Wnt Signaling in Cancer Metabolism and Immunity


Abstract
:1. Introduction
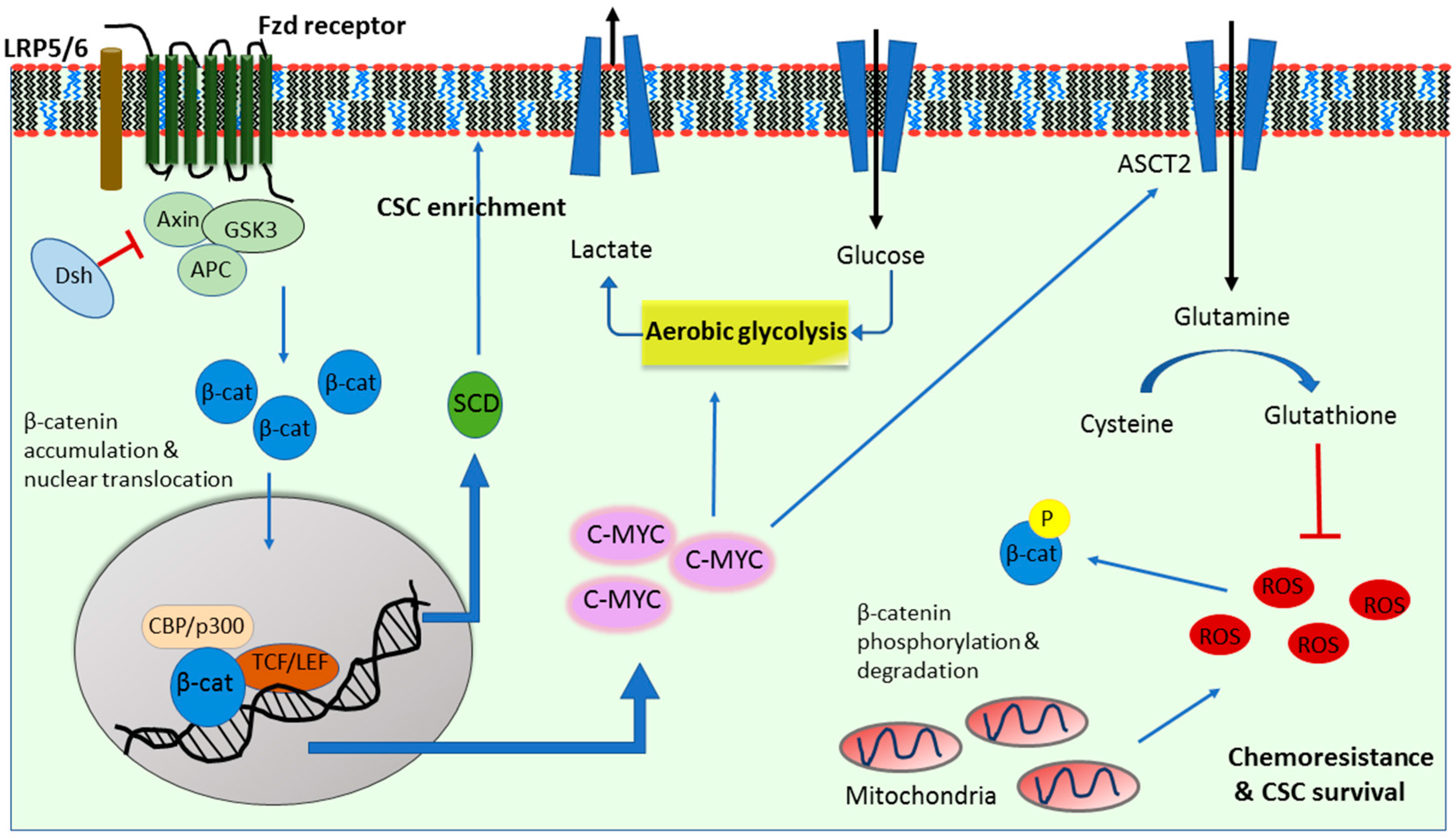
2. Wnt Signaling in Cancer Glycolysis
3. Wnt Signaling in Cancer Glutaminolysis
4. Wnt Signaling in Cancer Lipogenesis
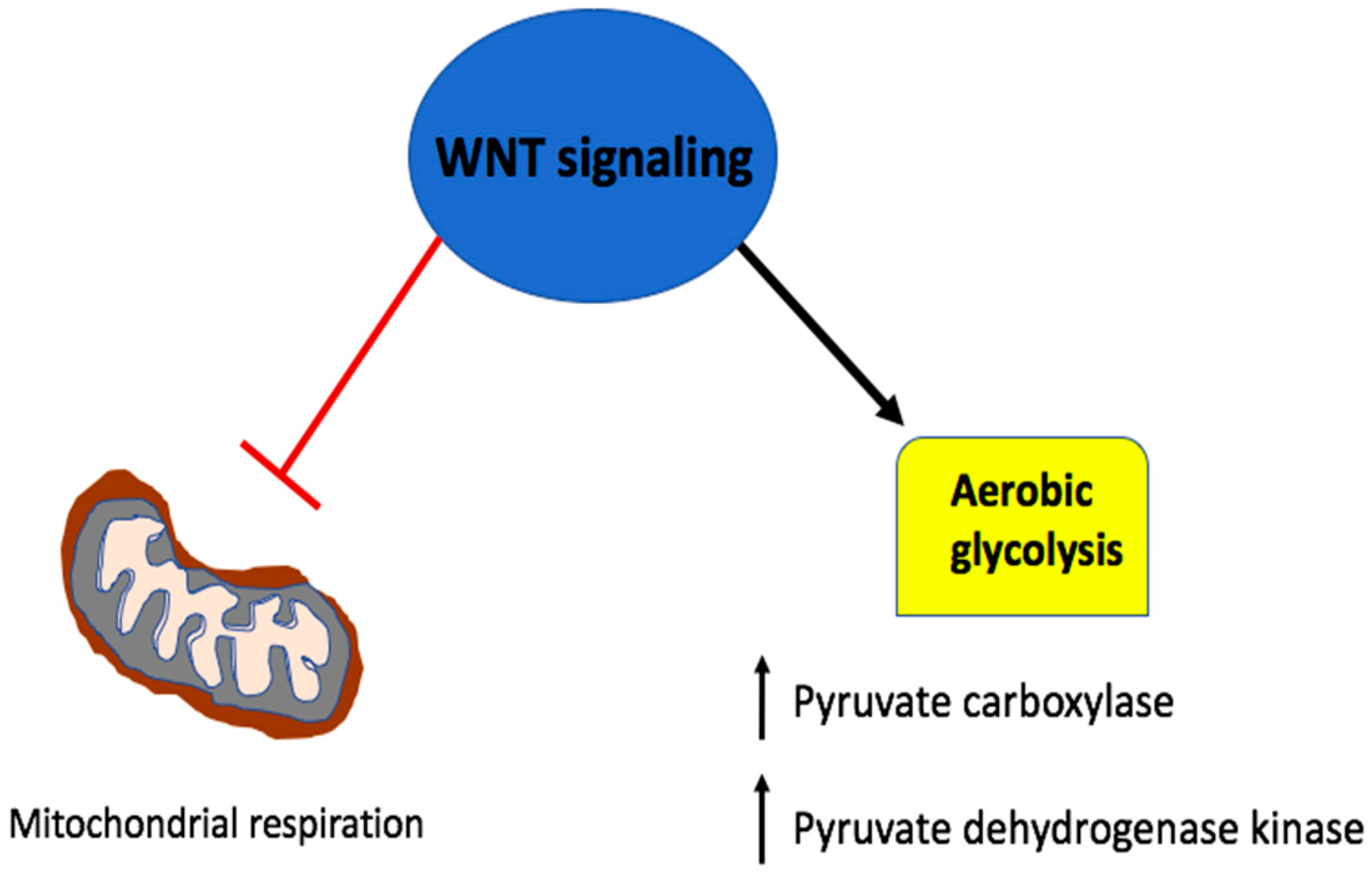
5. Wnt Signaling in A Metabolic Negative Feedback Loop
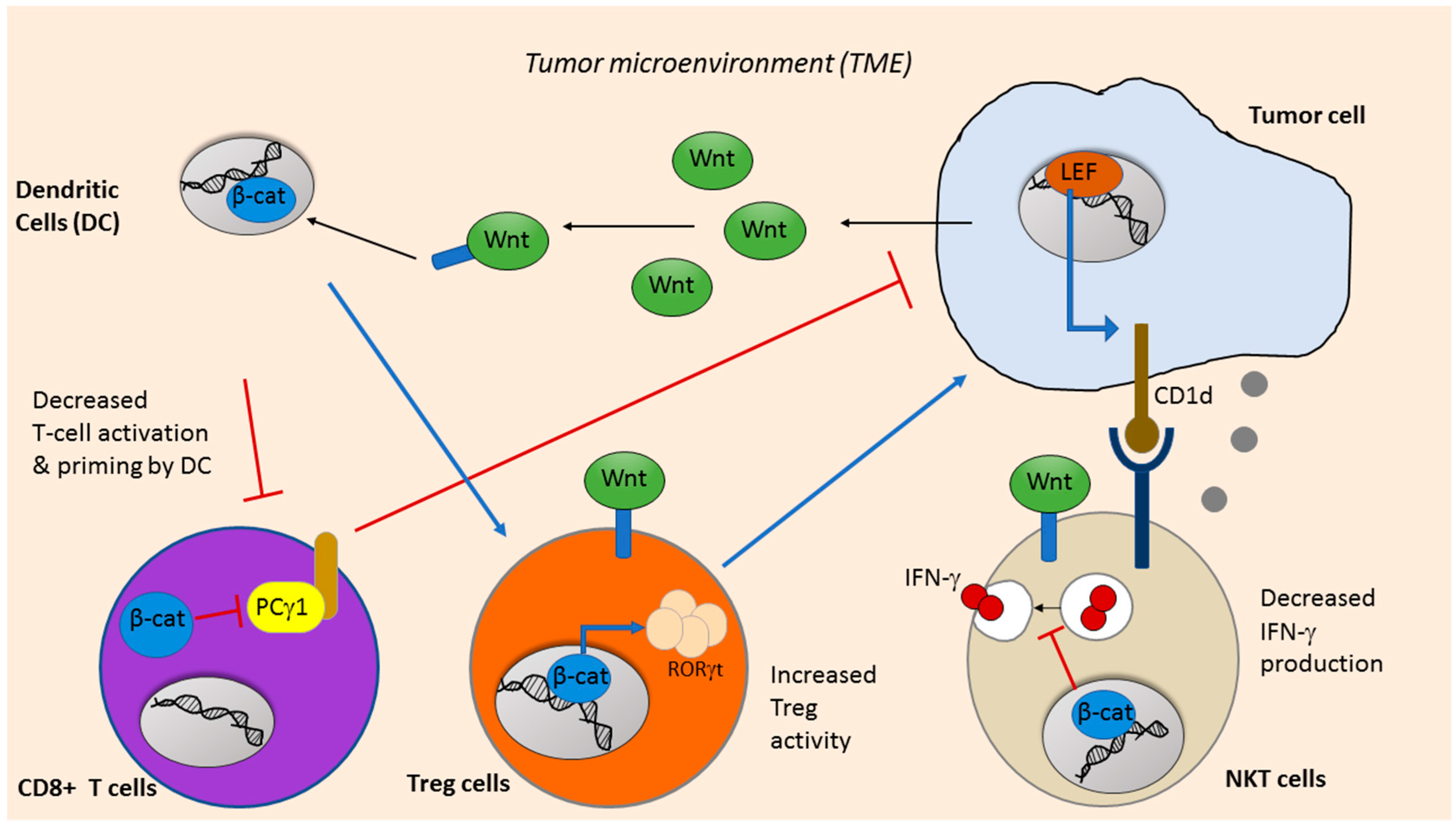
6. Wnt Signaling in Cancer Immunotherapy
7. Future Directions: Targeting Wnt Signaling to Inhibit Cancer Metabolism and Enhance Immune Response
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, A.; Benchabane, H.; Wang, Z.; Ahmed, Y. Regulation of Stem Cell Proliferation and Cell Fate Specification by Wingless/Wnt Signaling Gradients Enriched at Adult Intestinal Compartment Boundaries. PLoS Genet. 2016, 12, e1005822. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Schlange, T.; Oakeley, E.J.; Boulay, A.; Hynes, N.E. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 2009, 11, R32. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Yang, W.; Li, L.; Liu, H.; Tan, Y.; Ooi, S.; Chi, L.; Filion, L.G.; Figeys, D.; Wang, L. Beta-Catenin and NF-kappaB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. 2015, 22, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Tuupanen, S.; Turunen, M.; Lehtonen, R.; Hallikas, O.; Vanharanta, S.; Kivioja, T.; Bjorklund, M.; Wei, G.; Yan, J.; Niittymaki, I.; et al. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat. Genet. 2009, 41, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbosh, P.H.; Nephew, K.P. Multiple signaling pathways converge on beta-catenin in thyroid cancer. Thyroid 2005, 15, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sinha, T.; Wynshaw-Boris, A. Wnt signaling in mammalian development: Lessons from mouse genetics. Cold Spring Harb. Perspect. Biol. 2012, 4, a007963. [Google Scholar] [CrossRef]
- Ramos-Solano, M.; Meza-Canales, I.D.; Torres-Reyes, L.A.; Alvarez-Zavala, M.; Alvarado-Ruiz, L.; Rincon-Orozco, B.; Garcia-Chagollan, M.; Ochoa-Hernandez, A.B.; Ortiz-Lazareno, P.C.; Rosl, F.; et al. Expression of WNT genes in cervical cancer-derived cells: Implication of WNT7A in cell proliferation and migration. Exp. Cell Res. 2015, 335, 39–50. [Google Scholar] [CrossRef]
