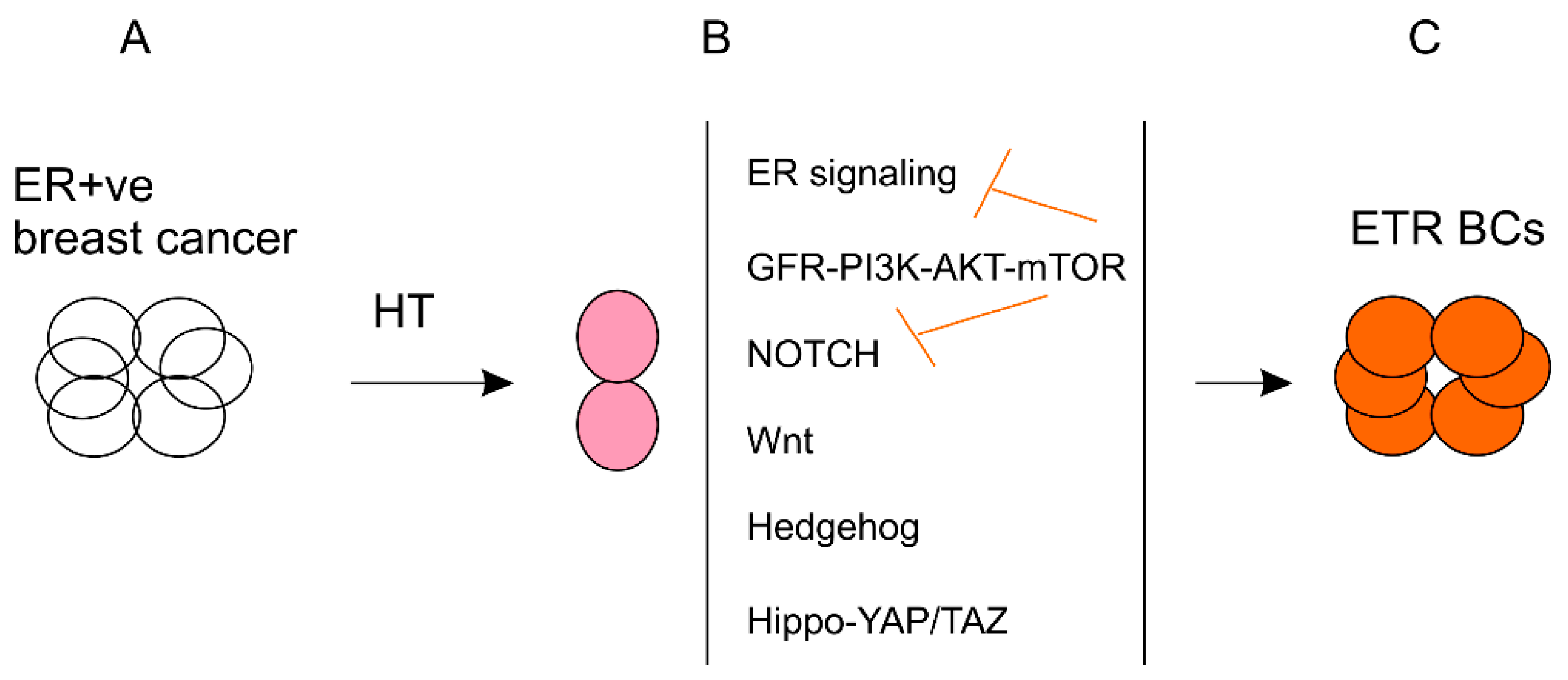
4. Mechanisms Underlying BCSC Enrichment Following the Development of Endocrine Resistance
4.1. The Relationship of ER Signaling and BCSCs in Resistance to Endocrine Therapy
4.1.1. Alterations of ER in Relapse Tumors Treated with ENDOCRINE Therapy
4.1.2. Elevation in ER Signaling via Its Co-Transcriptional Factors in Endocrine Resistance
4.1.3. The Impact of ER Signaling on Endocrine Resistance-Associated Enrichment of BCSCs
4.2. Growth Factor Signaling Stimulating BCSC Enrichment in Developing ETR
4.3. NOTCH Pathway Regulating BCSCs in Endocrine Therapy
4.4. The Wnt, Hedgehog, and Hippo-YAP/TAZ Pathways
4.5. Microenvironment Contributions to BCSC Evolvement Following Endocrine Resistance Development
4.6. Other Factors—PAK4 Stimulating BCSCs in Response to Endocrine Treatment
5. The Involvement of Core Stemness Genes in Regulating BCSCs during ETR Development
6. Strategy of Targeting BCSCs to Overcome Endocrine Resistance
7. A Dynamic Model of BCSC Regulation in the Settings of Hormone Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in globocan 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA: Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Wei, F.; Liu, Y.; Wang, E.; Zhang, H.; Lin, X.; Wong, N.; Bane, A.; Tang, D. Factors promoting tamoxifen resistance in breast cancer via stimulating breast cancer stem cell expansion. Curr. Med. Chem. 2015, 22, 2360–2374. [Google Scholar] [CrossRef] [PubMed]
- Clark, G.M.; Osborne, C.K.; McGuire, W.L. Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J. Clin. Oncol. 1984, 2, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Keeling, J.W.; Ozer, E.; King, G.; Walker, F. Oestrogen receptor alpha in female fetal, infant, and child mammary tissue. J. Pathol. 2000, 191, 449–451. [Google Scholar] [CrossRef]
- Korach, K.S.; Couse, J.F.; Curtis, S.W.; Washburn, T.F.; Lindzey, J.; Kimbro, K.S.; Eddy, E.M.; Migliaccio, S.; Snedeker, S.M.; Lubahn, D.B.; et al. Estrogen receptor gene disruption: Molecular characterization and experimental and clinical phenotypes. Recent Prog. Horm. Res. 1996, 51, 159–186. [Google Scholar] [PubMed]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein foxa1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin d as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Eeckhoute, J.; Carroll, J.S.; Geistlinger, T.R.; Torres-Arzayus, M.I.; Brown, M. A cell-type-specific transcriptional network required for estrogen regulation of cyclin d1 and cell cycle progression in breast cancer. Genes Dev. 2006, 20, 2513–2526. [Google Scholar] [CrossRef]
- Dong, L.; Wang, W.; Wang, F.; Stoner, M.; Reed, J.C.; Harigai, M.; Samudio, I.; Kladde, M.P.; Vyhlidal, C.; Safe, S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J. Biol. Chem. 1999, 274, 32099–32107. [Google Scholar] [CrossRef] [PubMed]
- Seeger, H.; Wallwiener, D.; Mueck, A.O. Different effects of estradiol and various antiestrogens on tnf-alpha-induced changes of biochemical markers for growth and invasion of human breast cancer cells. Life Sci. 2006, 78, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, E.E.; McDaniel, R.E.; Maximov, P.Y.; Fan, P.; Jordan, V.C. Models and mechanisms of acquired antihormone resistance in breast cancer: Significant clinical progress despite limitations. Horm. Mol. Biol. Clin. Investig. 2012, 9, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Dukes, M.; Bowler, J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991, 51, 3867–3873. [Google Scholar] [PubMed]
- Johnston, S.J.; Cheung, K.L. Fulvestrant - a novel endocrine therapy for breast cancer. Curr. Med. Chem. 2010, 17, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Aromatase inhibitors in relation to other forms of endocrine therapy for breast cancer. Endocr. Relat. Cancer 1999, 6, 271–276. [Google Scholar] [CrossRef]
- Buzdar, A.U. Aromatase inhibitors in breast cancer therapy. Clin. Breast Cancer 2003, 4 (Suppl. 2), S84–S88. [Google Scholar] [CrossRef]
- Osborne, C.K.; Schiff, R.; Arpino, G.; Lee, A.S.; Hilsenbeck, V.G. Endocrine responsiveness: Understanding how progesterone receptor can be used to select endocrine therapy. Breast 2005, 14, 458–465. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Shao, Z. Fulvestrant in the treatment of hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: A review. Cancer Med. 2019, 8, 1943–1957. [Google Scholar] [CrossRef]
- Arpino, G.; Weiss, H.; Lee, A.V.; Schiff, R.; De Placido, S.; Osborne, C.K.; Elledge, R.M. Estrogen receptor-positive, progesterone receptor-negative breast cancer: Association with growth factor receptor expression and tamoxifen resistance. J. Natl. Cancer Inst. 2005, 97, 1254–1261. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Paish, E.C.; Powe, D.G.; Gee, J.; Nicholson, R.I.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Biologic and clinical characteristics of breast cancer with single hormone receptor positive phenotype. J. Clin. Oncol. 2007, 25, 4772–4778. [Google Scholar] [CrossRef] [PubMed]
- Early Breast Cancer Trialists’ Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet 2005, 365, 1687–1717. [Google Scholar]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Badia, E.; Oliva, J.; Balaguer, P.; Cavailles, V. Tamoxifen resistance and epigenetic modifications in breast cancer cell lines. Curr. Med. Chem. 2007, 14, 3035–3045. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr. Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Schiff, R.; Massarweh, S.; Shou, J.; Osborne, C.K. Breast cancer endocrine resistance: How growth factor signaling and estrogen receptor coregulators modulate response. Clin. Cancer Res. 2003, 9, 447S–454S. [Google Scholar] [PubMed]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Giuliano, M.; Trivedi, M.V.; Schiff, R.; Osborne, C.K. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2013, 19, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Costa, A.; Norton, L.; Senkus, E.; Aapro, M.; Andre, F.; Barrios, C.H.; Bergh, J.; Biganzoli, L.; Blackwell, K.L.; et al. Eso-esmo 2nd international consensus guidelines for advanced breast cancer (abc2) dagger. Ann. Oncol. 2014, 25, 1871–1888. [Google Scholar] [CrossRef]
