Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approvals and Consent to Participate
2.2. K562, GBM Cell Culture and NK Cell Isolation
2.3. Proteasome Inhibitor
2.4. Treatment Groups In Vitro
2.5. Clonogenic Assay
2.6. Flow Cytometry, in Vitro Cytotoxicity Assays and Luminex ELISA
2.7. Tandem Fluorescent-Tagged LC3 (mCherry-EGFP-LC3B) Plasmid, Transfections and Gene Transduction
2.8. Autophagic Flux Assay
2.9. Seahorse Mitochondrial Stress Test
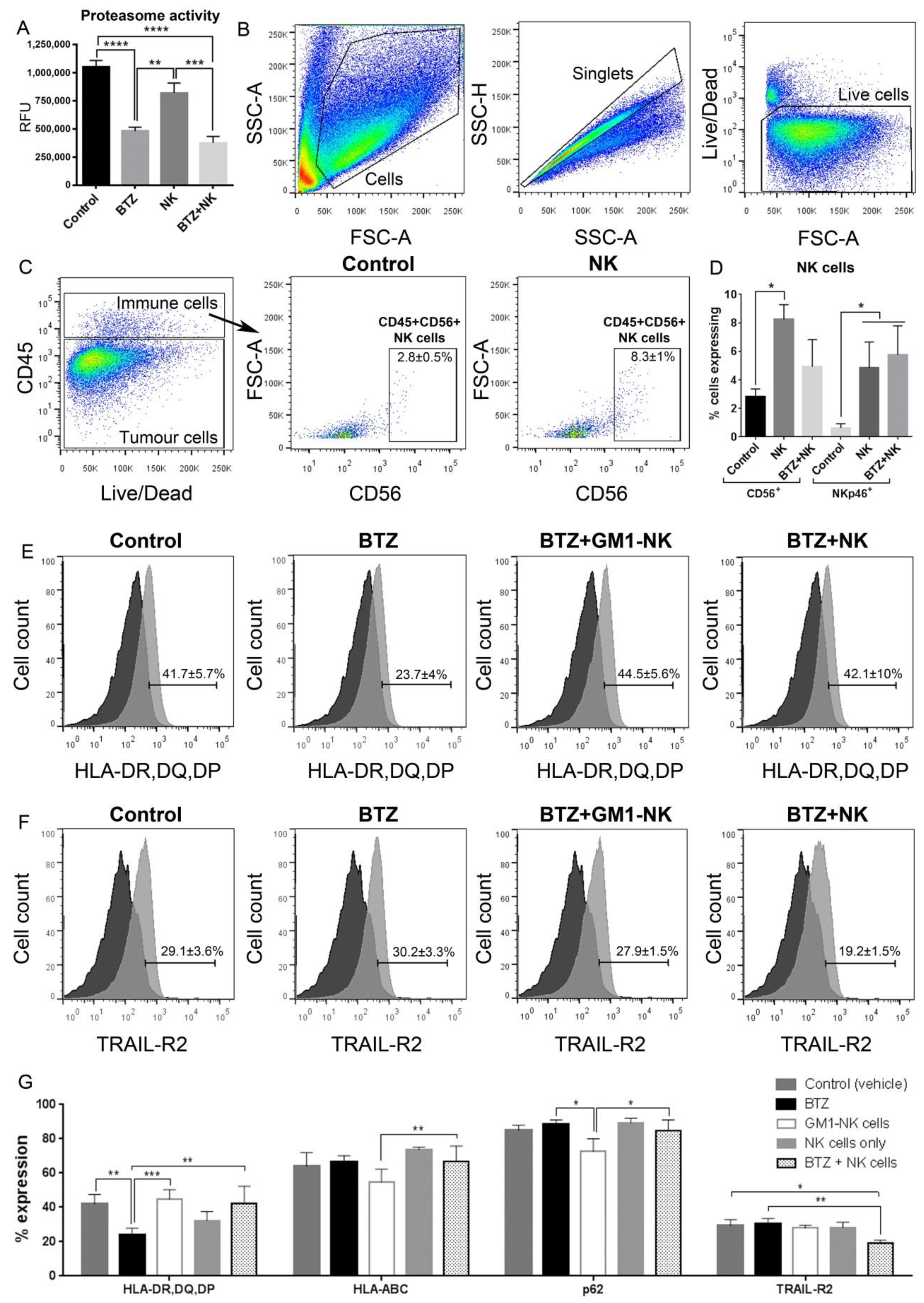
2.10. Proteasome Activity Assay
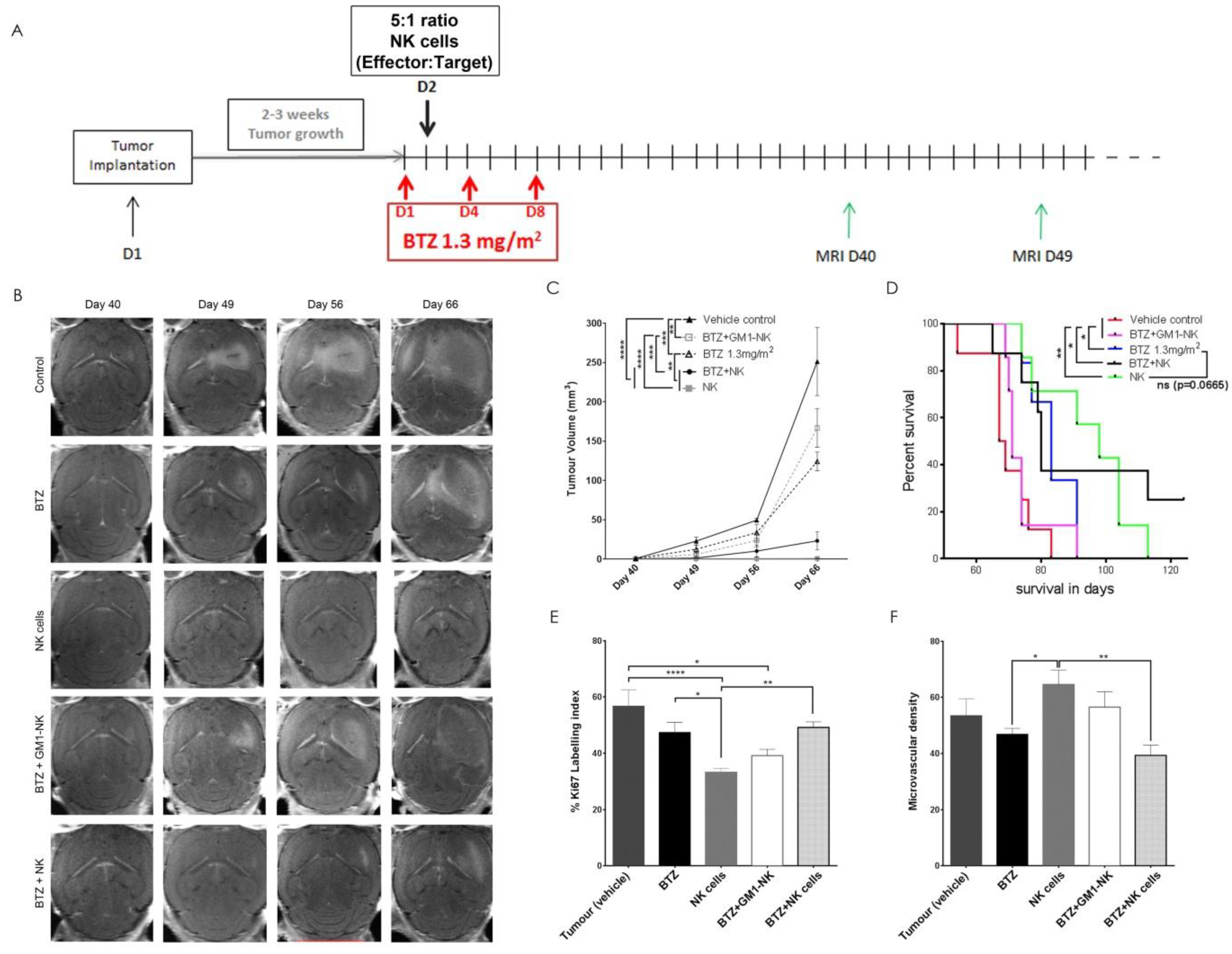
2.11. Treatment of GBM-Bearing Mice
2.12. LC-MS-MS Assay for Detection of Bortezomib in Mouse Brain Tissue