- Cui, N.; Yang, W.T.; Zheng, P.S. Slug inhibits the proliferation and tumor formation of human cervical cancer cells by up-regulating the p21/p27 proteins and down-regulating the activity of the Wnt/beta-catenin signaling pathway via the trans-suppression Akt1/p-Akt1 expression. Oncotarget 2016, 7, 26152–26167. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Mohseny, A.B.; Karperien, M.; Hogendoorn, P.C.; Zhou, G.; Cleton-Jansen, A.M. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J. Pathol. 2009, 220, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Knowles, E.; Zardawi, S.J.; McNeil, C.M.; Millar, E.K.; Crea, P.; Musgrove, E.A.; Sutherland, R.L.; O’Toole, S.A. Cytoplasmic localization of beta-catenin is a marker of poor outcome in breast cancer patients. Cancer Epidemiol. Prev. Biomark. 2010, 19, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Ambrosi, G.; Wandmacher, A.M.; Rauscher, B.; Betge, J.; Rindtorff, N.; Haussler, R.S.; Hinsenkamp, I.; Bamberg, L.; Hessling, B.; et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat. Commun. 2019, 10, 2197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hu, J.; Huang, Z.; Li, H.; Liu, X.; Feng, W. Effect of indomethacin on BCR/ABL-Wnt/beta-catenin pathway in K562 cells. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 998–1002. [Google Scholar] [PubMed]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londono-Joshi, A.I.; Gangrade, A.; Katre, A.A.; Kurpad, C.; Li, Y.; Samant, R.S.; Li, P.K.; Landen, C.N.; Yang, E.S.; et al. Niclosamide and its analogs are potent inhibitors of Wnt/beta-catenin, mTOR and STAT3 signaling in ovarian cancer. Oncotarget 2016, 7, 86803–86815. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lan, C.; Li, L.; Yang, D.; Xia, X.; Liao, Q.; Fu, W.; Chen, X.; An, S.; Wang, W.E.; et al. A novel porcupine inhibitor blocks WNT pathways and attenuates cardiac hypertrophy. Biochim Biophys. Acta Mol. Basis Dis. 2018, 1864, 3459–3467. [Google Scholar] [CrossRef]
- Huang, R.; Han, J.; Liang, X.; Sun, S.; Jiang, Y.; Xia, B.; Niu, M.; Li, D.; Zhang, J.; Wang, S.; et al. Androgen Receptor Expression and Bicalutamide Antagonize Androgen Receptor Inhibit beta-Catenin Transcription Complex in Estrogen Receptor-Negative Breast Cancer. Cell Physiol. Biochem. 2017, 43, 2212–2225. [Google Scholar] [CrossRef]
- Mina, A.; Yoder, R.; Sharma, P. Targeting the androgen receptor in triple-negative breast cancer: Current perspectives. Onco Targets Ther. 2017, 10, 4675–4685. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Schoffski, P.; Littlefield, B.A. Multiple modes of action of eribulin mesylate: Emerging data and clinical implications. Cancer Treat. Rev. 2018, 70, 190–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Lin, X.; Zhang, C.; An, G.; Li, Z.; Dong, B.; Shen, L.; Gao, J.; Zhang, X. Activated Wnt signaling promotes growth and progression of AFP-producing gastric cancer in preclinical models. Cancer Manag. Res. 2019, 11, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Chen, Y.; Liu, H.; Yang, J.; Song, X.; Zhao, J.; He, N.; Zhou, C.J.; Wang, Y.; Dong, Q. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 2017, 8, 115254–115269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, H.; Ong, R.; Goh, K.Y.; Lee, L.Y.; Puehler, F.; Scholz, A.; Politz, O.; Mumberg, D.; Ziegelbauer, K. Sorafenib/MEK inhibitor combination inhibits tumor growth and the Wnt/betacatenin pathway in xenograft models of hepatocellular carcinoma. Int. J. Oncol. 2019, 54, 1123–1133. [Google Scholar]
- Fischer, M.M.; Cancilla, B.; Yeung, V.P.; Cattaruzza, F.; Chartier, C.; Murriel, C.L.; Cain, J.; Tam, R.; Cheng, C.Y.; Evans, J.W.; et al. WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci. Adv. 2017, 3, e1700090. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Cao, L.; Zhu, J.; Tan, Z.; Tang, M.; Li, Z.; Hu, Y.; Yu, R.; Zhang, S.; Song, L.; et al. Loss of RBMS3 Confers Platinum Resistance in Epithelial Ovarian Cancer via Activation of miR-126-5p/beta-catenin/CBP signaling. Clin. Cancer Res. 2019, 25, 1022–1035. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Liu, J.X.; Endo, T.; Zhou, H.; Yao, S.; Willert, K.; Schmidt-Wolf, I.G.; Kipps, T.J.; Carson, D.A. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/beta-catenin pathway. PLoS ONE 2009, 4, e8294. [Google Scholar] [CrossRef]
- Liu, H.; Fergusson, M.M.; Wu, J.J.; Rovira, I.I.; Liu, J.; Gavrilova, O.; Lu, T.; Bao, J.; Han, D.; Sack, M.N.; et al. Wnt signaling regulates hepatic metabolism. Sci. Signal. 2011, 4, ra6. [Google Scholar] [CrossRef] [PubMed]
- Fevr, T.; Robine, S.; Louvard, D.; Huelsken, J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell Biol. 2007, 27, 7551–7559. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, M.; Reichert, T.E.; Benz, P.; Lehr, H.A.; Jeong, J.H.; Wieand, S.; Bartenstein, P.; Wagner, W.; Whiteside, T.L. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer 2003, 97, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Perez, A.A.; Fujie, S.; Warden, C.; Li, J.; Wang, Y.; Yung, B.; Chen, Y.R.; Liu, X.; Zhang, H.; et al. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer 2014, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers (Basel) 2018, 10, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef] [PubMed]