- Cardoso, F.; Costa, A.; Norton, L.; Senkus, E.; Aapro, M.; Andre, F.; Barrios, C.H.; Bergh, J.; Biganzoli, L.; Blackwell, K.L.; et al. Eso-esmo 2nd international consensus guidelines for advanced breast cancer (abc2). Breast 2014, 23, 489–502. [Google Scholar] [CrossRef]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [PubMed]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (prisma-p) 2015: Elaboration and explanation. BMJ 2015, 349, g7647. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (prisma-p) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- She, J.J.; Zhang, P.G.; Wang, Z.M.; Gan, W.M.; Che, X.M. Identification of side population cells from bladder cancer cells by dyecycle violet staining. Cancer Biol. Ther. 2008, 7, 1663–1668. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ovalle, S.; Gutierrez-Lopez, M.D.; Olmo, N.; Turnay, J.; Lizarbe, M.A.; Majano, P.; Molina-Jimenez, F.; Lopez-Cabrera, M.; Yanez-Mo, M.; Sanchez-Madrid, F.; et al. The tetraspanin cd9 inhibits the proliferation and tumorigenicity of human colon carcinoma cells. Int. J. Cancer 2007, 121, 2140–2152. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; Van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H.; Kandel, R.; Wunder, J.S.; Alman, B.A. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Inman, J.L.; Robertson, C.; Mott, J.D.; Bissell, M.J. Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development 2015, 142, 1028–1042. [Google Scholar] [CrossRef]
- Kordon, E.C.; Smith, G.H. An entire functional mammary gland may comprise the progeny from a single cell. Development 1998, 125, 1921–1930. [Google Scholar] [PubMed]
- Daniel, C.W.; Smith, G.H. The mammary gland: A model for development. J. Mammary Gland Biol. Neoplasia 1999, 4, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Bowman, A.N.; Nusse, R. Developmental stage and time dictate the fate of wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 2012, 11, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Fu, N.Y.; Lindeman, G.J.; Visvader, J.E. In situ identification of bipotent stem cells in the mammary gland. Nature 2014, 506, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.Y.; Rios, A.C.; Pal, B.; Law, C.W.; Jamieson, P.; Liu, R.; Vaillant, F.; Jackling, F.; Liu, K.H.; Smyth, G.K.; et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017, 19, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Kalisky, T.; Sahoo, D.; Dalerba, P.; Feng, W.; Lin, Y.; Qian, D.; Kong, A.; Yu, J.; Wang, F.; et al. A quiescent bcl11b high stem cell population is required for maintenance of the mammary gland. Cell Stem Cell 2017, 20, 247–260. [Google Scholar] [CrossRef]
- Eirew, P.; Stingl, J.; Raouf, A.; Turashvili, G.; Aparicio, S.; Emerman, J.T.; Eaves, C.J. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat. Med. 2008, 14, 1384–1389. [Google Scholar] [CrossRef]
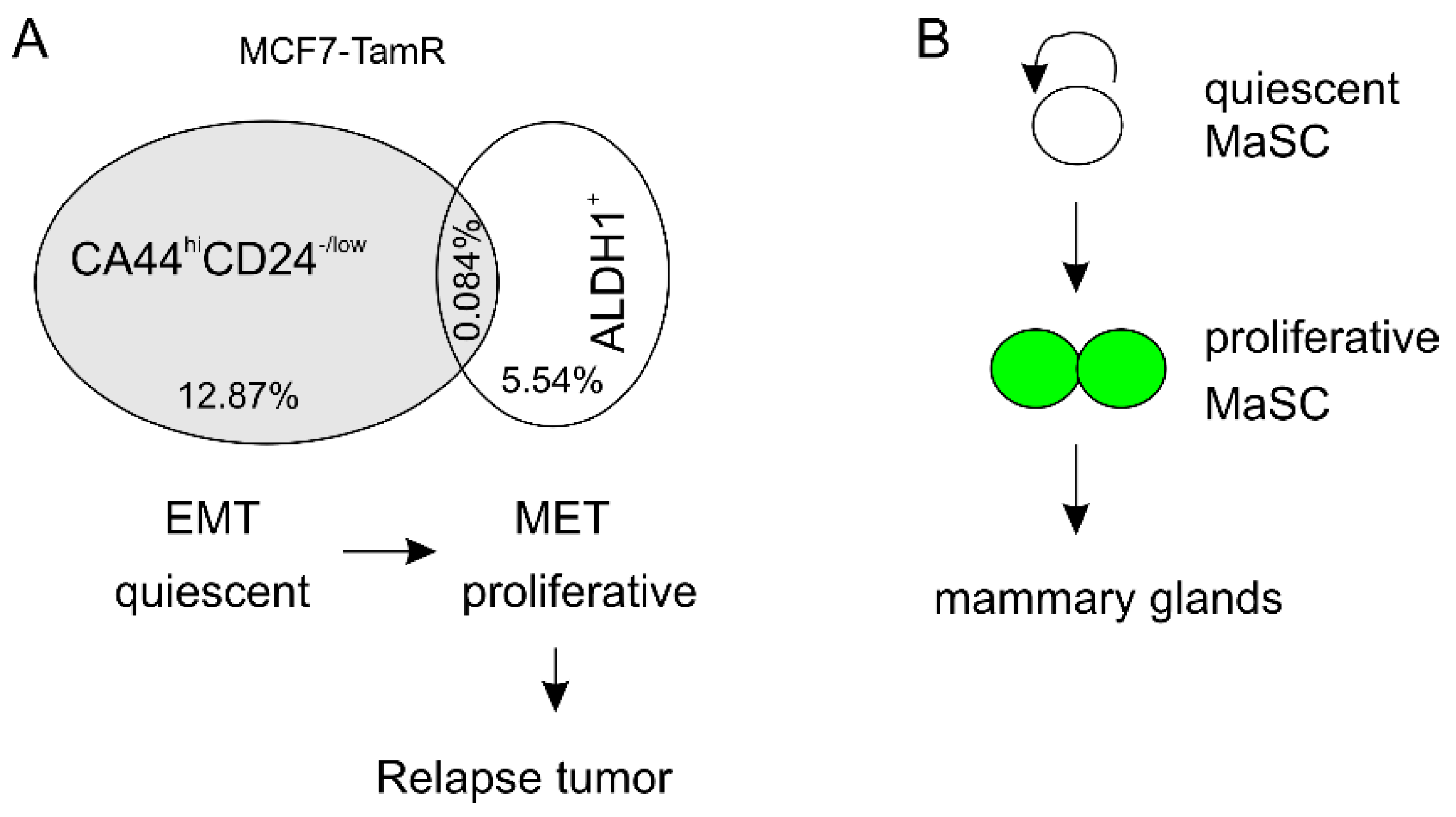
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Lee, E.; Piranlioglu, R.; Wicha, M.S.; Korkaya, H. Plasticity and potency of mammary stem cell subsets during mammary gland development. Int. J. Mol. Sci. 2019, 20, 2357. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Biehs, B.; Warming, S.; Leong, K.G.; Rangell, L.; Klein, O.D.; de Sauvage, F.J. A reserve stem cell population in small intestine renders lgr5-positive cells dispensable. Nature 2011, 478, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Miragaya, J.; Gonzalez-Suarez, E. Tumor-initiating cd49f cells are a hallmark of chemoresistant triple negative breast cancer. Mol. Cell. Oncol. 2017, 4, e1338208. [Google Scholar] [CrossRef]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of cd133(hi) cells by il6/notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. Cd133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef]
- Tsang, J.Y.; Huang, Y.H.; Luo, M.H.; Ni, Y.B.; Chan, S.K.; Lui, P.C.; Yu, A.M.; Tan, P.H.; Tse, G.M. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 136, 407–417. [Google Scholar] [CrossRef]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitao, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers cd44, cd24 and aldh1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Shien, T.; Ogiya, A.; Ishida, N.; Yamazaki, K.; Horii, R.; Horimoto, Y.; Masuda, N.; Yasojima, H.; Inao, T.; et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res. 2016, 18, 73. [Google Scholar] [CrossRef]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. Stat3 mediates resistance of cd44(+)cd24(-/low) breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef]
- Piva, M.; Domenici, G.; Iriondo, O.; Rabano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; Lopez-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Hartung, A.; Bouchez, L.C.; Walker, J.R.; Reddy, V.A.; Cho, C.Y.; Schultz, P.G. Cxcr4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through ahr signalling. Br. J. Cancer 2012, 107, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.W.; Sun, X.F.; Guo, X.H.; He, Y.N.; Cui, S.D.; Fan, Q.X. Tamoxifen-resistant breast cancer cells possess cancer stem-like cell properties. Chin. Med. J. 2013, 126, 3030–3034. [Google Scholar] [PubMed]
- Raffo, D.; Berardi, D.E.; Pontiggia, O.; Todaro, L.; de Kier Joffe, E.B.; Simian, M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res. Treat. 2013, 142, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoes, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alferez, D.G.; Spence, K.; Santiago-Gomez, A.; Chemi, F.; et al. Anti-estrogen resistance in human breast tumors is driven by jag1-notch4-dependent cancer stem cell activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Lin, X.; Wu, Y.; Cockburn, J.; Bane, A.; Tang, D. Polycomb complex protein bmi1 confers resistance to tamoxifen in estrogen receptor positive breast cancer. Cancer Lett. 2018, 426, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and ps2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995, 55, 3331–3338. [Google Scholar]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular changes in tamoxifen-resistant breast cancer: Relationship between estrogen receptor, her-2, and p38 mitogen-activated protein kinase. J. Clin. Oncol. 2005, 23, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M. Overexpression of her-2 as a resistance mechanism to hormonal therapy for breast cancer. Endocr. Relat. Cancer 2001, 8, 191–195. [Google Scholar] [CrossRef]