2.13. Magnetic Resonance Imaging and Tumour Volume
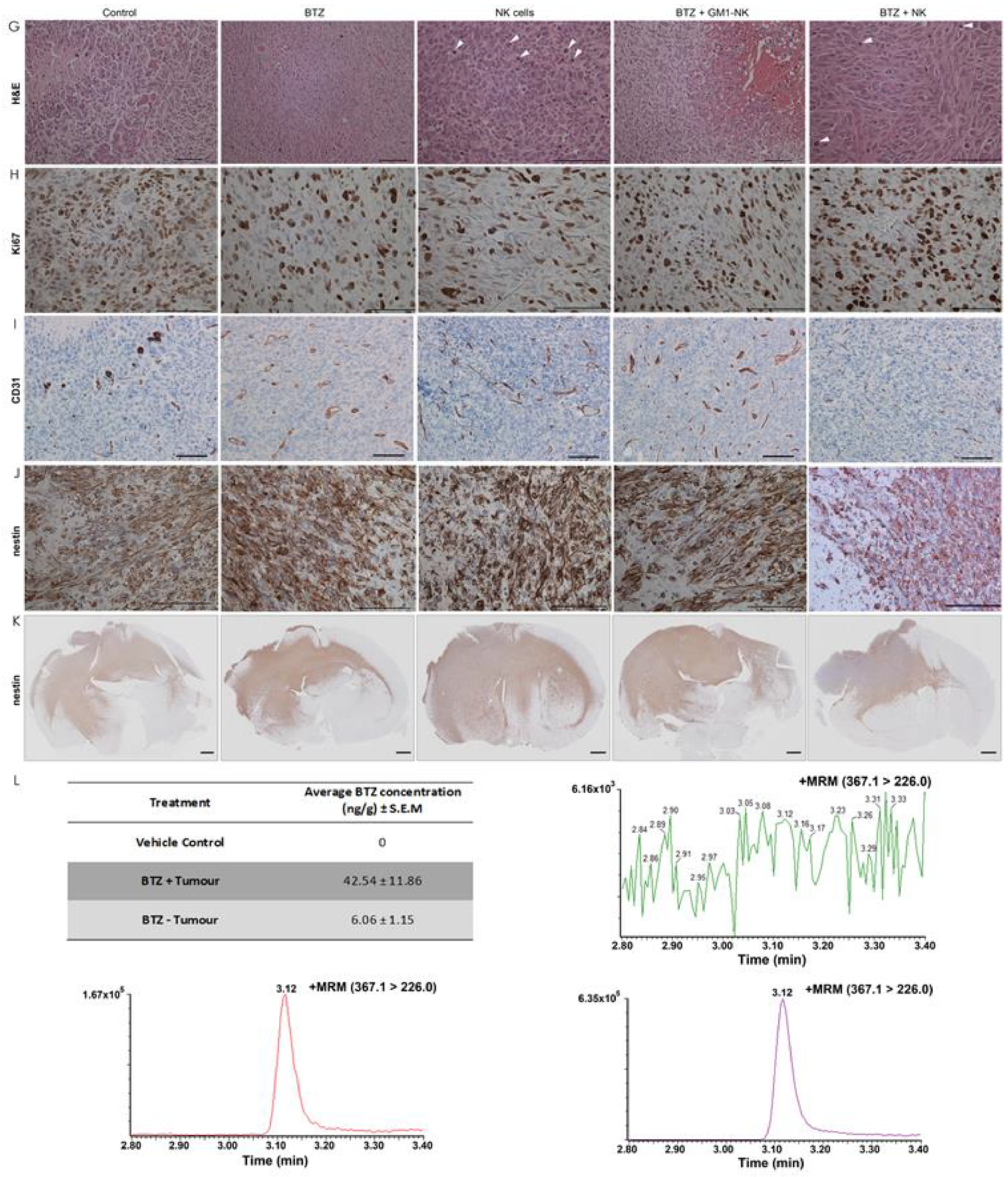
2.14. Immunohistochemistry
2.15. Statistical Analysis
3. Results
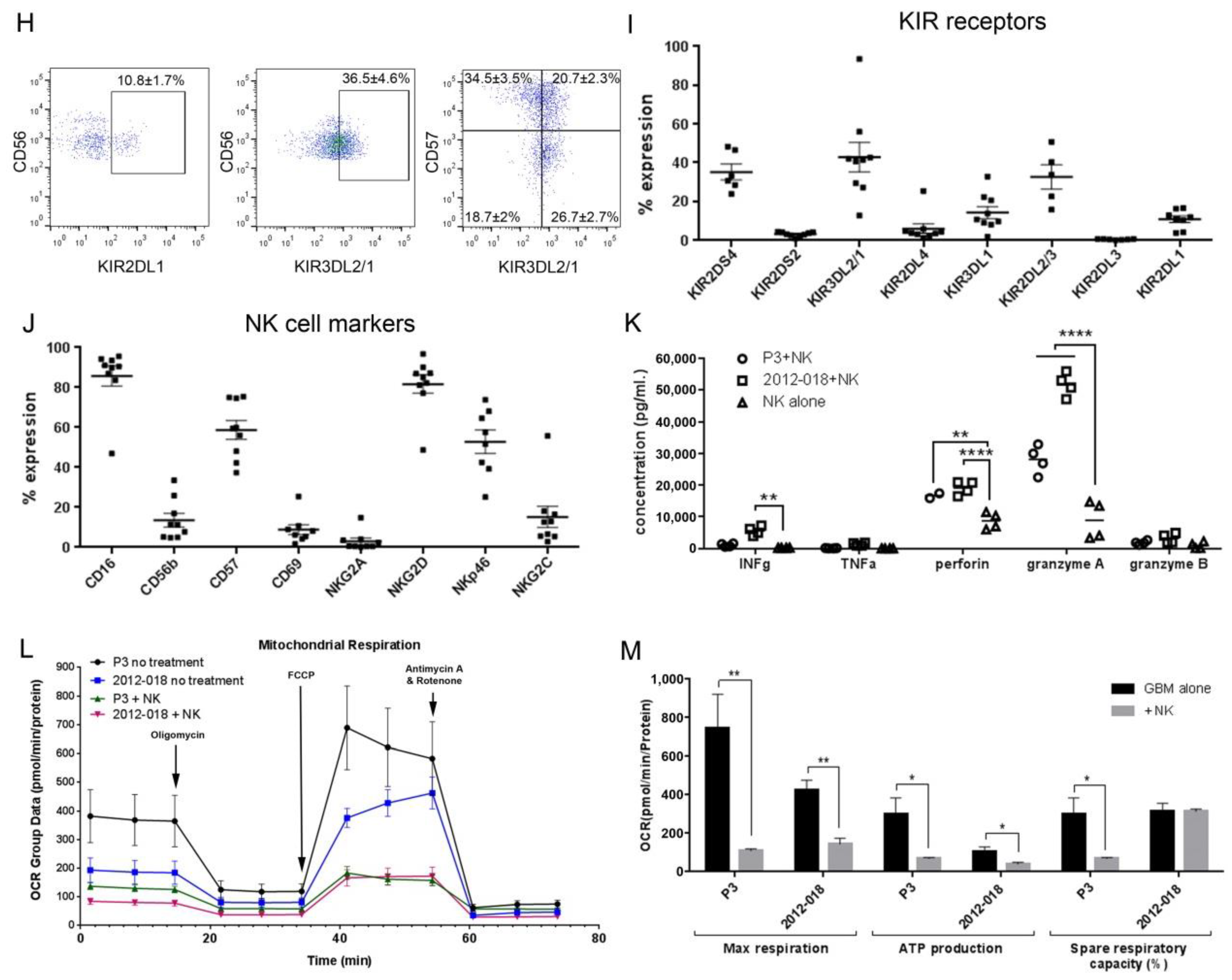
3.1. Heterogeneity in NK Cell Cytotoxic Potency against GBM Cells
3.2. NK Cells Secrete Cytotoxic Factors that Impair Mitochondrial Function in GBM Cells
3.3. Bortezomib Is Cytotoxic Against Patient-Derived GBM Cells In Vitro
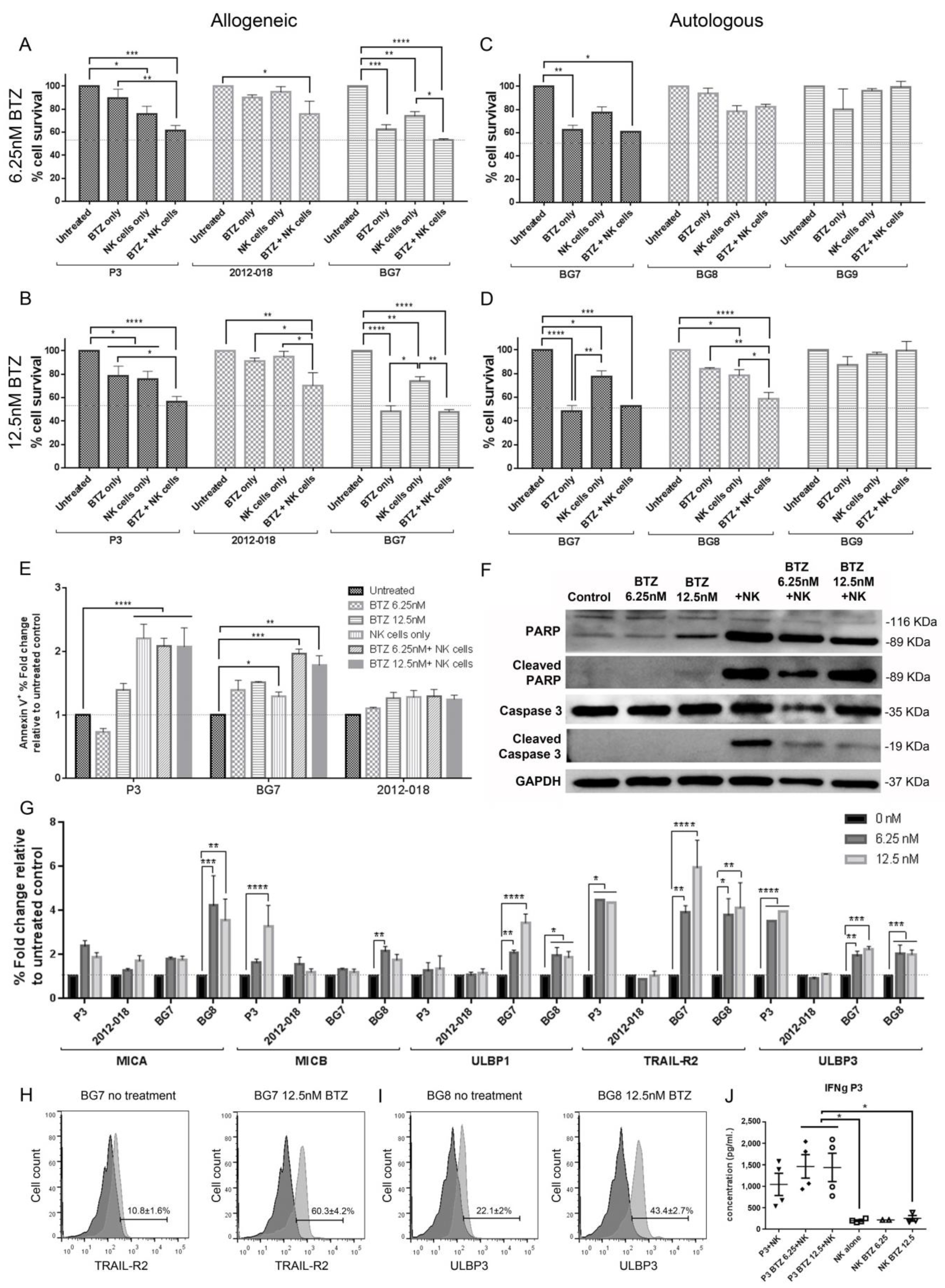
3.4. Combination BTZ+NK Cell Treatment Is More Effective against GBM than Monotherapy In Vitro
3.5. Combination of Bortezomib+NK Cell Treatment Kills GBM Cells by Apoptosis