- van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.N.; Marks, J.R.; Chi, J.T. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011, 7, e1002229. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Li, C.; Zhang, G.; Zhao, L.; Ma, Z.; Chen, H. Metabolic reprogramming in cancer cells: Glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer. World J. Surg. Oncol. 2016, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Terris, B.; Souil, E.; Levy, L.; Lamers, W.H.; Kitajewski, J.; Kahn, A.; Perret, C. New targets of beta-catenin signaling in the liver are involved in the glutamine metabolism. Oncogene 2002, 21, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Li, Z.; Yang, P.; Zhang, L.; Fan, Y. PKM2 depletion induces the compensation of glutaminolysis through beta-catenin/c-Myc pathway in tumor cells. Cell. Signal. 2014, 26, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through beta-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed]
- Mihm, S.; Galter, D.; Droge, W. Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply. FASEB J. 1995, 9, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Miran, T.; Vogg, A.T.J.; Drude, N.; Mottaghy, F.M.; Morgenroth, A. Modulation of glutathione promotes apoptosis in triple-negative breast cancer cells. FASEB J. 2018, 32, 2803–2813. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patel, M.R.; Tannir, N.M.; Owonikoko, T.K.; Haas, N.B.; Voss, M.H.; et al. CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. J. Clin. Oncol. 2019, 37, 549. [Google Scholar] [CrossRef]
- Jones, J.E.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; Lopez-Vilaro, L.; Nasarre, L.; Perez-Olabarria, M.; Vazquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortes, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Hardy, R.W.; Ren, C.; Ponnazhagan, S.; Welch, D.R.; McDonald, J.M.; Chen, Y. Free fatty acids enhance breast cancer cell migration through plasminogen activator inhibitor-1 and SMAD4. Lab. Investig. 2009, 89, 1221–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Sun, S.; Wang, J.; Fei, F.; Dong, Z.; Ke, A.W.; He, R.; Wang, L.; Zhang, L.; Ji, M.B.; et al. Canonical Wnt Signaling Remodels Lipid Metabolism in Zebrafish Hepatocytes following Ras Oncogenic Insult. Cancer Res. 2018, 78, 5548–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.P.; Fargnoli, J.; Henley, B.J.; Yang, W.P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 2011, 9, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Kenny, H.A.; Lengyel, E. Unsaturated Fatty Acids Maintain Cancer Cell Stemness. Cell Stem Cell 2017, 20, 291–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. beta-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e5. [Google Scholar] [CrossRef]
