Targeting Cancer Stem Cells in Triple-Negative Breast Cancer



Abstract

1. Introduction
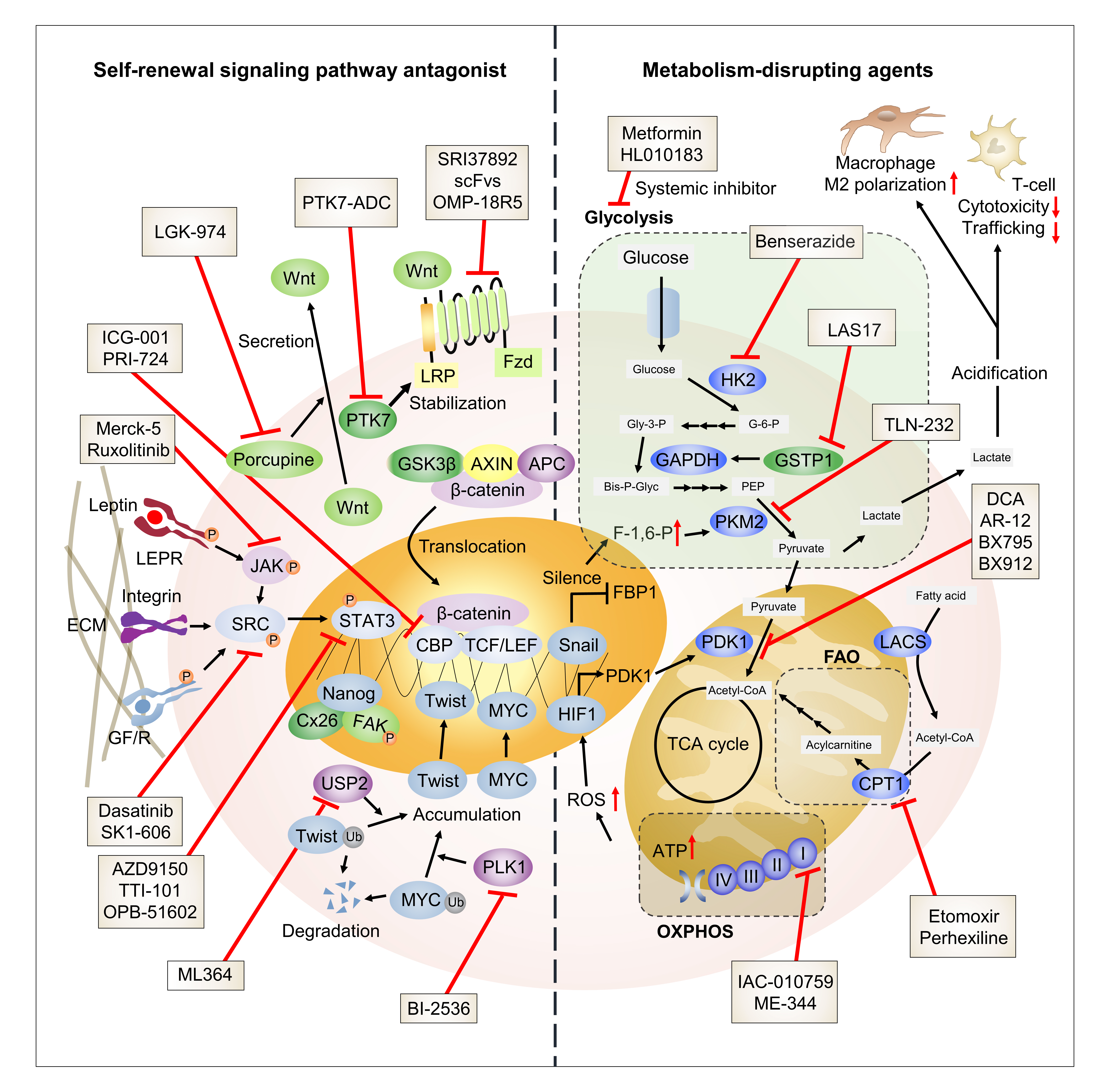
2. Approaches for Targeting the Self-Renewal Process in TNBC Cells
2.1. STAT3
2.1.1. STAT3 Signaling in BCSCs
2.1.2. STAT3 Signaling Dysregulation in TNBC Cells
2.1.3. STAT3 Signaling Inhibitors in Clinical Trials
2.2. Proto-Oncogene Tyrosine-Protein Kinase Src (SRC)Signaling
2.2.1. SRC Kinase Signaling in BCSCs
2.2.2. SRC Kinase Signaling Dysregulation in TNBC Cells
2.2.3. SRC Kinase Inhibitors in Clinical Trials
2.3. Wnt/β-Catenin Signaling
2.3.1. Wnt/β-Catenin Signaling in BCSCs
2.3.2. Dysregulation of Wnt/β-Catenin Signaling in TNBC Cells
2.3.3. Wnt/β-Catenin Signaling Inhibitors in Clinical Trials
2.4. Other Molecules Linked to TNBC Self-Renewal
2.4.1. Connexin (CX)
2.4.2. Ubiquitin-Specific Protease (USP)
2.4.3. Polo-Like Kinase (PLK)
3. Attempts to Target Metabolic Reprogramming in Triple Negative Breast Cancer (TNBC)
3.1. Anaerobic Glycolysis
3.1.1. Glycolysis in Breast Cancer Stem Cells (BCSCs)
3.1.2. Glycolysis in TNBC Cells
3.1.3. Glycolysis Inhibitors in Clinical Trials
3.2. OXPHOS
3.2.1. OXPHOS in BCSCs
3.2.2. OXPHOS in TNBC Cells
3.2.3. OXPHOS Inhibitors
3.3. FAO
3.3.1. FAO in BCSCs
3.3.2. FAO in TNBC Cells
3.3.3. FAO Inhibitors in Clinical Trials
3.4. Other Molecules Linked to TNBC Metabolism
Glutathione S-Transferase Pi 1 (GSTP1)
4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| 2DG | 2-Deoxyglucose |
| ACADM | Acyl-coenzyme A dehydrogenase medium chain |
| acetyl-CoA | Acetyl-coenzyme A |
| ALDH | Aldehyde dehydrogenase |
| APC | Adenomatous polyposis coli |
| ATP | Adenosine triphosphate |
| BCSC | Breast cancer stem cells |
| CBP | cAMP response element-binding protein (CREB)-binding protein |
| CD24 | Cluster of differentiation 24 |
| CDCP1 | CUB domain-containing protein 1 |
| CK1 | Casein kinase 1 |
| CPT1 | Carnitine palmitoyltransferase 1 |
| CREB | cAMP response element-binding protein |
| CSC | Cancer stem cell |
| CX | Connexin |
| CXCL | Chemokine (C-X-C motif) ligand |
| DCA | Dichloroacetate |
| ECAR | Extracellular acidification rate |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial to mesenchymal transition |
| ER | Estrogen receptor |
| FAK | Focal adhesion kinase |
| FAO | Fatty acid oxidation |
| FBP1 | Fructose-1,6-biphosphatase 1 |
| FDR | False discovery rate |
| FGFR | Fibroblast growth factor receptor |
| FZD | Frizzled |
| G-6-P | Glucose-6-phosphate |
| GEO | Gene expression omnibus |
| GF/R | Growth factor and receptor |
| GJ | Gap junction |
| GSE | Gene set enrichment |
| GSEA | Gene set enrichment analysis |
| GSK3β | Glycogen synthase kinase 3 beta |
| GSTO1 | Glutathione S-transferase omega 1 |
| GSTP1 | Glutathione S-transferase pi 1 |
| HAS1 | Hyaluronan synthase 1 |
| HER2 | Human epidermal growth factor receptor 2 |
| HIF1α | Hypoxia-inducible factor 1 alpha |
| HK | Hexokinase |
| HN1L | Hematological and neurological expressed 1-like |
| IL | Interleukin |
| IL6RA | Interleukin 6 receptor, alpha |
| JAG1 | Jagged canonical Notch ligand 1 |
| JAK | Janus kinase |
| KLF4 | Kruppel-like factor 4 |
| LACS | Long-chain acyl-CoA synthetase |
| LEF | Lymphoid enhancer-binding factor |
| LEPR | Leptin receptor |
| LIF | Leukemia inhibitory factor |
| LRP | Low-density lipoprotein receptor-related protein |
| MMTV-PyMT | Mouse mammary tumor virus-polyoma middle tumor-antigen |
| MSI1 | Musashi RNA binding protein 1 |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NES | Normalized enrichment score |
| NICD | Notch1 intracellular domain |
| OCR | Oxygen consumption rate |
| OCT4 | Octamer-binding transcription factor 4 |
| OXPHOS | Oxidative phosphorylation |
| PCR | Polymerase chain reaction |
| PDH | Pyruvate dehydrogenase |
| PDX | Patient-derived tumor xenograft |
| p-FAK | Activated form of focal adhesion kinase |
| PFKFB | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase |
| PI3K | Phosphoinositide 3-kinase |
| PK | Pyruvate kinase |
| PKM | M isoform of PK |
| PLK | Polo-like kinase |
| PR | Progesterone receptor |
| PTGIS | Prostaglandin I2 (prostacyclin) synthase |
| PTK7 | Tyrosine-protein kinase-like 7 |
| PYK2 | Pyruvate kinase 2 |
| ROS | Reactive oxygen species |
| RYR1 | Type 1 ryanodine receptor |
| SOX | SRY (sex determining region Y)-box |
| SPSP | Self-renewal signaling pathway |
| STAT | Signal transducer and activator of transcription |
| TCF | T cell factor |
| TNBC | Triple-negative breast cancer |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| USP | Ubiquitin-specific protease |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Glass, J. The phenotypic radiation resistance of CD44+/CD24− or low breast cancer cells is mediated through the enhanced activation of ATM signaling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef] [PubMed]