3.6. Bortezomib Pretreatment Enhances Expression of Stress Ligands in GBM Cells
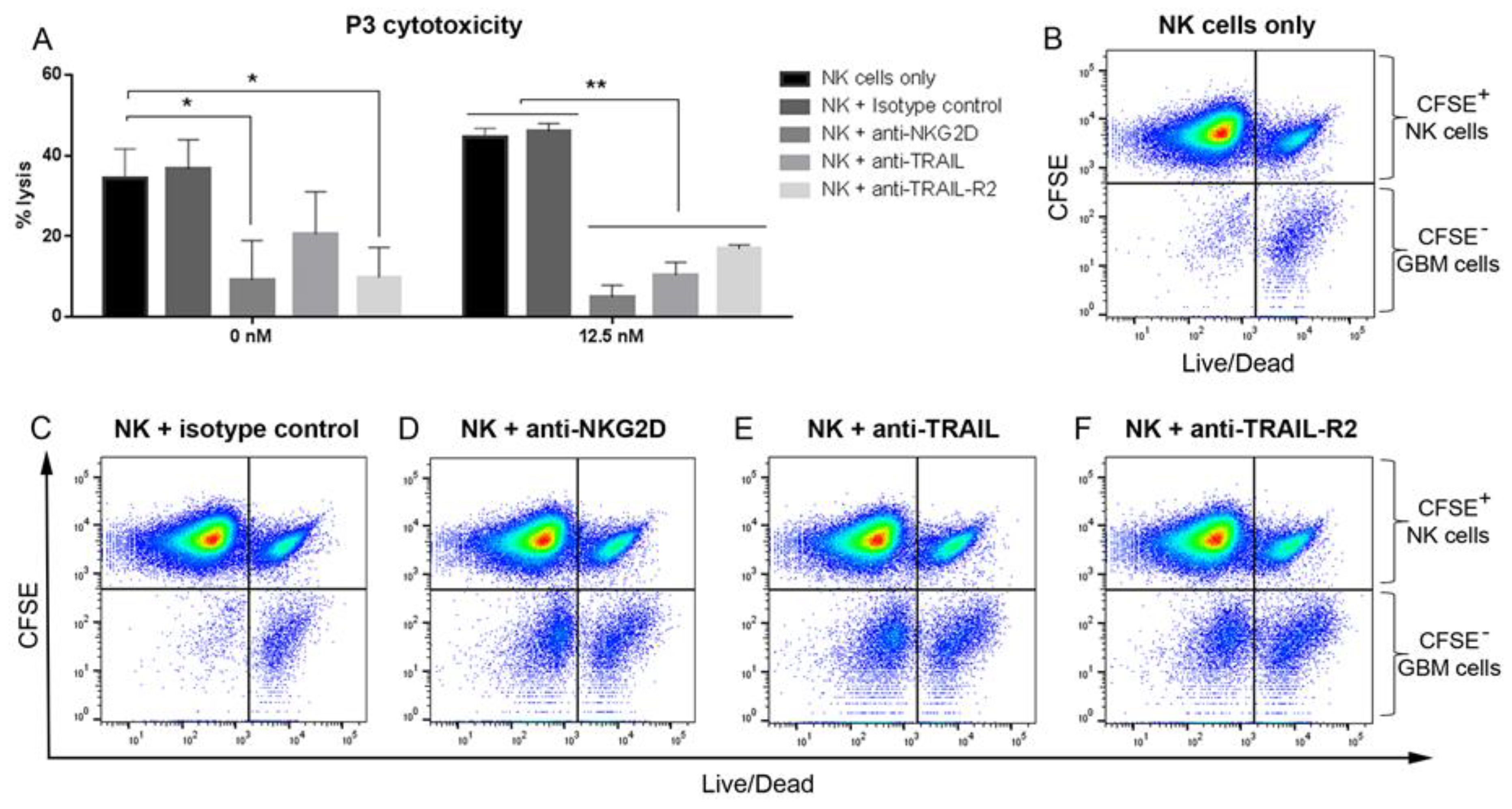
3.7. Blocking NKG2D, TRAIL and TRAIL-R2 Rescues GBM Cells from NK Lysis
3.8. Combination Bortezomib+NK Cell Treatment of GBM Enhances NK Cell Secretion of IFNγ
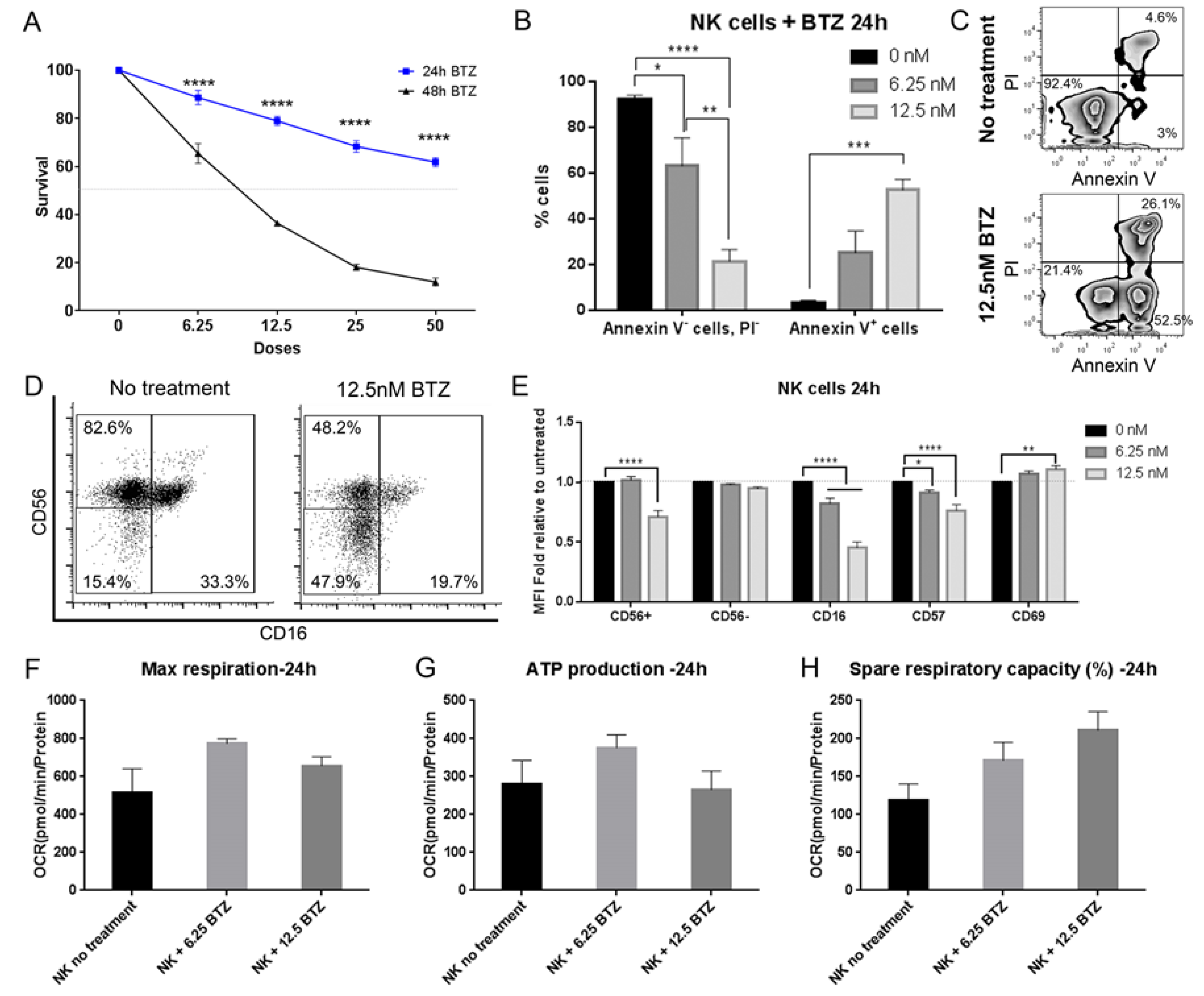
3.9. Bortezomib Promotes Maturation of NK Cells into an Activated Phenotype
3.10. Autologous NK Cells Alone or in Combination with Bortezomib Diminishes Tumour Volume and Prolongs Survival in Tumour Bearing Animals
3.11. NK Cell Treatment Attenuates Tumour Proliferation while BTZ Treatment Diminishes Angiogenesis
3.12. BTZ Penetrates the Blood-Brain Barrier
