Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
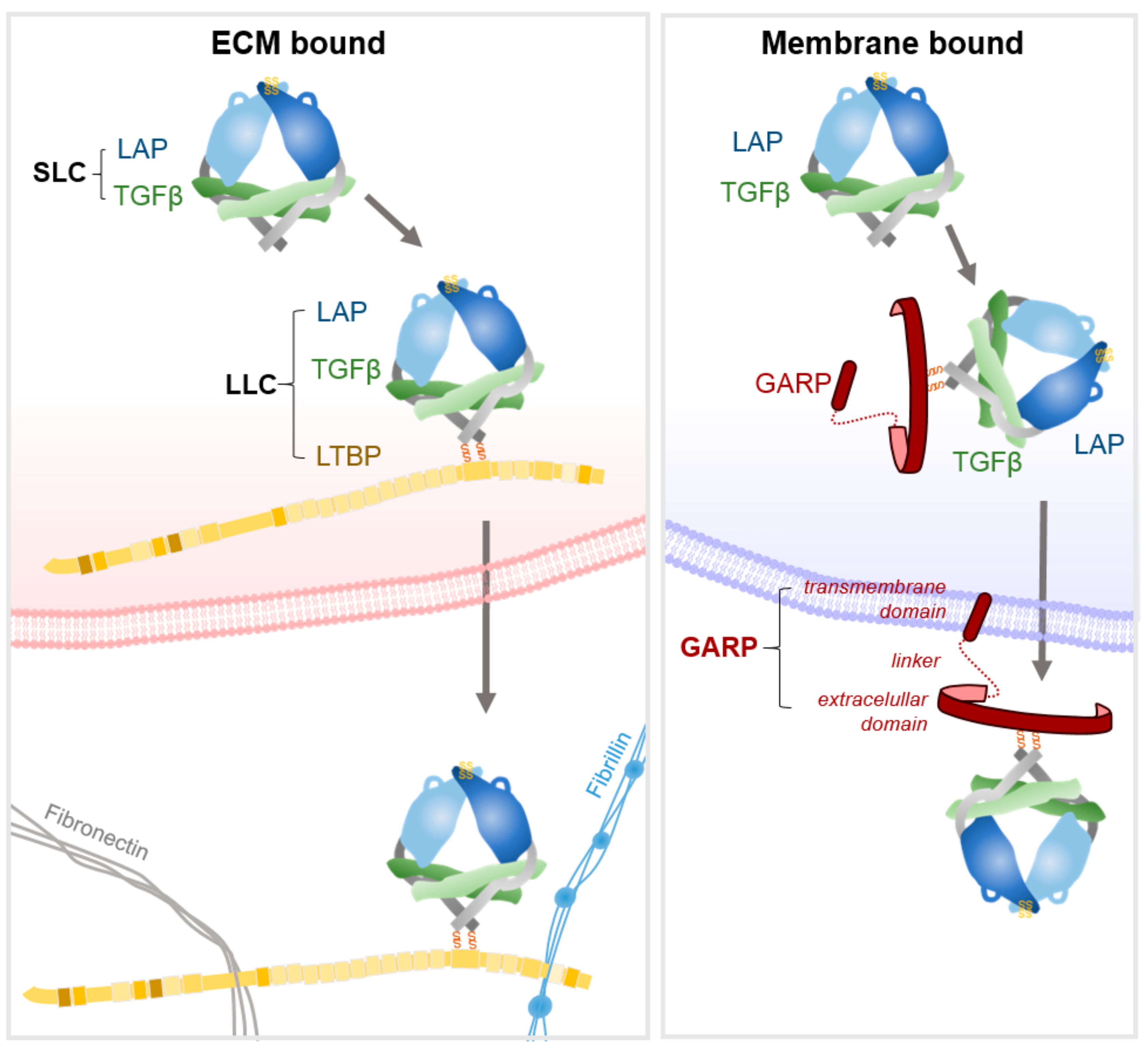