- Van Phuc, P.; Nhan, P.L.C.; Nhung, T.H.; Tam, N.T.; Hoang, N.M.; Tue, V.G.; Thuy, D.T.; Ngoc, P.K. Downregulation of CD44 reduces doxorubicin resistance of CD44+ CD24− breast cancer cells. Oncotargets Ther. 2011, 4, 71–78. [Google Scholar] [CrossRef]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Di Benedetto, A.; Todaro, M.; Stassi, G.; Sperati, F. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44+ CD24−/low breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44+/CD24− population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef]
- Li, H.; Ma, F.; Wang, H.; Lin, C.; Fan, Y.; Zhang, X.; Qian, H.; Xu, B. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int. J. Biol. Mark. 2013, 28, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Honeth, G.; Bendahl, P.-O.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, Å.; Hegardt, C. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Song, C.; Liu, P.; Wang, J.; Chen, B.; Huang, X.; Pei, X.; Liu, L. SOX2 promotes cell proliferation and metastasis in triple negative breast cancer. Front. Pharmacol. 2018, 9, 942. [Google Scholar]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Yehiely, F.; Moyano, J.V.; Evans, J.R.; Nielsen, T.O.; Cryns, V.L. Deconstructing the molecular portrait of basal-like breast cancer. Trends Mol. Med. 2006, 12, 537–544. [Google Scholar] [CrossRef]
- Raz, R.; Lee, C.-K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of STAT3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsky, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Karihtala, P.; Auvinen, P.; Kauppila, S.; Haapasaari, K.-M.; Jukkola-Vuorinen, A.; Soini, Y. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: Association with an aggressive tumour phenotype. Breast Cancer Res. Treat. 2013, 138, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Shang, M.; Zhang, Y.; Xia, B.; Niu, M.; Liu, Y.; Pang, D. Dysregulated expression of Slug, vimentin, and E-cadherin correlates with poor clinical outcome in patients with basal-like breast cancer. J. Surg. Oncol. 2013, 107, 188–194. [Google Scholar] [CrossRef]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef]
- Sarrió, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]
- Yamashita, N.; Tokunaga, E.; Kitao, H.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; Aishima, S.; Morita, M.; Maehara, Y. Vimentin as a poor prognostic factor for triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Nalla, L.V.; Kalia, K.; Khairnar, A. Self-renewal signaling pathways in breast cancer stem cells. Int. J. Biochem. Cell Biol. 2019, 107, 140–153. [Google Scholar] [CrossRef]
- Hirai, H.; Karian, P.; Kikyo, N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem. J. 2011, 438, 11–23. [Google Scholar] [CrossRef]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Qin, S.; Schulte, B.A.; Ethier, S.P.; Tew, K.D.; Wang, G.Y. MYC inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res. 2017, 77, 6641–6650. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Tian, J.; Al Raffa, F.; Dai, M.; Moamer, A.; Khadang, B.; Hachim, I.Y.; Bakdounes, K.; Ali, S.; Jean-Claude, B.; Lebrun, J.-J. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. Br. J. Cancer 2018, 119, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Dunlap, S.M.; Zhu, J.; Downs-Kelly, E.; Rich, J.; Hursting, S.D.; Berger, N.A.; Reizes, O. Leptin deficiency suppresses MMTV-Wnt-1 mammary tumor growth in obese mice and abrogates tumor initiating cell survival. Endocr. Relat. Cancer 2011, 18, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, P.S.; Zheng, Q.; Bhagrath, M.; Mulkearns-Hubert, E.E.; Myers, M.G.; Lathia, J.D.; Reizes, O. STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr. Relat. Cancer 2017, 24, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Liu, Y.; Choi, D.S.; Sheng, J.; Ensor, J.E.; Liang, D.H.; Rodriguez-Aguayo, C.; Polley, A.; Benz, S.; Elemento, O.; Verma, A. HN1L promotes triple-negative breast cancer stem cells through LEPR-STAT3 pathway. Stem Cell Rep. 2018, 10, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Müller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: A rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Bernaciak, T.M.; Zareno, J.; Parsons, J.T.; Silva, C.M. A novel role for signal transducer and activator of transcription 5b (STAT5b) in beta1-integrinmediated human breast cancer cell migration. Breast Cancer Res. 2009, 11, R52. [Google Scholar] [CrossRef]