- Hull, D.F.; Clark, G.M.; Osborne, C.K.; Chamness, G.C.; Knight, W.A.; McGuire, W.L. Multiple estrogen receptor assays in human breast cancer. Cancer Res. 1983, 43, 413–416. [Google Scholar] [PubMed]
- Herman, M.E.; Katzenellenbogen, B.S. Response-specific antiestrogen resistance in a newly characterized mcf-7 human breast cancer cell line resulting from long-term exposure to trans-hydroxytamoxifen. J. Steroid Biochem. Mol. Biol. 1996, 59, 121–134. [Google Scholar] [CrossRef]
- Kabos, P.; Finlay-Schultz, J.; Li, C.; Kline, E.; Finlayson, C.; Wisell, J.; Manuel, C.A.; Edgerton, S.M.; Harrell, J.C.; Elias, A.; et al. Patient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res. Treat. 2012, 135, 415–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K.; Pippen, J.; Jones, S.E.; Parker, L.M.; Ellis, M.; Come, S.; Gertler, S.Z.; May, J.T.; Burton, G.; Dimery, I.; et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a north american trial. J. Clin. Oncol. 2002, 20, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Howell, A.; Pippen, J.; Elledge, R.M.; Mauriac, L.; Vergote, I.; Jones, S.E.; Come, S.E.; Osborne, C.K.; Robertson, J.F. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma: A prospectively planned combined survival analysis of two multicenter trials. Cancer 2005, 104, 236–239. [Google Scholar] [CrossRef]
- Fuqua, S.A. The role of estrogen receptors in breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 2001, 6, 407–417. [Google Scholar] [CrossRef]
- Giguere, V. Estrogen receptor mutations in breast cancer-an anticipated “rediscovery?”. Mol. Endocrinol. 2014, 28, 427–428. [Google Scholar] [CrossRef]
- Shaw, L.E.; Sadler, A.J.; Pugazhendhi, D.; Darbre, P.D. Changes in oestrogen receptor-alpha and -beta during progression to acquired resistance to tamoxifen and fulvestrant (faslodex, ici 182,780) in mcf7 human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2006, 99, 19–32. [Google Scholar] [CrossRef]
- Holst, F.; Stahl, P.R.; Ruiz, C.; Hellwinkel, O.; Jehan, Z.; Wendland, M.; Lebeau, A.; Terracciano, L.; Al-Kuraya, K.; Janicke, F.; et al. Estrogen receptor alpha (esr1) gene amplification is frequent in breast cancer. Nat. Genet. 2007, 39, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.A.; Hoog, J.; Chin, S.F.; Tao, Y.; Zayed, A.A.; Chin, K.; Teschendorff, A.E.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; et al. Esr1 gene amplification in breast cancer: A common phenomenon? Nat. Genet. 2008, 40, 806–807. [Google Scholar] [CrossRef] [PubMed]
- Szostakowska, M.; Trebinska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor alpha gene esr1 amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, K.V.; Ejlertsen, B.; Muller, S.; Moller, S.; Rasmussen, B.B.; Balslev, E.; Laenkholm, A.V.; Christiansen, P.; Mouridsen, H.T. Amplification of esr1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 345–355. [Google Scholar] [CrossRef]
- Lin, C.H.; Liu, J.M.; Lu, Y.S.; Lan, C.; Lee, W.C.; Kuo, K.T.; Wang, C.C.; Chang, D.Y.; Huang, C.S.; Cheng, A.L. Clinical significance of esr1 gene copy number changes in breast cancer as measured by fluorescence in situ hybridisation. J. Clin. Pathol. 2013, 66, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Basudan, A.; Priedigkeit, N.; Hartmaier, R.J.; Sokol, E.S.; Bahreini, A.; Watters, R.J.; Boisen, M.M.; Bhargava, R.; Weiss, K.R.; Karsten, M.M.; et al. Frequent esr1 and cdk pathway copy-number alterations in metastatic breast cancer. Mol. Cancer Res. 2019, 17, 457–468. [Google Scholar] [CrossRef]
- Magnani, L.; Frige, G.; Gadaleta, R.M.; Corleone, G.; Fabris, S.; Kempe, M.H.; Verschure, P.J.; Barozzi, I.; Vircillo, V.; Hong, S.P.; et al. Acquired cyp19a1 amplification is an early specific mechanism of aromatase inhibitor resistance in eralpha metastatic breast cancer. Nat. Genet. 2017, 49, 444–450. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Barozzi, I.; Faronato, M.; Lombardo, Y.; Steel, J.H.; Patel, N.; Darbre, P.; Castellano, L.; Gyorffy, B.; Woodley, L.; et al. Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat. Commun. 2015, 6, 10044. [Google Scholar] [CrossRef]
- Pejerrey, S.M.; Dustin, D.; Kim, J.A.; Gu, G.; Rechoum, Y.; Fuqua, S.A.W. The impact of esr1 mutations on the treatment of metastatic breast cancer. Horm. Cancer 2018, 9, 215–228. [Google Scholar] [CrossRef]
- Weis, K.E.; Ekena, K.; Thomas, J.A.; Lazennec, G.; Katzenellenbogen, B.S. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol. 1996, 10, 1388–1398. [Google Scholar] [PubMed]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. Esr1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating esr1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Merenbakh-Lamin, K.; Ben-Baruch, N.; Yeheskel, A.; Dvir, A.; Soussan-Gutman, L.; Jeselsohn, R.; Yelensky, R.; Brown, M.; Miller, V.A.; Sarid, D.; et al. D538g mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013, 73, 6856–6864. [Google Scholar] [CrossRef] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Bahreini, A.; Gyanchandani, R.; Lucas, P.C.; Hartmaier, R.J.; Watters, R.J.; Jonnalagadda, A.R.; Trejo Bittar, H.E.; Berg, A.; Hamilton, R.L.; et al. Sensitive detection of mono- and polyclonal esr1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin. Cancer Res. 2016, 22, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Ando, S. Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Rayala, S.K.; Talukder, A.H.; Balasenthil, S.; Tharakan, R.; Barnes, C.J.; Wang, R.A.; Aldaz, C.M.; Khan, S.; Kumar, R. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006, 66, 1694–1701. [Google Scholar] [CrossRef]
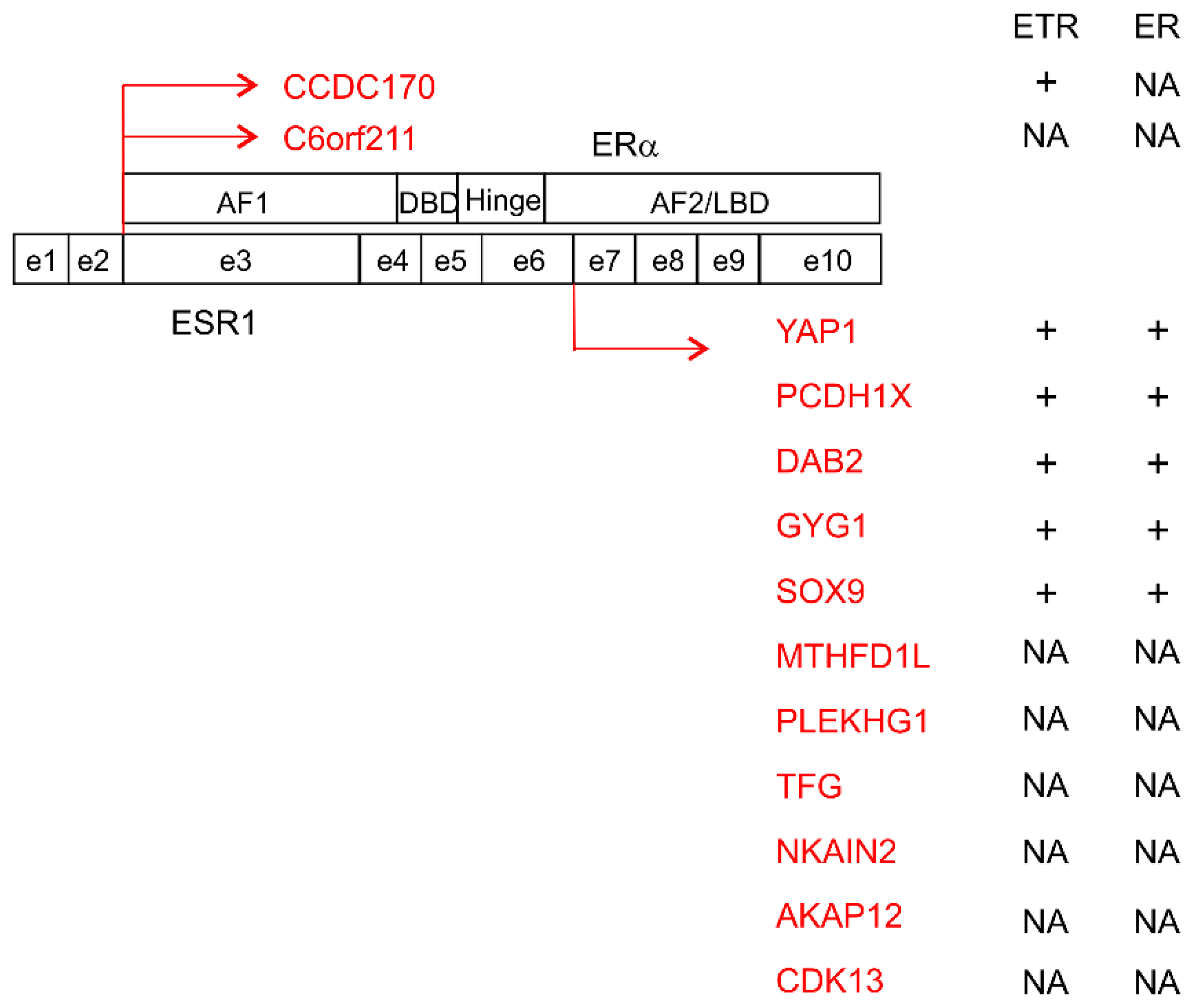
- Veeraraghavan, J.; Tan, Y.; Cao, X.X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.; Edwards, D.P.; Contreras, A.; et al. Recurrent esr1-ccdc170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 4577. [Google Scholar] [CrossRef]
- Giltnane, J.M.; Hutchinson, K.E.; Stricker, T.P.; Formisano, L.; Young, C.D.; Estrada, M.V.; Nixon, M.J.; Du, L.; Sanchez, V.; Ericsson, P.G.; et al. Genomic profiling of er (+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci. Transl. Med. 2017, 9, eaai7993. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant esr1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; et al. Functional annotation of esr1 gene fusions in estrogen receptor-positive breast cancer. Cell Rep. 2018, 24, 1434–1444. [Google Scholar] [CrossRef]