- Colacino, J.A.; McDermott, S.P.; Sartor, M.A.; Wicha, M.S.; Rozek, L.S. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res. Treat. 2016, 158, 29–41. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Lai, K.K.Y.; Kweon, S.M.; Chi, F.; Hwang, E.; Kabe, Y.; Higashiyama, R.; Qin, L.; Yan, R.; Wu, R.P.; Lai, K.; et al. Stearoyl-CoA Desaturase Promotes Liver Fibrosis and Tumor Development in Mice via a Wnt Positive-Signaling Loop by Stabilization of Low-Density Lipoprotein-Receptor-Related Proteins 5 and 6. Gastroenterology 2017, 152, 1477–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, L.; Zhang, H.; Wang, D.; Wang, M.; Zhu, J.; Pang, C.; Wang, C. Succinate dehydrogenase 5 (SDH5) regulates glycogen synthase kinase 3beta-beta-catenin-mediated lung cancer metastasis. J. Biol. Chem. 2013, 288, 29965–29973. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Takemaru, K.; Yamaguchi, S.; Lee, Y.S.; Zhang, Y.; Carthew, R.W.; Moon, R.T. Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway. Nature 2003, 422, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.F.; Ye, G.D.; Shen, D.Y.; Zhang, W.; Chen, M.L.; Chen, X.X.; Han, D.X.; Mi, Y.J.; Luo, Q.C.; Cai, W.Y.; et al. Chibby suppresses aerobic glycolysis and proliferation of nasopharyngeal carcinoma via the Wnt/beta-catenin-Lin28/let7-PDK1 cascade. J. Exp. Clin. Cancer Res. 2018, 37, 104. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xiong, X.; Scott, T.; Li, A.T.; Wang, C.; Weiss, H.L.; Tan, L.; Bradford, E.; Fan, T.W.M.; Chandel, N.S.; et al. The mitochondrial retrograde signaling regulates Wnt signaling to promote tumorigenesis in colon cancer. Cell Death Differ. 2019. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Shi, Y.B. An essential and evolutionarily conserved role of protein arginine methyltransferase 1 for adult intestinal stem cells during postembryonic development. Stem Cells 2010, 28, 2073–2083. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2014, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.T.; Adams, S.F.; Tahirovic, E.; Hagemann, I.S.; Coukos, G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2012, 124, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Role of dendritic cells in the induction of regulatory T cells. Cell Biosci. 2011, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Bao, R.; Sweis, R.F.; Spranger, S.; Gajewski, T.F. WNT/beta-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clin. Cancer Res. 2019, 25, 3074–3083. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Fu, C.; Cui, W.; Ober-Blobaum, J.L.; Zahner, S.P.; Shrikant, P.A.; Clausen, B.E.; Flavell, R.A.; Mellman, I.; Jiang, A. beta-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8(+) T cells. J. Leukoc. Biol. 2014, 95, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Shanmugam, A.; Swafford, D.; Ahmad, S.; Chinnadurai, R.; Manicassamy, B.; He, Y.; Mellor, A.L.; et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology 2016, 5, e1115941. [Google Scholar] [CrossRef]
- Kerdidani, D.; Chouvardas, P.; Arjo, A.R.; Giopanou, I.; Ntaliarda, G.; Guo, Y.A.; Tsikitis, M.; Kazamias, G.; Potaris, K.; Stathopoulos, G.T.; et al. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat. Commun. 2019, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Keerthivasan, S.; Aghajani, K.; Dose, M.; Molinero, L.; Khan, M.W.; Venkateswaran, V.; Weber, C.; Emmanuel, A.O.; Sun, T.; Bentrem, D.J.; et al. beta-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl. Med. 2014, 6, 225ra28. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jin, J.Q.; Xia, L.; Xiao, T.; Mei, S.; Wang, X.; Huang, X.; Chen, J.; Liu, M.; Chen, C.; et al. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci. Adv. 2019, 5, eaau5240. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.M.; Yu, S.; Zhou, X.; Haring, J.S.; Held, W.; Badovinac, V.P.; Harty, J.T.; Xue, H.H. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J. Immunol. 2010, 184, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Driessens, G.; Zheng, Y.; Locke, F.; Cannon, J.L.; Gounari, F.; Gajewski, T.F. Beta-catenin inhibits T cell activation by selective interference with linker for activation of T cells-phospholipase C-gamma1 phosphorylation. J. Immunol. 2011, 186, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Wu, J.; Wang, W.J.; Chen, S.; Zheng, Y.; Yu, X.; Meeth, K.; Sahraei, M.; Bothwell, A.L.M.; Chen, L.; et al. DKK2 imparts tumor immunity evasion through beta-catenin-independent suppression of cytotoxic immune-cell activation. Nat. Med. 2018, 24, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Dhodapkar, M.V. Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.Y.; Zhang, T.; Pincus, S.H.; Wu, S.; Ricks, D.; Liu, D.; Sun, Z.; Maclaren, N.; Lan, M.S. Human CD1D gene expression is regulated by LEF-1 through distal promoter regulatory elements. J. Immunol. 2010, 184, 5047–5054. [Google Scholar] [CrossRef] [PubMed]