- Walker, S.R.; Nelson, E.A.; Zou, L.; Chaudhury, M.; Signoretti, S.; Richardson, A.; Frank, D.A. Reciprocal Effects of STAT5 and STAT3 in Breast Cancer. Mol. Cancer Res. 2009, 7, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.S.; Brim, H.; Sherif, Z.A. Co-overexpression of Janus kinase 2 and signal transducer and activator of transcription 5a promotes differentiation of mammary cancer cells through reversal of epithelial–mesenchymal transition. Cancer Sci. 2008, 99, 272–279. [Google Scholar] [CrossRef]
- Sultan, A.S.; Xie, J.; LeBaron, M.J.; Ealley, E.L.; Nevalainen, M.T.; Rui, H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene 2005, 24, 746–760. [Google Scholar] [CrossRef]
- Tvorogov, D.; Sundvall, M.; Kurppa, K.; Hollmén, M.; Repo, S.; Johnson, M.S.; Elenius, K. Somatic mutations of ErbB4: Selective loss-of-function phenotype affecting signal transduction pathways in cancer. J. Biol. Chem. 2009, 284, 5582–5591. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Tweardy, D.J. STAT3 inhibitors in cancer: A comprehensive update. In STAT Inhibitors in Cancer; Springer: Berlin, Germany, 2016; pp. 95–161. [Google Scholar]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Trivedi, R.; Rastogi, N.; Singh, M.; Mishra, D.P. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci. Rep. 2015, 5, 10194. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.K.; Cui, Y.H.; Yoo, K.C.; Kim, I.G.; Lee, M.; Choi, Y.H.; Suh, Y.; Lee, S.J. Radiation promotes malignant phenotypes through SRC in breast cancer cells. Cancer Sci. 2015, 106, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Gilani, R.A.; Phadke, S.; Bao, L.W.; Lachacz, E.J.; Dziubinski, M.L.; Brandvold, K.R.; Steffey, M.E.; Kwarcinski, F.E.; Graveel, C.R.; Kidwell, K.M. UM-164: A potent c-Src/p38 kinase inhibitor with in vivo activity against triple-negative breast cancer. Clin. Cancer Res. 2016, 22, 5087–5096. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef]
- Herrmann, A.; Vogt, M.; Mönnigmann, M.; Clahsen, T.; Sommer, U.; Haan, S.; Poli, V.; Heinrich, P.C.; Müller-Newen, G. Nucleocytoplasmic shuttling of persistently activated STAT3. J. Cell Sci. 2007, 120, 3249–3261. [Google Scholar] [CrossRef]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca2+ release stimulates breast cancer stem cell enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef]
- Qian, X.-L.; Zhang, J.; Li, P.-Z.; Lang, R.-G.; Li, W.-D.; Sun, H.; Liu, F.-F.; Guo, X.-J.; Gu, F.; Fu, L. Dasatinib inhibits c-src phosphorylation and prevents the proliferation of Triple-Negative Breast Cancer (TNBC) cells which overexpress Syndecan-Binding Protein (SDCBP). PLoS ONE 2017, 12, e0171169. [Google Scholar] [CrossRef]
- Vultur, A.; Buettner, R.; Kowolik, C.; Liang, W.; Smith, D.; Boschelli, F.; Jove, R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol. Cancer Ther. 2008, 7, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Jallal, H.; Valentino, M.-L.; Chen, G.; Boschelli, F.; Ali, S.; Rabbani, S.A. A Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, growth, and metastasis in vitro and in vivo. Cancer Res. 2007, 67, 1580–1588. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/β-catenin signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef] [PubMed]
- Hrckulak, D.; Kolar, M.; Strnad, H.; Korinek, V. TCF/LEF transcription factors: An update from the internet resources. Cancers 2016, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Kim, Y.-M.; Kahn, M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. Rep. 2014, 10, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Vermeulen, L. Wnt signaling in cancer stem cell biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Hong, I.-S.; Kim, R.-J.; Lee, S.-Y.; Park, S.-J.; Lee, E.-S.; Park, J.H.; Yun, C.-H.; Chung, J.-U.; Lee, K.-J. Wnt/β-catenin small-molecule inhibitor CWP232,228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Kim, J.-Y.; Cho, S.-D.; Park, K.-S.; Jung, J.-Y.; Lee, H.-Y.; Hong, I.-S.; Nam, J.-S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef] [PubMed]
- Green, J.L.; La, J.; Yum, K.W.; Desai, P.; Rodewald, L.-W.; Zhang, X.; Leblanc, M.; Nusse, R.; Lewis, M.T.; Wahl, G.M. Paracrine Wnt signaling both promotes and inhibits human breast tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 6991–6996. [Google Scholar] [CrossRef] [PubMed]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.; Nguyen, C.; Smbatyan, G.; Tripathy, D.; Yu, M.; Press, M.; Kahn, M.; Lang, J. CBP/β-Catenin/FOXM1 Is a Novel Therapeutic Target in Triple Negative Breast Cancer. Cancers 2018, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Corda, G.; Sala, G.; Lattanzio, R.; Iezzi, M.; Sallese, M.; Fragassi, G.; Lamolinara, A.; Mir, H.; Barcaroli, D.; Ermler, S. Functional and prognostic significance of the genomic amplification of frizzled 6 (FZD6) in breast cancer. J. Pathol. 2017, 241, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.; Deng, X.; Chen, L.; Kim, C.; Lau, S. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef]