- Hartmaier, R.J.; Trabucco, S.E.; Priedigkeit, N.; Chung, J.H.; Parachoniak, C.A.; Vanden Borre, P.; Morley, S.; Rosenzweig, M.; Gay, L.M.; Goldberg, M.E.; et al. Recurrent hyperactive esr1 fusion proteins in endocrine therapy-resistant breast cancer. Ann. Oncol. 2018, 29, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. Emt, cscs, and drug resistance: The mechanistic link and clinical implications. Nature reviews. Clin. Oncol. 2017, 14, 611–629. [Google Scholar]
- Bernardo, G.M.; Lozada, K.L.; Miedler, J.D.; Harburg, G.; Hewitt, S.C.; Mosley, J.D.; Godwin, A.K.; Korach, K.S.; Visvader, J.E.; Kaestner, K.H.; et al. Foxa1 is an essential determinant of eralpha expression and mammary ductal morphogenesis. Development 2010, 137, 2045–2054. [Google Scholar] [CrossRef]
- Hurtado, A.; Holmes, K.A.; Ross-Innes, C.S.; Schmidt, D.; Carroll, J.S. Foxa1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 2011, 43, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Samanthapudi, V.S.; Gajulapalli, V.N. Estrogen receptor coregulators and pioneer factors: The orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014, 2, 34. [Google Scholar] [CrossRef]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef]
- Magnani, L.; Patten, D.K.; Nguyen, V.T.; Hong, S.P.; Steel, J.H.; Patel, N.; Lombardo, Y.; Faronato, M.; Gomes, A.R.; Woodley, L.; et al. The pioneer factor pbx1 is a novel driver of metastatic progression in eralpha-positive breast cancer. Oncotarget 2015, 6, 21878–21891. [Google Scholar] [CrossRef]
- Magnani, L.; Ballantyne, E.B.; Zhang, X.; Lupien, M. Pbx1 genomic pioneer function drives eralpha signaling underlying progression in breast cancer. PLoS Genet. 2011, 7, e1002368. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438 e426. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, T.M.; Wardell, S.E.; Jasper, J.S.; Stice, J.P.; Safi, R.; Nelson, E.R.; McDonnell, D.P. Delineation of a foxa1/eralpha/agr2 regulatory loop that is dysregulated in endocrine therapy-resistant breast cancer. Mol. Cancer Res. 2014, 12, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. Foxa1 overexpression mediates endocrine resistance by altering the er transcriptome and il-8 expression in er-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef] [PubMed]
- Leonard, M.; Zhang, X. Estrogen receptor coactivator mediator subunit 1 (med1) as a tissue-specific therapeutic target in breast cancer. J. Zhejiang Univ. Sci. B 2019, 20, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Krutchinsky, A.; Fukuda, A.; Chen, W.; Yamamura, S.; Chait, B.T.; Roeder, R.G. Med1/trap220 exists predominantly in a trap/mediator subpopulation enriched in rna polymerase ii and is required for er-mediated transcription. Mol. Cell 2005, 19, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Germer, K.; Wu, T.; Wang, J.; Luo, J.; Wang, S.C.; Wang, Q.; Zhang, X. Cross-talk between her2 and med1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res. 2012, 72, 5625–5634. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, J.; Leonard, M.; Nephew, K.; Li, Y.; Zhang, X. Silencing med1 sensitizes breast cancer cells to pure anti-estrogen fulvestrant in vitro and in vivo. PLoS ONE 2013, 8, e70641. [Google Scholar] [CrossRef]
- Zhang, Y.; Leonard, M.; Shu, Y.; Yang, Y.; Shu, D.; Guo, P.; Zhang, X. Overcoming tamoxifen resistance of human breast cancer by targeted gene silencing using multifunctional prna nanoparticles. ACS Nano 2016, 11, 335–346. [Google Scholar] [CrossRef]
- Anzick, S.L.; Kononen, J.; Walker, R.L.; Azorsa, D.O.; Tanner, M.M.; Guan, X.Y.; Sauter, G.; Kallioniemi, O.P.; Trent, J.M.; Meltzer, P.S. AIb1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 1997, 277, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Dihge, L.; Bendahl, P.O.; Grabau, D.; Isola, J.; Lovgren, K.; Ryden, L.; Ferno, M. Epidermal growth factor receptor (egfr) and the estrogen receptor modulator amplified in breast cancer (aib1) for predicting clinical outcome after adjuvant tamoxifen in breast cancer. Breast Cancer Res. Treat. 2008, 109, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Percharde, M.; Lavial, F.; Ng, J.H.; Kumar, V.; Tomaz, R.A.; Martin, N.; Yeo, J.C.; Gil, J.; Prabhakar, S.; Ng, H.H.; et al. Ncoa3 functions as an essential esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012, 26, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Rohira, A.D.; Yan, F.; Wang, L.; Wang, J.; Zhou, S.; Lu, A.; Yu, Y.; Xu, J.; Lonard, D.M.; O’Malley, B.W. Targeting src coactivators blocks the tumor-initiating capacity of cancer stem-like cells. Cancer Res. 2017, 77, 4293–4304. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Hu, H.; Temiz, N.A.; Hagen, K.M.; Girard, B.J.; Brady, N.J.; Schwertfeger, K.L.; Lange, C.A.; Ostrander, J.H. Cancer stem cell phenotypes in er (+) breast cancer models are promoted by pelp1/aib1 complexes. Mol. Cancer Res. 2018, 16, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Simoes, B.M.; Vivanco, M.D. Cancer stem cells in the human mammary gland and regulation of their differentiation by estrogen. Future Oncol. 2011, 7, 995–1006. [Google Scholar] [CrossRef]
- Morimoto, K.; Kim, S.J.; Tanei, T.; Shimazu, K.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Terada, N.; Noguchi, S. Stem cell marker aldehyde dehydrogenase 1-positive breast cancers are characterized by negative estrogen receptor, positive human epidermal growth factor receptor type 2, and high ki67 expression. Cancer Sci. 2009, 100, 1062–1068. [Google Scholar] [CrossRef]
- Harrison, H.; Simoes, B.M.; Rogerson, L.; Howell, S.J.; Landberg, G.; Clarke, R.B. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine egfr and notch signalling. Breast Cancer Res. 2013, 15, R21. [Google Scholar] [CrossRef]
- Tachi, K.; Shiraishi, A.; Bando, H.; Yamashita, T.; Tsuboi, I.; Kato, T.; Hara, H.; Ohneda, O. Foxa1 expression affects the proliferation activity of luminal breast cancer stem cell populations. Cancer Sci. 2016, 107, 281–289. [Google Scholar] [CrossRef]