3.13. Long-Term Persistence of Adoptively Transferred Human CD56+ NK Cells in Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NK | natural killer |
| GBM | glioblastoma |
| BTZ | bortezomib |
| GM1 | monosialotetrahexosylganglioside |
| TMZ | temozolomide |
| HLA | human leucocyte antigen |
| KIR | killer immunoglobulin-like receptor |
| SHP-1/-2 | Src-Homology 2 domain Phosphatase |
| MICA/B | MHC chain-related antigen |
| ULBP | UL16-binding protein |
| TNF | tumour necrosis factor |
| IFN | interferon |
| TRAIL | TNF-related apoptosis-inducing ligand |
| DNAM-1 | DNAX accessory molecule-1 |
| ATP | adenosine triphosphate |
| IC50 | Half maximal inhibitory concentration |
| PARP-1 | poly ADP-ribose polymerase-1 |
| CQ | chloroquine |
| GFP | green fluorescent protein |
| MRI | magnetic resonance imaging |
| Mlc-1 | myeloid leukaemia cell differentiation protein |
| DMEM | Dulbecco’s Modified Eagle’s medium |
| FBS | fetal bovine serum |
| NEAA | non-essential Amino Acid |
| REK | regional committees for medical and health research ethics |
| NB | Neural Basal medium |
| PBMCs | Peripheral blood mononuclear cells |
| SC | Stem Cell Growth Medium |
| ELISA | Enzyme-linked immunosorbent assay |
| NHA | normal human astrocytes |
| E:T | effector:target |
| PBS | phosphate-buffered saline |
| IR | infrared |
| FMO | fluorescent minus one |
| DAPI | 4′,6-diamidino-2-phenylindole |
| OCR | oxygen consumption rate |
| NOD/SCID | non-obese diabetic/severe combined immunodeficiency |
| H&E | hematoxylin and eosin |
| IHC | immunohistochemistry |
| SEM | standard error of the mean |
| ANOVA | analysis of variance |
| UiB | University of Bergen |
| PI | propidium iodide |
| MFI | mean of fluorescence intensity |
| IDH | isocitrate dehydrogenase |
References
- Louis, D.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W. World Health Organization Histological Classification of Tumours of the Central Nervous System; International Agency for Research on Cancer: Lyon, France, 2016.