- Yin, S.; Xu, L.; Bonfil, R.D.; Banerjee, S.; Sarkar, F.H.; Sethi, S.; Reddy, K.B. Tumor-initiating cells and FZD8 play a major role in drug resistance in triple-negative breast cancer. Mol. Cancer Ther. 2013, 12, 491–498. [Google Scholar] [CrossRef]
- Liu, C.-C.; Prior, J.; Piwnica-Worms, D.; Bu, G. LRP6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5141. [Google Scholar] [CrossRef]
- Ma, J.; Lu, W.; Chen, D.; Xu, B.; Li, Y. Role of Wnt co-receptor LRP6 in triple negative breast cancer cell migration and invasion. J. Cell. Biochem. 2017, 118, 2968–2976. [Google Scholar] [CrossRef]
- Lin, C.-C.; Lo, M.-C.; Moody, R.; Jiang, H.; Harouaka, R.; Stevers, N.; Tinsley, S.; Gasparyan, M.; Wicha, M.; Sun, D. Targeting LRP8 inhibits breast cancer stem cells in triple-negative breast cancer. Cancer Lett. 2018, 438, 165–173. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Liu, Q.; Lu, W.; Bu, G. Wnt signaling activation and mammary gland hyperplasia in MMTV–LRP6 transgenic mice: Implication for breast cancer tumorigenesis. Oncogene 2010, 29, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Bin-Nun, N.; Lichtig, H.; Malyarova, A.; Levy, M.; Elias, S.; Frank, D. PTK7 modulates Wnt signaling activity via LRP6. Development 2014, 141, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, S.; Gunesch, A.; Knyazeva, T.; Wolf, P.; Högel, B.; Eiermann, W.; Ullrich, A.; Knyazev, P.; Ataseven, B. PTK 7 is a transforming gene and prognostic marker for breast cancer and nodal metastasis involvement. PLoS ONE 2014, 9, e84472. [Google Scholar] [CrossRef] [PubMed]
- Ataseven, B.; Angerer, R.; Kates, R.; Gunesch, A.; Knyazev, P.; Hoegel, B.; Becker, C.; Eiermann, W.; Harbeck, N. PTK7 expression in triple-negative breast cancer. Anticancer Res. 2013, 33, 3759–3763. [Google Scholar] [PubMed]
- Damelin, M.; Bankovich, A.; Bernstein, J.; Lucas, J.; Chen, L.; Williams, S.; Park, A.; Aguilar, J.; Ernstoff, E.; Charati, M. A PTK7-targeted antibody-drug conjugate reduces tumor-initiating cells and induces sustained tumor regressions. Sci. Transl. Med. 2017, 9, eaag2611. [Google Scholar] [CrossRef] [PubMed]
- Lenz, H.J.; Kahn, M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014, 105, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Barrott, J.J.; Cash, G.M.; Smith, A.P.; Barrow, J.R.; Murtaugh, L.C. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 12752–12757. [Google Scholar] [CrossRef] [PubMed]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayán, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef]
- Thiagarajan, P.S.; Sinyuk, M.; Turaga, S.M.; Mulkearns-Hubert, E.E.; Hale, J.S.; Rao, V.; Demelash, A.; Saygin, C.; China, A.; Alban, T.J. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nat. Commun. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Mao, Y.; Xiao, G.; Fei, X.; Wang, J.; Zhang, Y.; Liu, J.; Cheng, G.; Chen, X.; Wang, J. USP2 promotes cell migration and invasion in triple negative breast cancer cell lines. Tumor Biol. 2015, 36, 5415–5423. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lee, H.-J.; Saha, S.; Ruan, D.; Guo, H.; Chan, C.-H. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Pragani, R.; Fox, J.T.; Shen, M.; Parmar, K.; Gaudiano, E.F.; Liu, L.; Tanega, C.; McGee, L.; Hall, M.D. Small molecule inhibition of the ubiquitin-specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J. Biol. Chem. 2016, 291, 24628–24640. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Z.; Lee, P.L.; Guan, P.; Aau, M.Y.; Lee, S.T.; Feng, M.; Lim, C.Z.; Lee, E.Y.J.; Wee, Z.N. PDK1 signaling toward PLK1–MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov. 2013, 3, 1156–1171. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Schülein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat. Cell Biol. 2010, 12, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Maire, V.; Némati, F.; Richardson, M.; Vincent-Salomon, A.; Tesson, B.; Rigaill, G.; Gravier, E.; Marty-Prouvost, B.; De Koning, L.; Lang, G. Polo-like kinase 1: A potential therapeutic option in combination with conventional chemotherapy for the management of patients with triple-negative breast cancer. Cancer Res. 2013, 73, 813–823. [Google Scholar] [CrossRef]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol. Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- Hirpara, J.; Eu, J.Q.; Tan, J.K.M.; Wong, A.L.; Clement, M.-V.; Kong, L.R.; Ohi, N.; Tsunoda, T.; Qu, J.; Goh, B.C. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2018, 101076. [Google Scholar] [CrossRef]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kasembeli, M.M.; Jiang, X.; Tweardy, B.J.; Tweardy, D.J. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS ONE 2009, 4, e4783. [Google Scholar] [CrossRef] [PubMed]