- Nasr, M.; Farghaly, M.; Elsaba, T.; El-Mokhtar, M.; Radwan, R.; Elsabahy, M.; Abdelkareem, A.; Fakhry, H.; Mousa, N. Resistance of primary breast cancer cells with enhanced pluripotency and stem cell activity to sex hormonal stimulation and suppression. Int. J. Biochem. Cell Biol. 2018, 105, 84–93. [Google Scholar] [CrossRef]
- Yang, Y.; Leonard, M.; Zhang, Y.; Zhao, D.; Mahmoud, C.; Khan, S.; Wang, J.; Lower, E.E.; Zhang, X. Her2-driven breast tumorigenesis relies upon interactions of the estrogen receptor with coactivator med1. Cancer Res. 2018, 78, 422–435. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, R.; Neefjes, J.; Michalides, R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. Int. J. Breast Cancer 2011, 2011, 232435. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Cufi, S.; Lopez-Bonet, E.; Corominas-Faja, B.; Cuyas, E.; Vellon, L.; Iglesias, J.M.; Leis, O.; Martin, A.G.; Menendez, J.A. Reprogramming of non-genomic estrogen signaling by the stemness factor sox2 enhances the tumor-initiating capacity of breast cancer cells. Cell Cycle 2013, 12, 3471–3477. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Karthik, G.M.; Lovrot, J.; Haglund, F.; Rosin, G.; Katchy, A.; Zhang, X.; Viberg, L.; Frisell, J.; Williams, C.; et al. Estrogen receptor beta as a therapeutic target in breast cancer stem cells. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, M.; Carlisi, D.; Giuliano, M.; Calvaruso, G.; Cernigliaro, C.; Vento, R.; D’Anneo, A. The analysis of estrogen receptor-alpha positive breast cancer stem-like cells unveils a high expression of the serpin proteinase inhibitor pi-9: Possible regulatory mechanisms. Int. J. Oncol. 2016, 49, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of eralpha36(+) breast cancer by upregulating aldh1a1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Fry, E.A. Aberrant splicing of estrogen receptor, her2, and cd44 genes in breast cancer. Genet. Epigenet. 2015, 7, 19–32. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, X.T.; Wang, M.L.; Zheng, H.Y.; Liu, L.J.; Wang, Z.Y. Er-alpha36-mediated rapid estrogen signaling positively regulates er-positive breast cancer stem/progenitor cells. PLoS ONE 2014, 9, e88034. [Google Scholar]
- Deng, H.; Yin, L.; Zhang, X.T.; Liu, L.J.; Wang, M.L.; Wang, Z.Y. Er-alpha variant er-alpha36 mediates antiestrogen resistance in er-positive breast cancer stem/progenitor cells. J. Steroid Biochem. Mol. Biol. 2014, 144 Pt B, 417–426. [Google Scholar] [CrossRef]
- Knowlden, J.M.; Hutcheson, I.R.; Barrow, D.; Gee, J.M.; Nicholson, R.I. Insulin-like growth factor-i receptor signaling in tamoxifen-resistant breast cancer: A supporting role to the epidermal growth factor receptor. Endocrinology 2005, 146, 4609–4618. [Google Scholar] [CrossRef] [PubMed]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbb2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant mcf-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Santen, R.J.; Song, R.X.; Zhang, Z.; Kumar, R.; Jeng, M.H.; Masamura, A.; Lawrence, J., Jr.; Berstein, L.; Yue, W. Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr. Relat. Cancer 2005, 12 (Suppl. 1), S61–S73. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, R.I.; Staka, C.; Boyns, F.; Hutcheson, I.R.; Gee, J.M. Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: New opportunities for therapy. Endocr. Relat. Cancer 2004, 11, 623–641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Slingerland, J.M. Links between oestrogen receptor activation and proteolysis: Relevance to hormone-regulated cancer therapy. Nat. Rev. Cancer 2014, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Tamoxifen—What next? Oncologist 2004, 9, 378–384. [Google Scholar] [CrossRef]
- Garcia-Becerra, R.; Santos, N.; Diaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, mirnas and genetically based resistance. Int. J. Mol. Sci. 2012, 14, 108–145. [Google Scholar] [CrossRef]
- Foley, J.; Nickerson, N.K.; Nam, S.; Allen, K.T.; Gilmore, J.L.; Nephew, K.P.; Riese, D.J., II. EGFR signaling in breast cancer: Bad to the bone. Semin. Cell Dev. Biol. 2010, 21, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Piasecka, D.; Braun, M.; Kitowska, K.; Mieczkowski, K.; Kordek, R.; Sadej, R.; Romanska, H. Fgfs/fgfrs-dependent signalling in regulation of steroid hormone receptors—Implications for therapy of luminal breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 230. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Endocrine-responsive breast cancer and strategies for combating resistance. Nat. Rev. Cancer 2002, 2, 101–112. [Google Scholar] [CrossRef]
- Hardt, O.; Wild, S.; Oerlecke, I.; Hofmann, K.; Luo, S.; Wiencek, Y.; Kantelhardt, E.; Vess, C.; Smith, G.P.; Schroth, G.P.; et al. Highly sensitive profiling of cd44+/cd24- breast cancer stem cells by combining global mrna amplification and next generation sequencing: Evidence for a hyperactive pi3k pathway. Cancer Lett. 2012, 325, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Pommier, S.J.; Hernandez, A.; Han, E.; Massimino, K.; Muller, P.; Diggs, B.; Chamberlain, E.; Murphy, J.; Hansen, J.; Naik, A.; et al. Fresh surgical specimens yield breast stem/progenitor cells and reveal their oncogenic abnormalities. Ann. Surg. Oncol. 2012, 19, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Donovan, C.A.; Pommier, R.F.; Schillace, R.; O’Neill, S.; Muller, P.; Alabran, J.L.; Hansen, J.E.; Murphy, J.A.; Naik, A.M.; Vetto, J.T.; et al. Correlation of breast cancer axillary lymph node metastases with stem cell mutations. JAMA Surg. 2013, 148, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Karthik, G.M.; Ma, R.; Lovrot, J.; Kis, L.L.; Lindh, C.; Blomquist, L.; Fredriksson, I.; Bergh, J.; Hartman, J. Mtor inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015, 367, 76–87. [Google Scholar] [CrossRef]
- Vilquin, P.; Donini, C.F.; Villedieu, M.; Grisard, E.; Corbo, L.; Bachelot, T.; Vendrell, J.A.; Cohen, P.A. Microrna-125b upregulation confers aromatase inhibitor resistance and is a novel marker of poor prognosis in breast cancer. Breast Cancer Res. 2015, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Labat, M.L.; Shackleton, M.; Stingl, J.; Vaillant, F.; Forrest, N.C.; Eaves, C.J.; Visvader, J.E.; Lindeman, G.J. Steroid hormone receptor status of mouse mammary stem cells. J. Natl. Cancer Inst. 2006, 98, 1011–1014. [Google Scholar] [CrossRef]