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Tambussi, G.; Bottino, C.; Tripodi, G.; Merli, A.; Ciccone, E.; Pantaleo, G.; Moretta, L. A novel surface antigen expressed by a subset of human CD3-CD16 natural killer cells. Role in cell activation and regulation of cytolytic function. J. Exp. Med. 1990, 171, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Carrington, M.; Martin, M.P. The impact of variation at the KIR gene cluster on human disease. Curr. Top. Microbiol. Immunol. 2006, 298, 225–257. [Google Scholar] [PubMed]
- Parham, P.; Norman, P.J.; Abi-Rached, L.; Guethlein, L.A. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O. Regulation of immune responses through inhibitory receptors. Annu. Rev. Immunol. 1999, 17, 875–904. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Nunes, J.A.; Vely, F. Natural killer cell signaling pathways. Science 2004, 306, 1517–1519. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Karre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Gras Navarro, A.; Bjorklund, A.T.; Chekenya, M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front. Immunol. 2015, 6, 202. [Google Scholar] [CrossRef] [PubMed]
- Townsend, A.R.; Gotch, F.M.; Davey, J. Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell 1985, 42, 457–467. [Google Scholar] [CrossRef]
- Cooley, S.; Weisdorf, D.J.; Guethlein, L.A.; Klein, J.P.; Wang, T.; Le, C.T.; Marsh, S.G.; Geraghty, D.; Spellman, S.; Haagenson, M.D.; et al. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood 2010, 116, 2411–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood 1999, 94, 333–339. [Google Scholar] [PubMed]
- Shi, J.; Tricot, G.J.; Garg, T.K.; Malaviarachchi, P.A.; Szmania, S.M.; Kellum, R.E.; Storrie, B.; Mulder, A.; Shaughnessy, J.D., Jr.; Barlogie, B.; et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood 2008, 111, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Namazi, A.; Reger, R.; Levy, E.; Berg, M.; St Hilaire, C.; Childs, R.W. Bortezomib sensitizes multiple myeloma to NK cells via ER-stress-induced suppression of HLA-E and upregulation of DR5. Oncoimmunology 2019, 8, e1534664. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Unterkircher, T.; Cristofanon, S.; Vellanki, S.H.; Nonnenmacher, L.; Karpel-Massler, G.; Wirtz, C.R.; Debatin, K.M.; Fulda, S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin. Cancer Res. 2011, 17, 4019–4030. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Riccione, K.A.; Sampson, J.H. Immunotherapy Gone Viral: Bortezomib and oHSV Enhance Antitumor NK-Cell Activity. Clin. Cancer Res. 2016, 22, 5164–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armeanu, S.; Krusch, M.; Baltz, K.M.; Weiss, T.S.; Smirnow, I.; Steinle, A.; Lauer, U.M.; Bitzer, M.; Salih, H.R. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.I.; Grossenbacher, S.K.; Sturgill, I.R.; Ames, E.; Judge, S.J.; Bouzid, L.A.; Darrow, M.A.; Murphy, W.J.; Canter, R.J. Bortezomib Augments Natural Killer Cell Targeting of Stem-Like Tumor Cells. Cancers 2019, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Chao, A.; Tsai, C.L.; Chuang, W.C.; Huang, W.P.; Chen, G.C.; Lin, C.Y.; Wang, T.H.; Wang, H.S.; Lai, C.H. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis. 2014, 5, e1510. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [Green Version]
- Anfossi, N.; Andre, P.; Guia, S.; Falk, C.S.; Roetynck, S.; Stewart, C.A.; Breso, V.; Frassati, C.; Reviron, D.; Middleton, D.; et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 2006, 25, 331–342. [Google Scholar] [CrossRef]
- Kim, S.; Poursine-Laurent, J.; Truscott, S.M.; Lybarger, L.; Song, Y.J.; Yang, L.; French, A.R.; Sunwoo, J.B.; Lemieux, S.; Hansen, T.H.; et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 2005, 436, 709–713. [Google Scholar] [CrossRef]
- Haspels, H.N.; Rahman, M.A.; Joseph, J.V.; Gras Navarro, A.; Chekenya, M. Glioblastoma Stem-Like Cells Are More Susceptible Than Differentiated Cells to Natural Killer Cell Lysis Mediated Through Killer Immunoglobulin-Like Receptors–Human Leukocyte Antigen Ligand Mismatch and Activation Receptor–Ligand Interactions. Front. Immunol. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Bahador, M.; Gras Navarro, A.; Rahman, M.A.; Dominguez-Valentin, M.; Sarowar, S.; Ulvestad, E.; Njolstad, G.; Lie, S.A.; Kristoffersen, E.K.; Bratland, E.; et al. Increased infiltration and tolerised antigen-specific CD8(+) TEM cells in tumor but not peripheral blood have no impact on survival of HCMV(+) glioblastoma patients. Oncoimmunology 2017, 6, e1336272. [Google Scholar] [CrossRef] [PubMed]
- Gras Navarro, A.; Kmiecik, J.; Leiss, L.; Zelkowski, M.; Engelsen, A.; Bruserud, O.; Zimmer, J.; Enger, P.O.; Chekenya, M. NK cells with KIR2DS2 immunogenotype have a functional activation advantage to efficiently kill glioblastoma and prolong animal survival. J. Immunol. 2014, 193, 6192–6206. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.; Verhoeff, J.J.; Immervoll, H.; Brogger, J.C.; Kmiecik, J.; Poli, A.; Netland, I.A.; Prestegarden, L.; Planaguma, J.; Torsvik, A.; et al. Expression of the progenitor marker NG2/CSPG4 predicts poor survival and resistance to ionising radiation in glioblastoma. Acta Neuropathol. 2011, 122, 495–510. [Google Scholar] [CrossRef] [Green Version]
- U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry Estimating the Maximum Safe Starting dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Department of Health and Human Services Food and Drug Administration: Silver Spring, MD, USA, 2005. [Google Scholar]