- Arensman, M.D.; Telesca, D.; Lay, A.R.; Kershaw, K.M.; Wu, N.; Donahue, T.R.; Dawson, D.W. The CREB binding protein inhibitor ICG-001 suppresses pancreatic cancer growth. Mol. Cancer Ther. 2014, 13, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Inada, T.; Kouji, H.; McWilliams, R.R. Final results of a phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2016, 34, e315721. [Google Scholar] [CrossRef]
- Solzak, J.P.; Atale, R.V.; Hancock, B.A.; Sinn, A.L.; Pollok, K.E.; Jones, D.R.; Radovich, M. Dual PI3K and Wnt pathway inhibition is a synergistic combination against triple negative breast cancer. NPJ Breast Cancer 2017, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459–91470. [Google Scholar] [PubMed]
- Nickho, H.; Younesi, V.; Aghebati-Maleki, L.; Motallebnezhad, M.; Majidi Zolbanin, J.; Movassagh Pour, A.; Yousefi, M. Developing and characterization of single chain variable fragment (scFv) antibody against frizzled 7 (Fzd7) receptor. Bioengineered 2017, 8, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Zarei, N.; Fazeli, M.; Mohammadi, M.; Nejatollahi, F. Cell growth inhibition and apoptosis in breast cancer cells induced by anti-FZD7 scfvs: Involvement of bioinformatics-based design of novel epitopes. Breast Cancer Res. Treat. 2018, 169, 427–436. [Google Scholar] [CrossRef]
- Mita, M.M.; Becerra, C.; Richards, D.A.; Mita, A.C.; Shagisultanova, E.; Osborne, C.R.C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; et al. Phase 1b study of WNT inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel (P) in patients (pts) with 1st- to 3rd-line metastatic HER2-negative breast cancer (BC). J. Clin. Oncol. 2016, 34, 2516. [Google Scholar] [CrossRef]
- Schöffski, P.; Blay, J.Y.; De Greve, J.; Brain, E.; Machiels, J.P.; Soria, J.C.; Sleijfer, S.; Wolter, P.; Ray-Coquard, I.; Fontaine, C.; et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur. J. Cancer 2010, 46, 2206–2215. [Google Scholar]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.-J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018, 27, 136–150. [Google Scholar] [CrossRef]
- Hammoudi, N.; Ahmed, K.B.R.; Garcia-Prieto, C.; Huang, P. Metabolic alterations in cancer cells and therapeutic implications. Chin. J. Cancer 2011, 30, 508–523. [Google Scholar] [CrossRef]
- Palorini, R.; Votta, G.; Balestrieri, C.; Monestiroli, A.; Olivieri, S.; Vento, R.; Chiaradonna, F. Energy Metabolism Characterization of a Novel Cancer Stem Cell-L ike Line 3 AB-OS. J. Cell. Biochem. 2014, 115, 368–379. [Google Scholar] [CrossRef]
- Pecqueur, C.; Oliver, L.; Oizel, K.; Lalier, L.; Vallette, F.M. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int. J. Cell Biol. 2013, 2013, 805975. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’agostino, D. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; Rathmell, J.C. Metabolic barriers to T cell function in tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.-H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H. EGFR signaling enhances aerobic glycolysis in triple-negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, J.-H.; Fan, W.-J.; Meng, Y.-T.; Li, M.-M.; Li, T.-T.; Cui, B.; Wang, H.-F.; Zhao, Y.; An, F. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Salani, B.; Del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, cancer and glucose metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.; Edgerton, S.; Salem, H.; Thor, A. Metformin targets glucose metabolism in triple negative breast cancer. J. Oncol. Transl. Res. 2018, 4, 129. [Google Scholar] [PubMed]
- Salani, B.; Marini, C.; Del Rio, A.; Ravera, S.; Massollo, M.; Orengo, A.M.; Amaro, A.; Passalacqua, M.; Maffioli, S.; Pfeffer, U. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci. Rep. 2013, 3, 2070. [Google Scholar] [CrossRef]
- Koh, M.; Lee, J.-C.; Min, C.; Moon, A. A novel metformin derivative, HL010183, inhibits proliferation and invasion of triple-negative breast cancer cells. Bioorg. Med. Chem. 2013, 21, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, M.; Wu, S.; Gao, S.; Yang, M.; Li, Z.; Min, Q.; Sun, W.; Chen, L.; Xiang, G. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36, 58. [Google Scholar] [CrossRef]