- Hebbard, L.; Steffen, A.; Zawadzki, V.; Fieber, C.; Howells, N.; Moll, J.; Ponta, H.; Hofmann, M.; Sleeman, J. Cd44 expression and regulation during mammary gland development and function. J. Cell Sci. 2000, 113 Pt 14, 2619–2630. [Google Scholar]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. Her2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef]
- Padua, M.B.; Bhat-Nakshatri, P.; Anjanappa, M.; Prasad, M.S.; Hao, Y.; Rao, X.; Liu, S.; Wan, J.; Liu, Y.; McElyea, K.; et al. Dependence receptor unc5a restricts luminal to basal breast cancer plasticity and metastasis. Breast Cancer Res. 2018, 20, 35. [Google Scholar] [CrossRef]
- Choi, H.J.; Joo, H.S.; Won, H.Y.; Min, K.W.; Kim, H.Y.; Son, T.; Oh, Y.H.; Lee, J.Y.; Kong, G. Role of rbp2-induced er and igf1r-erbb signaling in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 2018, 110. [Google Scholar] [CrossRef]
- Acar, A.; Simoes, B.M.; Clarke, R.B.; Brennan, K. A role for notch signalling in breast cancer and endocrine resistance. Stem Cells Int. 2016, 2016, 2498764. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Raouf, A.; Zhao, Y.; To, K.; Stingl, J.; Delaney, A.; Barbara, M.; Iscove, N.; Jones, S.; McKinney, S.; Emerman, J.; et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell 2008, 3, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Yalcin-Ozuysal, O.; Fiche, M.; Guitierrez, M.; Wagner, K.U.; Raffoul, W.; Brisken, C. Antagonistic roles of notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010, 17, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Pannuti, A.; Espinoza, I.; Zhu, H.; Hicks, C.; Zhu, X.; Caskey, M.; Rizzo, P.; D’Souza, G.; Backus, K.; et al. Crosstalk between pkcalpha and notch-4 in endocrine-resistant breast cancer cells. Oncogenesis 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, Y.; Faronato, M.; Filipovic, A.; Vircillo, V.; Magnani, L.; Coombes, R.C. Nicastrin and notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in mcf7 breast cancer cells. Breast Cancer Res. 2014, 16, R62. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miao, H.; D’Souza, G.; Osipo, C.; Song, L.L.; Yun, J.; Zhao, H.; Mascarenhas, J.; Wyatt, D.; Antico, G.; et al. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008, 68, 5226–5235. [Google Scholar] [CrossRef] [PubMed]
- McClements, L.; Annett, S.; Yakkundi, A.; O’Rourke, M.; Valentine, A.; Moustafa, N.; Alqudah, A.; Simoes, B.M.; Furlong, F.; Short, A.; et al. Fkbpl and its peptide derivatives inhibit endocrine therapy resistant cancer stem cells and breast cancer metastasis by downregulating dll4 and notch4. BMC Cancer 2019, 19, 351. [Google Scholar] [CrossRef]
- Haughian, J.M.; Pinto, M.P.; Harrell, J.C.; Bliesner, B.S.; Joensuu, K.M.; Dye, W.W.; Sartorius, C.A.; Tan, A.C.; Heikkila, P.; Perou, C.M.; et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of notch. Proc. Natl. Acad. Sci. USA 2012, 109, 2742–2747. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the wnt pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boman, B.M.; Fields, J.Z. An APC: WNT counter-current-like mechanism regulates cell division along the human colonic crypt axis: A mechanism that explains how apc mutations induce proliferative abnormalities that drive colon cancer development. Front. Oncol. 2013, 3, 244. [Google Scholar] [CrossRef] [PubMed]
- Atlasi, Y.; Looijenga, L.; Fodde, R. Cancer stem cells, pluripotency, and cellular heterogeneity: A WNTer perspective. Curr. Top Dev. Biol. 2014, 107, 373–404. [Google Scholar] [PubMed]
- Angeloni, V.; Tiberio, P.; Appierto, V.; Daidone, M.G. Implications of stemness-related signaling pathways in breast cancer response to therapy. Semin. Cancer Biol. 2015, 31, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.N.; Hedditch, E.L.; Baker, L.A.; Jary, E.; Ward, R.L.; Ford, C.E. The wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer 2013, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine therapy of estrogen receptor-positive breast cancer cells: Early differential effects on stem cell markers. Front. Oncol. 2017, 7, 184. [Google Scholar] [CrossRef]
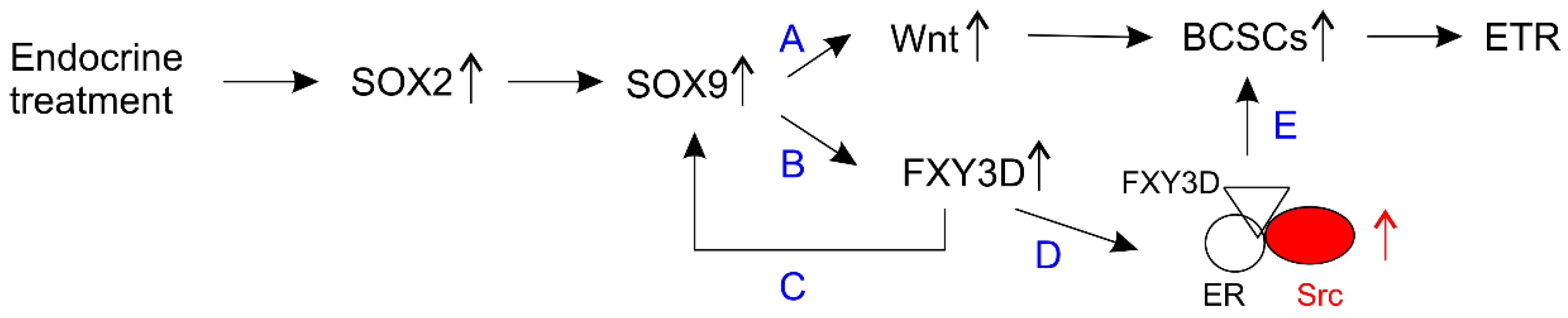
- Domenici, G.; Aurrekoetxea-Rodriguez, I.; Simoes, B.M.; Rabano, M.; Lee, S.Y.; Millan, J.S.; Comaills, V.; Oliemuller, E.; Lopez-Ruiz, J.A.; Zabalza, I.; et al. A sox2-sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yin, W.; Yu, Z.H.; Zhou, Y.J.; Chi, J.R.; Ge, J.; Cao, X.C. Mir-190 enhances endocrine therapy sensitivity by regulating sox9 expression in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 22. [Google Scholar] [CrossRef]
- Petrova, R.; Joyner, A.L. Roles for hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef]
- Campbell, V.; Copland, M. Hedgehog signaling in cancer stem cells: A focus on hematological cancers. Stem Cells Cloning Adv. Appl. 2015, 8, 27–38. [Google Scholar]
- Riobo-Del Galdo, N.A.; Lara Montero, A.; Wertheimer, E.V. Role of hedgehog signaling in breast cancer: Pathogenesis and therapeutics. Cells 2019, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by pi3k/akt pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, J.E.; Park, H.W. The role of hippo pathway in cancer stem cell biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of yap and taz in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The hippo transducer taz confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Guan, K.L. Hippo signaling at a glance. J. Cell Sci. 2010, 123, 4001–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardi, D.E.; Raffo, D.; Todaro, L.B.; Simian, M. Laminin modulates the stem cell population in lm05-e murine breast cancer cells through the activation of the mapk/erk pathway. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2017, 49, 869–879. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef]
- Castellaro, A.M.; Rodriguez-Baili, M.C.; Di Tada, C.E.; Gil, G.A. Tumor-associated macrophages induce endocrine therapy resistance in er+ breast cancer cells. Cancers 2019, 11, 189. [Google Scholar] [CrossRef]