- Berga-Bolanos, R.; Sharma, A.; Steinke, F.C.; Pyaram, K.; Kim, Y.H.; Sultana, D.A.; Fang, J.X.; Chang, C.H.; Xue, H.H.; Heller, N.M.; et al. beta-Catenin is required for the differentiation of iNKT2 and iNKT17 cells that augment IL-25-dependent lung inflammation. BMC Immunol. 2015, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Pyaram, K.; Sen, J.M.; Chang, C.H. Temporal regulation of Wnt/beta-catenin signaling is important for invariant NKT cell development and terminal maturation. Mol. Immunol. 2017, 85, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kling, J.C.; Jordan, M.A.; Pitt, L.A.; Meiners, J.; Thanh-Tran, T.; Tran, L.S.; Nguyen, T.T.K.; Mittal, D.; Villani, R.; Steptoe, R.J.; et al. Temporal Regulation of Natural Killer T Cell Interferon Gamma Responses by beta-Catenin-Dependent and -Independent Wnt Signaling. Front. Immunol. 2018, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Yu, H.; Zhang, J.; Jing, H.; Zhu, W.; Li, X.; Kong, L.; Xing, L.; Yu, J.; Meng, X. Correlation of cancer stem cell markers and immune cell markers in resected non-small cell lung cancer. J. Cancer 2017, 8, 3190–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Robitaille, A.M.; Berndt, J.D.; Davidson, K.C.; Fischer, K.A.; Mathieu, J.; Potter, J.C.; Ruohola-Baker, H.; Moon, R.T. Wnt/beta-catenin signaling promotes self-renewal and inhibits the primed state transition in naive human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6382–E6390. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2009, 70, 68–77. [Google Scholar] [CrossRef]
- Van de Velde, L.A.; Subramanian, C.; Smith, A.M.; Barron, L.; Qualls, J.E.; Neale, G.; Alfonso-Pecchio, A.; Jackowski, S.; Rock, C.O.; Wynn, T.A.; et al. T Cells Encountering Myeloid Cells Programmed for Amino Acid-dependent Immunosuppression Use Rictor/mTORC2 Protein for Proliferative Checkpoint Decisions. J. Biol. Chem. 2017, 292, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A.; Filipazzi, P.; De Milito, A.; Fais, S.; Rivoltini, L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology 2013, 2, e22058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sprowl-Tanio, S.; Habowski, A.N.; Pate, K.T.; McQuade, M.M.; Wang, K.; Edwards, R.A.; Grun, F.; Lyou, Y.; Waterman, M.L. Lactate/pyruvate transporter MCT-1 is a direct Wnt target that confers sensitivity to 3-bromopyruvate in colon cancer. Cancer Metab. 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Vincent, E.E.; Cronin, J.G.; Panetti, S.; Chambers, M.; Holm, S.R.; Owens, S.E.; Francis, N.J.; Finlay, D.K.; Thornton, C.A. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat. Commun. 2019, 10, 2042. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Jope, R.S. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008, 283, 21934–21944. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Mijn, J.C.; Panka, D.J.; Geissler, A.K.; Verheul, H.M.; Mier, J.W. Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. 2016, 4, 14. [Google Scholar] [CrossRef]