- Ho, N.; Coomber, B.L. Pyruvate dehydrogenase kinase expression and metabolic changes following dichloroacetate exposure in anoxic human colorectal cancer cells. Exp. Cell Res. 2015, 331, 73–81. [Google Scholar] [CrossRef]
- Sun, R.C.; Fadia, M.; Dahlstrom, J.E.; Parish, C.R.; Board, P.G.; Blackburn, A.C. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 2010, 120, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, V.; Agriesti, F.; Scrima, R.; Laurenzana, I.; Perrone, D.; Tataranni, T.; Mazzoccoli, C.; Muzio, L.L.; Capitanio, N.; Piccoli, C. Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: A metabolic perspective of treatment. Oncotarget 2015, 6, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.; Jung, K.; Levine, H.; Kaipparettu, B. Elucidating the metabolic plasticity of cancer: Mitochondrial reprogramming and hybrid metabolic states. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017, 26, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Jansen, V.M.; Koch, J.P.; Li, H.; Formisano, L.; Williams, J.A.; Grandis, J.R.; Arteaga, C.L. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res. 2016, 76, 440–452. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Deribe, Y.L.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [CrossRef]
- Holubarsch, C.J.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef]
- Crawford, L.; Weerapana, E. A tyrosine-reactive irreversible inhibitor for glutathione S-transferase Pi (GSTP1). Mol. Biosyst. 2016, 12, 1768–1771. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 is a driver of triple-negative breast cancer cell metabolism and pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Liu, W.; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; Chen, W. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017, 3, 17010. [Google Scholar] [CrossRef] [PubMed]
- Kéri, G.; Erchegyi, J.; Horvath, A.; Mezo, I.; Idei, M.; Vantus, T.; Balogh, A.; Vadász, Z.; Bökönyi, G.; Seprodi, J. A tumor-selective somatostatin analog (TT-232) with strong in vitro and in vivo antitumor activity. Proc. Natl. Acad. Sci. USA 1996, 93, 12513–12518. [Google Scholar] [CrossRef] [PubMed]
- Butler, E.B.; Zhao, Y.; Muñoz-Pinedo, C.; Lu, J.; Tan, M. Stalling the engine of resistance: Targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013, 73, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.-C.; Kashida, Y.; Kulp, S.K.; Wang, D.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.-S. Sensitizing estrogen receptor–negative breast cancer cells to tamoxifen with OSU-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol. Cancer Ther. 2008, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Feldman, R.I.; Wu, J.M.; Polokoff, M.A.; Kochanny, M.J.; Dinter, H.; Zhu, D.; Biroc, S.L.; Alicke, B.; Bryant, J.; Yuan, S. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005, 280, 19867–19874. [Google Scholar] [CrossRef]
- Lim, S.C.; Carey, K.T.; McKenzie, M. Anti-cancer analogues ME-143 and ME-344 exert toxicity by directly inhibiting mitochondrial NADH: Ubiquinone oxidoreductase (Complex I). Am. J. Cancer Res. 2015, 5, 689–701. [Google Scholar]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mourón, S.; Muñoz, J.; Gómez-López, G.; Jimenez-Renard, V.; Mulero, F. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef]
- Christenson, J.L.; Butterfield, K.T.; Spoelstra, N.S.; Norris, J.D.; Josan, J.S.; Pollock, J.A.; McDonnell, D.P.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Richer, J.K. MMTV-PyMT and derived Met-1 mouse mammary tumor cells as models for studying the role of the androgen receptor in triple-negative breast cancer progression. Horm. Cancer 2017, 8, 69–77. [Google Scholar] [CrossRef]
- Senanayake, E.L.; Howell, N.J.; Ranasinghe, A.M.; Drury, N.E.; Freemantle, N.; Frenneaux, M.; Oelofse, T.; Green, D.; Wilson, I.C.; Rooney, S.J.; et al. Multicentre double-blind randomized controlled trial of perhexiline as a metabolic modulator to augment myocardial protection in patients with left ventricular hypertrophy undergoing cardiac surgery. Eur. J. Cardio Thorac. Surg. 2015, 48, 354–362. [Google Scholar] [CrossRef][Green Version]



{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Preclinical Results | Clinical Trial Status and Results |
|---|---|---|---|
| STAT3 signaling pathway inhibitors | |||
| JAK | Merck-5 (JAK inhibitor I) | It reduces the growth of basal-like tumor cells through the inhibition of STAT3 activity in vitro [39]. | Preclinical |
| Ruxolitinib | It reduces the proliferation, invasion, and mammosphere formation in HCC38 cells and increases apoptosis [98]. | Phase II -NCT01562873: The trial was terminated because of the limited responses to continue treatment despite on-target activity in refractory, metastatic TNBC patients [99]. -NCT02876302: The trial is currently recruiting patients to test the combinatory effect of ruxolitinib with paclitaxel on triple-negative inflammatory breast cancer patients. | |