- Recouvreux, S.; Sampayo, R.; Bessone, M.I.; Simian, M. Microenvironment and endocrine resistance in breast cancer: Friend or foe? World J. Clin. Oncol. 2015, 6, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Callari, M.; Guffanti, A.; Solda, G.; Merlino, G.; Fina, E.; Brini, E.; Moles, A.; Cappelletti, V.; Daidone, M.G. In-depth characterization of breast cancer tumor-promoting cell transcriptome by rna sequencing and microarrays. Oncotarget 2016, 7, 976–994. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. Cxcr1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.K.; Farnie, G.; Bundred, N.J.; Simoes, B.M.; Shergill, A.; Landberg, G.; Howell, S.J.; Clarke, R.B. Targeting cxcr1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting her2 via her2-dependent and -independent mechanisms. Clin. Cancer Res. 2013, 19, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The rb-il-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Sun, J.; Wang, C.; Bu, X.; Liu, X.; Mao, Y.; Wang, H. Il-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem. Biophys. Res. Commun. 2017, 485, 643–650. [Google Scholar] [CrossRef]
- Pichaud, F.; Walther, R.F.; Nunes de Almeida, F. Regulation of cdc42 and its effectors in epithelial morphogenesis. J. Cell Sci. 2019, 132, jcs217869. [Google Scholar] [CrossRef]
- Bi, Y.; Tian, M.; Le, J.; Wang, L.; Liu, X.; Qu, J.; Hao, M. Study on the expression of pak4 and p54 protein in breast cancer. World J. Surg. Oncol. 2016, 14, 160. [Google Scholar] [CrossRef]
- He, L.F.; Xu, H.W.; Chen, M.; Xian, Z.R.; Wen, X.F.; Chen, M.N.; Du, C.W.; Huang, W.H.; Wu, J.D.; Zhang, G.J. Activated-pak4 predicts worse prognosis in breast cancer and promotes tumorigenesis through activation of pi3k/akt signaling. Oncotarget 2017, 8, 17573–17585. [Google Scholar] [CrossRef] [PubMed]
- Dart, A.E.; Box, G.M.; Court, W.; Gale, M.E.; Brown, J.P.; Pinder, S.E.; Eccles, S.A.; Wells, C.M. Pak4 promotes kinase-independent stabilization of rhou to modulate cell adhesion. J. Cell Biol. 2015, 211, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Zhu, J.; Li, Z.; Lorent, J.; Zhao, C.; Dahlman-Wright, K.; Stromblad, S. P21-activated kinase group ii small compound inhibitor gne-2861 perturbs estrogen receptor alpha signaling and restores tamoxifen-sensitivity in breast cancer cells. Oncotarget 2015, 6, 43853–43868. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Gomez, A.; Kedward, T.; Simoes, B.M.; Dragoni, I.; NicAmhlaoibh, R.; Trivier, E.; Sabin, V.; Gee, J.M.; Sims, A.H.; Howell, S.J.; et al. Pak4 regulates stemness and progression in endocrine resistant er-positive metastatic breast cancer. Cancer Lett. 2019, 458, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yoon, S.Y.; Kim, C.N.; Joo, J.H.; Moon, S.K.; Choe, I.S.; Choe, Y.K.; Kim, J.W. The bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16ink4a/p14arf proteins. Cancer Lett. 2004, 203, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.H.; Feng, Y.; Zhang, R.; Xu, L.H.; Li, M.Z.; Kung, H.F.; Song, L.B.; Zeng, M.S. Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol. Cancer 2011, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef]
- Zacharek, S.J.; Fillmore, C.M.; Lau, A.N.; Gludish, D.W.; Chou, A.; Ho, J.W.; Zamponi, R.; Gazit, R.; Bock, C.; Jager, N.; et al. Lung stem cell self-renewal relies on bmi1-dependent control of expression at imprinted loci. Cell Stem Cell 2011, 9, 272–281. [Google Scholar] [CrossRef]
- Siddique, H.R.; Saleem, M. Role of bmi1, a stem cell factor, in cancer recurrence and chemoresistance: Preclinical and clinical evidences. Stem Cells 2012, 30, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Garban, H.; Bonavida, B. Yin yang 1 is associated with cancer stem cell transcription factors (sox2, oct4, bmi1) and clinical implication. J. Exp. Clin. Cancer Res. 2016, 35, 84. [Google Scholar] [CrossRef]
- Arif, K.; Hussain, I.; Rea, C.; El-Sheemy, M. The role of nanog expression in tamoxifen-resistant breast cancer cells. Onco Targets Ther. 2015, 8, 1327–1334. [Google Scholar] [PubMed]
- Mehta, G.A.; Khanna, P.; Gatza, M.L. Emerging role of sox proteins in breast cancer development and maintenance. J. Mammary Gland Biol. Neoplasia 2019. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Weina, K.; Utikal, J. Sox2 and cancer: Current research and its implications in the clinic. Clin. Transl. Med. 2014, 3, 19. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Cornwell, M.; Pun, M.; Buchwalter, G.; Nguyen, M.; Bango, C.; Huang, Y.; Kuang, Y.; Paweletz, C.; Fu, X.; et al. Embryonic transcription factor sox9 drives breast cancer endocrine resistance. Proc. Natl. Acad. Sci. USA 2017, 114, E4482–E4491. [Google Scholar] [CrossRef]
- Xue, Y.; Lai, L.; Lian, W.; Tu, X.; Zhou, J.; Dong, P.; Su, D.; Wang, X.; Cao, X.; Chen, Y.; et al. Sox9/fxyd3/src axis is critical for er(+) breast cancer stem cell function. Mol. Cancer Res. 2019, 17, 238–249. [Google Scholar] [CrossRef]
- Crambert, G.; Li, C.; Claeys, D.; Geering, K. Fxyd3 (mat-8), a new regulator of Na,K-Atpase. Mol. Biol. Cell 2005, 16, 2363–2371. [Google Scholar] [CrossRef]
- Hiscox, S.; Barrett-Lee, P.; Borley, A.C.; Nicholson, R.I. Combining src inhibitors and aromatase inhibitors: A novel strategy for overcoming endocrine resistance and bone loss. Eur. J. Cancer 2010, 46, 2187–2195. [Google Scholar] [CrossRef]
- Anbalagan, M.; Rowan, B.G. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 264–272. [Google Scholar] [CrossRef]
- Piggott, L.; Silva, A.; Robinson, T.; Santiago-Gomez, A.; Simoes, B.M.; Becker, M.; Fichtner, I.; Andera, L.; Young, P.; Morris, C.; et al. Acquired resistance of er-positive breast cancer to endocrine treatment confers an adaptive sensitivity to trail through posttranslational downregulation of c-flip. Clin. Cancer Res. 2018, 24, 2452–2463. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, L.; Hu, C.; Liang, S.; Fei, X.; Yan, N.; Zhang, Y.; Zhang, F. Wnt pathway inhibitor pyrvinium pamoate inhibits the self-renewal and metastasis of breast cancer stem cells. Int. J. Oncol. 2016, 48, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Bertini, R.; Allegretti, M.; Bizzarri, C.; Moriconi, A.; Locati, M.; Zampella, G.; Cervellera, M.N.; Di Cioccio, V.; Cesta, M.C.; Galliera, E.; et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors cxcr1 and cxcr2: Prevention of reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 11791–11796. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.; Desgrosellier, J.S. Combined bcl-2/src inhibition synergize to deplete stem-like breast cancer cells. Cancer Lett. 2019, 457, 40–46. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.S.; Farnie, G.; Howell, S.J.; Clarke, R.B. Breast cancer stem cells and their role in resistance to endocrine therapy. Horm. Cancer 2011, 2, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Hoque, M.O. Targeting cancer stem cells: A strategy for effective eradication of cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef]