- Mantel, N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966, 50, 163–170. [Google Scholar]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef]
- Keppel, M.P.; Saucier, N.; Mah, A.Y.; Vogel, T.P.; Cooper, M.A. Activation-specific metabolic requirements for NK Cell IFN-gamma production. J. Immunol. 2015, 194, 1954–1962. [Google Scholar] [CrossRef]
- Hunter, P. The fourth pillar: Despite some setbacks in the clinic, immunotherapy has made notable progress toward becoming an additional therapeutic option against cancer. EMBO Rep. 2017, 18, 1889–1892. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; De Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Hallett, W.H.; Ames, E.; Motarjemi, M.; Barao, I.; Shanker, A.; Tamang, D.L.; Sayers, T.J.; Hudig, D.; Murphy, W.J. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J. Immunol. 2008, 180, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Abrams, S.I.; Schrump, D.S.; Alvarez, G.; Suffredini, D.; Berg, M.; Childs, R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: A novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006, 66, 7317–7325. [Google Scholar] [CrossRef] [PubMed]
- Poe, M.; Blake, J.T.; Boulton, D.A.; Gammon, M.; Sigal, N.H.; Wu, J.K.; Zweerink, H.J. Human cytotoxic lymphocyte granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J. Biol. Chem. 1991, 266, 98–103. [Google Scholar] [PubMed]
- Heibein, J.A.; Goping, I.S.; Barry, M.; Pinkoski, M.J.; Shore, G.C.; Green, D.R.; Bleackley, R.C. Granzyme B-mediated cytochrome c release is regulated by the Bcl-2 family members bid and Bax. J. Exp. Med. 2000, 192, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Beresford, P.J.; Xia, Z.; Greenberg, A.H.; Lieberman, J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999, 10, 585–594. [Google Scholar] [CrossRef]
- Lieberman, J.; Fan, Z. Nuclear war: The granzyme A-bomb. Curr. Opin. Immunol. 2003, 15, 553–559. [Google Scholar] [CrossRef]
- Reddy, P.G.; Graham, G.M.; Datta, S.; Guarini, L.; Moulton, T.A.; Jiang, H.P.; Gottesman, M.M.; Ferrone, S.; Fisher, P.B. Effect of recombinant fibroblast interferon and recombinant immune interferon on growth and the antigenic phenotype of multidrug-resistant human glioblastoma multiforme cells. J. Natl. Cancer Inst. 1991, 83, 1307–1315. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Becker, H.J.; Kondo, E.; Shimabukuro-Vornhagen, A.; Theurich, S.; von Bergwelt-Baildon, M.S. Processing and MHC class II presentation of exogenous soluble antigen involving a proteasome-dependent cytosolic pathway in CD40-activated B cells. Eur. J. Haematol. 2016, 97, 166–174. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Tsuji, T.; Luescher, I.; Old, L.J.; Shrikant, P.; Gnjatic, S.; Odunsi, K. Nonclassical antigen-processing pathways are required for MHC class II-restricted direct tumor recognition by NY-ESO-1-specific CD4(+) T cells. Cancer Immunol. Res. 2014, 2, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.J.; Bowers, G.; Zhang, Z. Proteasome Inhibitor Therapy in a Brain Tumor Model; Humana Press: Totowa, NJ, USA, 2004. [Google Scholar]
- Raizer, J.J.; Chandler, J.P.; Ferrarese, R.; Grimm, S.A.; Levy, R.M.; Muro, K.; Rosenow, J.; Helenowski, I.; Rademaker, A.; Paton, M.; et al. A phase II trial evaluating the effects and intra-tumoral penetration of bortezomib in patients with recurrent malignant gliomas. J. Neurooncol. 2016, 129, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jaime-Ramirez, A.C.; Bolyard, C.; Dai, H.; Nallanagulagari, T.; Wojton, J.; Hurwitz, B.S.; Relation, T.; Lee, T.J.; Lotze, M.T.; et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin. Cancer Res. 2016, 22, 5265–5276. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [Green Version]
- Natsumeda, M.; Aoki, H.; Miyahara, H.; Yajima, N.; Uzuka, T.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Fujii, Y. Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 2011, 31, 486–493. [Google Scholar] [CrossRef]
- Wurstle, S.; Schneider, F.; Ringel, F.; Gempt, J.; Lammer, F.; Delbridge, C.; Wu, W.; Schlegel, J. Temozolomide induces autophagy in primary and established glioblastoma cells in an EGFR independent manner. Oncol. Lett. 2017, 14, 322–328. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gras Navarro, A.; Espedal, H.; Joseph, J.V.; Trachsel-Moncho, L.; Bahador, M.; Tore Gjertsen, B.; Klæboe Kristoffersen, E.; Simonsen, A.; Miletic, H.; Øyvind Enger, P.; et al. Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival. Cancers 2019, 11, 996. https://doi.org/10.3390/cancers11070996
Gras Navarro A, Espedal H, Joseph JV, Trachsel-Moncho L, Bahador M, Tore Gjertsen B, Klæboe Kristoffersen E, Simonsen A, Miletic H, Øyvind Enger P, et al. Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival. Cancers. 2019; 11(7):996. https://doi.org/10.3390/cancers11070996
Chicago/Turabian StyleGras Navarro, Andrea, Heidi Espedal, Justin Vareecal Joseph, Laura Trachsel-Moncho, Marzieh Bahador, Bjørn Tore Gjertsen, Einar Klæboe Kristoffersen, Anne Simonsen, Hrvoje Miletic, Per Øyvind Enger, and et al. 2019. "Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival" Cancers 11, no. 7: 996. https://doi.org/10.3390/cancers11070996
APA StyleGras Navarro, A., Espedal, H., Joseph, J. V., Trachsel-Moncho, L., Bahador, M., Tore Gjertsen, B., Klæboe Kristoffersen, E., Simonsen, A., Miletic, H., Øyvind Enger, P., Rahman, M. A., & Chekenya, M. (2019). Pretreatment of Glioblastoma with Bortezomib Potentiates Natural Killer Cell Cytotoxicity through TRAIL/DR5 Mediated Apoptosis and Prolongs Animal Survival. Cancers, 11(7), 996. https://doi.org/10.3390/cancers11070996