| STAT3 | OPB-51602 | It reduces mammosphere formation and CD44+/CD24− BCSC populations in MDA-MB-231 cells [100]. | Phase I -NCT01184807: The trial has been completed in patients with malignant solid cancer, and OPB-51602 demonstrated promising antitumor activity, particularly in non-small-cell lung cancer. It has a long half-life and poor tolerability to continuous dosing compared with intermittent dosing [101]. |
| AZD9150 | It shows anti-proliferative efficacy as a single-agent in lymphoma and lung cancer preclinical mouse models. Its clinical trial is currently recruiting patients with advanced solid tumors [102]. | Phase I and II -NCT03421353: The trial is currently recruiting patients with advanced cancer. Phase I/Ib -NCT01839604: The trial has been completed in patients with advanced/metastatic hepatocellular carcinoma. | |
| TTI-101 (STAT3 inhibitor XIII) | It reduces the in vitro cell proliferation more potently in TNBC cells (MDA-MB-468 and MDA-MB-231) than in non-TNBC cells (MDA-MB-435 and MCF7). This clinical trial is currently recruiting patients with advanced cancers, including breast cancer [103]. | Phase I -NCT03195699: The trial is currently recruiting advanced cancer patients. | |
| SRC kinase signaling pathway inhibitors | |||
| SRC | Dasatinib | It reduces the proliferation of TNBC cells in vitro and their tumorigenic potential in vivo. It sensitizes TNBC cells to paclitaxel [41,61]. | Phase II -NCT02720185: The trial was planned for the study of TNBC patients with nuclear translocation of EGFR; however, the trial has been suspended for protocol modifications. |
| SKI-606 (Bosutinib) | It reduces tumor growth, invasion, and metastasis in MDA-MB-231 xenografts [62]. | Phase I -NCT03854093: The trial is currently recruiting breast cancer patients. -NCT03023319: The trial is currently recruiting patients with metastatic solid cancers to assess SKI-606 in combination with pemetrexed. -NCT02810990: The trial is currently recruiting elderly chronic myeloid leukemia patients. | |
| Wnt/β-catenin signaling inhibitors | |||
| CBP | ICG-001 | It reduces mammosphere formation and sensitizes TNBC cells to paclitaxel [75]. | Preclinical |
| PRI-724 | It is an ICG-001 derivative for clinical trials in patients with pancreatic cancer and myeloid leukemia [104]. | Phase I and II -NCT01606579: The trial has been completed in subjects with advanced myeloid malignancies, but the results have not yet been reported. Phase I -NCT01764477: The trial examined patients with advanced or metastatic pancreatic adenocarcinoma to assess PRI-724 in combination with gemcitabine (GEM), and the results show that this combination is safe and demonstrates modest clinical activity [105]. | |
| Porcupine | LGK-974 | It reduces tumor growth in metastatic MDA-MB-231 cell (TMD-231) xenografts. It sensitizes TMD-231 cells to buparlisib [106]. | Phase I -NCT01351103: The trial is currently recruiting patients with malignancies dependent on Wnt ligands. |
| FZD7 | SRI37892 | It reduces tumor growth and tumor-initiating potential in TNBC patient tissue- and cell line-derived xenografts [107]. | Preclinical |
| scFvs | It inhibits cell growth inhibition and promotes apoptosis in MDA-MB-231 cells without affecting SK-BR3 cells [108,109]. | Preclinical | |
| OMP-18R5 (vantictumab) | It promotes tumor growth regression by Taxol and prevents recurrent growth after Taxol treatment in breast cancer patient tissue-derived xenografts [89]. | Phase Ib -NCT01973309: The trial has been completed in patients with recurrent or metastatic breast cancer to evaluate OMP-18R5 in combination with paclitaxel. This combination was demonstrated to be well tolerated. Bone toxicity was encountered early in the study [110]. | |
| PTK7 | PTK7-ADC | It reduces tumor growth and tumor-initiating potential in TNBC patient tissue- and cell line-derived xenografts [86]. | Phase I -NCT03243331: The trial is currently recruiting metastatic TNBC patients to assess a combination drug regimen with gedatolisib. |
| Other molecules linked to the self-renewal process in TNBC | |||
| CX26 | - | The specific inhibitors have not been developed yet. | - |
| USP2 | ML364 | It reduces tumorsphere formation in vitro and tumor growth in vivo. It sensitizes TNBC cells to doxorubicin and paclitaxel [93]. | Preclinical |
| PLK1 | BI-2536 | It reduces tumor growth in TNBC xenografts [97]. | Phase II -NCT00526149: The trial has been completed in patients with recurrent or metastatic solid cancer, and BI-2536 showed limited antitumor activity [111]. |