- Hassanein, M.; Qian, J.; Hoeksema, M.D.; Wang, J.; Jacobovitz, M.; Ji, X.; Harris, F.T.; Harris, B.K.; Boyd, K.L.; Chen, H.; et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int. J. Cancer 2015, 137, 1587–1597. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Curi, R.; Pithon Curi, T.C.; Murphy, C.J.; Garcia, C.; Pires de Melo, M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: Its importance in health and disease. J. Nutr. Biochem. 1999, 10, 316–324. [Google Scholar] [CrossRef]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef]
- Wang, W.; Kryczek, I.; Dostal, L.; Lin, H.; Tan, L.; Zhao, L.; Lu, F.; Wei, S.; Maj, T.; Peng, D.; et al. Effector T Cells Abrogate Stroma-Mediated Chemoresistance in Ovarian Cancer. Cell 2016, 165, 1092–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Cancer Type | Phase | Component Targeted | References |
|---|---|---|---|---|
| NCT02950259 | Breast cancer | I | β-catenin | [16] |
| NCT02807805 | Prostate cancer | II | Dvl & β-catenin | [17,18] |
| NCT02675946 | GI cancer | I | Wnt ligands | [19] |
| NCT03090165 | TNBC | II | β-catenin | [20,21] |
| NCT02513472 | Breast cancer | I | β-catenin | [22] |
| NCT03355066 | Advanced solid tumors | I | Unknown | [23] |
| NCT01351103 | Lung cancer, colorectal cancer, TNBC... etc. | I | Wnt ligands | [24] |
| NCT02429427 | Breast cancer | III | GSK3 | [25] |
| NCT02346032 | Biliary Tract Cancer | II | Wnt3 & LRP6 | [26] |
| NCT02005315 | Pancreatic Cancer | I | FZD | [27] |
| NCT01302405 | Advanced solid tumors | I | β-catenin/CBP | [28] |
| NCT02402764 | Breast cancer | II | APC | [29] |
| NCT02426723 | Multiple myeloma | I | β-catenin | NCT02426723 |
| NCT02852564 | Bladder cancer | I | LEF-1/βcatenin | [30] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. https://doi.org/10.3390/cancers11070904
El-Sahli S, Xie Y, Wang L, Liu S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers. 2019; 11(7):904. https://doi.org/10.3390/cancers11070904
Chicago/Turabian StyleEl-Sahli, Sara, Ying Xie, Lisheng Wang, and Sheng Liu. 2019. "Wnt Signaling in Cancer Metabolism and Immunity" Cancers 11, no. 7: 904. https://doi.org/10.3390/cancers11070904
APA StyleEl-Sahli, S., Xie, Y., Wang, L., & Liu, S. (2019). Wnt Signaling in Cancer Metabolism and Immunity. Cancers, 11(7), 904. https://doi.org/10.3390/cancers11070904