- Saeg, F.; Anbalagan, M. Breast cancer stem cells and the challenges of eradication: A review of novel therapies. Stem Cell Investig. 2018, 5, 39. [Google Scholar] [CrossRef]
- Dey, P.; Rathod, M.; De, A. Targeting stem cells in the realm of drug-resistant breast cancer. Breast Cancer 2019, 11, 115–135. [Google Scholar] [CrossRef]
- Osipo, C.; Patel, P.; Rizzo, P.; Clementz, A.G.; Hao, L.; Golde, T.E.; Miele, L. Erbb-2 inhibition activates notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene 2008, 27, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, G.; Davaakhuu, G.; Chung, W.C.; Zhu, H.; Rana, A.; Filipovic, A.; Green, A.R.; Atfi, A.; Pannuti, A.; Miele, L.; et al. Akt and 14-3-3 regulate notch4 nuclear localization. Sci. Rep. 2015, 5, 8782. [Google Scholar] [CrossRef] [PubMed]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. Pi3k pathway regulates er-dependent transcription in breast cancer through the epigenetic regulator kmt2d. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. Pi3k inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 2015, 7, 283ra251. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of pi3k and androgen receptor signaling in pten-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.G.; Somer, R.A. Intratumor heterogeneity: Novel approaches for resolving genomic architecture and clonal evolution. Mol. Cancer Res. 2017, 15, 1127–1137. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA damage in stem cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Miraki-Moud, F.; Anjos-Afonso, F.; Pearce, D.J.; Allen, K.; Ridler, C.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Anti-cd38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 2008, 112, 568–575. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. Cd133(+) and cd133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. Emt transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-type-specific chromatin states differentially prime squamous cell carcinoma tumor-initiating cells for epithelial to mesenchymal transition. Cell Stem Cell 2017, 20, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during emt. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Xu, L.; Mao, X.; Guo, T.; Chan, P.Y.; Shaw, G.; Hines, J.; Stankiewicz, E.; Wang, Y.; Oliver, R.T.D.; Ahmad, A.S.; et al. The novel association of circulating tumor cells and circulating megakaryocytes with prostate cancer prognosis. Clin. Cancer Res. 2017, 23, 5112–5122. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.A.; Stoupis, G.; Theodoropoulos, P.A.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Circulating tumor cells with stemness and epithelial-to-mesenchymal transition features are chemoresistant and predictive of poor outcome in metastatic breast cancer. Mol. Cancer Ther. 2019, 18, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Tokunaga, E.; Iimori, M.; Inoue, Y.; Tanaka, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Epithelial paradox: Clinical significance of coexpression of e-cadherin and vimentin with regard to invasion and metastasis of breast cancer. Clin. Breast Cancer 2018, 18, e1003–e1009. [Google Scholar] [CrossRef]
- Rybak, A.P.; He, L.; Kapoor, A.; Cutz, J.C.; Tang, D. Characterization of sphere-propagating cells with stem-like properties from du145 prostate cancer cells. Biochim. Biophys. Acta 2011, 1813, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R. Cancer: Tumour stem-cell surprises. Nature 2017, 543, 626–627. [Google Scholar] [CrossRef] [PubMed]
- de Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A distinct role for lgr5(+) stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Peterse, J.L.; van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining emt during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (met) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | System 1 | BCSC 2 | Ref. |
|---|---|---|---|
| Tamoxifen | MCF7-TamR | CD44+CD24−/low | [70] |
| Tamoxifen | MCF7-TamR | CD44+CD24−/low Mammosphere | [71] |
| Tamoxifen | MCF7-TamR | ALDH1+ | [72] |
| Tamoxifen | MCF7-TamR | CD133+ Mammosphere | [73] |
| Tamoxifen | MCF7 and LM05-E, 5 days treatment | Mammosphere | [74] |
| Tamoxifen | MCF7-TamR | CD44+CD24−/low | [74] |
| Tamoxifen | LM05-E xenografts 3 | CD29hiCD24low | [74] |
| Letrozole | Patient BCs | CD44+CD24−/low | [75] |
| Tamoxifen 4 Fulvestrant 4 | Patient-derived mammosphere, 7–9 days Patient-derived xenograft, 14 days | ALDH1+ | [76] |
| Method | BCSC | Model | Action | Ref. |
|---|---|---|---|---|
| TRAIL | Tamoxifen 1 | Xenograft and PDX | Death receptor | [232] |
| Pyrvinium pamoate | CD44+CD24−/low ALDH+ | Xenograft | Wnt inhibitor | [233] |
| Reparixin | ALDH+ | Xenograft | Agonist of CXCR1/2 | [205] |
| Dasatinib + venetoclaz | BCSCs | In vitro | Src inhibitor BCL2 inhibitor | [235] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, D.; Ramkairsingh, M.; Lin, X.; Kapoor, A.; Major, P.; Tang, D. The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer. Cancers 2019, 11, 1028. https://doi.org/10.3390/cancers11071028
Rodriguez D, Ramkairsingh M, Lin X, Kapoor A, Major P, Tang D. The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer. Cancers. 2019; 11(7):1028. https://doi.org/10.3390/cancers11071028
Chicago/Turabian StyleRodriguez, David, Marc Ramkairsingh, Xiaozeng Lin, Anil Kapoor, Pierre Major, and Damu Tang. 2019. "The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer" Cancers 11, no. 7: 1028. https://doi.org/10.3390/cancers11071028
APA StyleRodriguez, D., Ramkairsingh, M., Lin, X., Kapoor, A., Major, P., & Tang, D. (2019). The Central Contributions of Breast Cancer Stem Cells in Developing Resistance to Endocrine Therapy in Estrogen Receptor (ER)-Positive Breast Cancer. Cancers, 11(7), 1028. https://doi.org/10.3390/cancers11071028