| Target | Drug | Preclinical Results | Clinical Trial Status and Results |
|---|---|---|---|
| Glycolysis inhibitors | |||
| HK2 | Metformin | A systemic glycolysis inhibitor; it suppresses TNBC stem cells and reduces the tumor-initiating potential in TNBC xenografts [137,153]. | Phase III -NCT02201381: The trial is currently recruiting cancer patients; overall survival is the primary outcome measure. |
| HL010183 | A metformin derivative; it inhibits proliferation and invasion of TNBC cells and reduces tumor growth in MDA-MB-231 xenografts [139]. | Preclinical | |
| Benserazide | FDA-approved drug for Parkinson’s disease. It reduces anaerobic glycolysis in breast cancer cells and inhibits tumor growth [140]. | Preclinical | |
| PKM2 | TLN-232 | It has anti-proliferative effects on diverse cancer cells [154,155]. | Phase II -NCT00422786: The trial has been completed in patients with refractory metastatic renal cell carcinoma, but the results have not yet been reported. -NCT00735332): The trial was conducted in recurring metastatic melanoma patients, but it was stopped because of license termination. |
| PDK1 | DCA | It inhibits metastatic breast cancer cell growth in vitro and in vivo [142]. | Phase II -NCT01029925: The trial was conducted in patients with metastatic breast cancer or with lung cancer, but it was terminated early due to higher than expected risk/safety concerns. |
| AR-12 (OSU-03012) | It reduces proliferation and induces apoptosis of MDA-MB-231 cells in vitro and in vivo. Additionally, it sensitizes MDA-MB-231 cells to tamoxifen [156]. | Phase I -NCT00978523: The trial was conducted in patients with advanced or recurrent solid tumors or with lymphoma. | |
| BX795/BX912 | It reduces the cell viability of MYC-expressing TNBC cells (MDA-MB-231, SUM159PT, Hs578T) but does not affect non-TNBC cells (BT474 and T47D). In addition, it attenuates the CD44+/CD24- population in MDA-MB-231 cells [95,157]. | Preclinical | |
| OXPHOS inhibitors | |||
| Mitochondrial complex I | IACS-010759 | It reduces cell growth and viability across a panel of cancer cell lines, including TNBC without affecting normal cells [147]. | Phase I -NCT03291938: The trial is currently recruiting patients with advanced cancer. -NCT02882321: The trial is currently recruiting subjects with relapsed or refractory acute myeloid leukemia. |
| ME-344 | It sensitizes breast tumors to tyrosine kinase inhibitors in mouse mammary tumor virus-polyoma middle tumor-antigen (MMTV-PyMT) mouse model [158,159,160]. | Phase I -NCT02100007: The trial was terminated because of the lack of efficacy in solid tumor patients as a combinatory agent with Hycamtin® -NCT01544322: The trial was completed in patients with refractory solid cancer; however, the results have not yet been released. -NCT02806817: The trial was recruiting HER2-negative breast cancer patients with antiangiogenic-induced mitochondrial metabolism, but the trial status has not been verified in over two years. | |
| FAO inhibitors | |||
| CPT1 | Etomoxir | It reduces the ATP production in MYC-expressing TNBC cells, thus leads to tumor regression in vitro and in vivo. Moreover, it reduces the CSC proliferation and their self-renewing activity in TNBC cells [116,117]. | Preclinical |
| Perhexiline | It reduces tumor growth, CSC population, and Sox2 expression in MMTV-PyMT tumors. Additionally, it restores the efficacy of paclitaxel in the paclitaxel-resistant MDA-MB-231 cells [117]. | Phase II and III -NCT00845364: The trial was conducted to assess whether an anti-anginal agent could protect the myocardium in patients undergoing coronary artery surgery (CASPER). The role of perhexiline in cardiac surgery is limited [161]. | |
| Other molecules linked to TNBC metabolism | |||
| GSTP1 | LAS17 | It reduces survival in TNBC cells and tumor growth in TNBC xenografts [152]. | Preclinical |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Choi, J.-H.; Nam, J.-S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers 2019, 11, 965. https://doi.org/10.3390/cancers11070965
Park S-Y, Choi J-H, Nam J-S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers. 2019; 11(7):965. https://doi.org/10.3390/cancers11070965
Chicago/Turabian StylePark, So-Yeon, Jang-Hyun Choi, and Jeong-Seok Nam. 2019. "Targeting Cancer Stem Cells in Triple-Negative Breast Cancer" Cancers 11, no. 7: 965. https://doi.org/10.3390/cancers11070965
APA StylePark, S.-Y., Choi, J.-H., & Nam, J.-S. (2019). Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers, 11(7), 965. https://doi.org/10.3390/cancers11070965