2. TGFβ Structure and Secretion
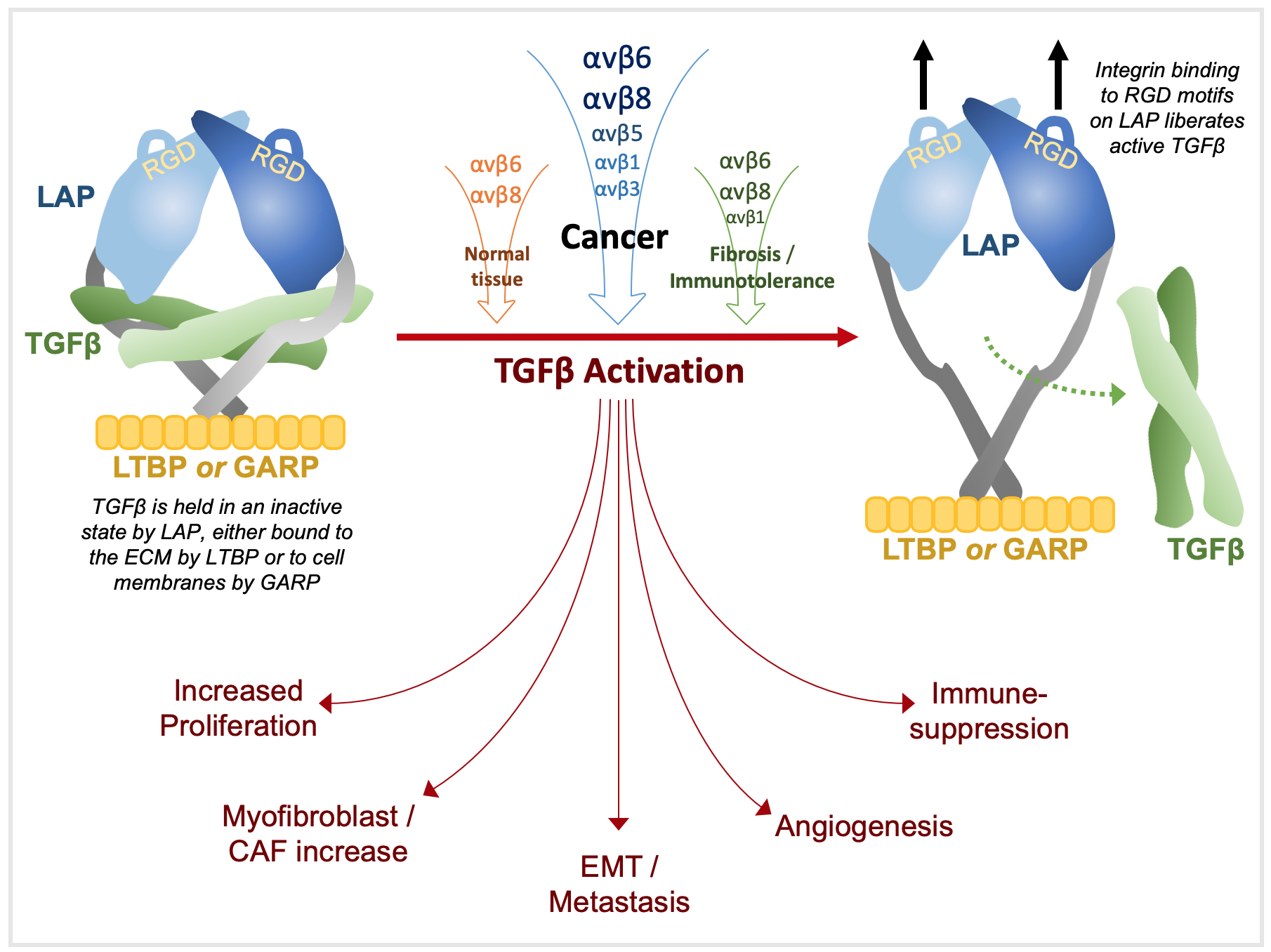
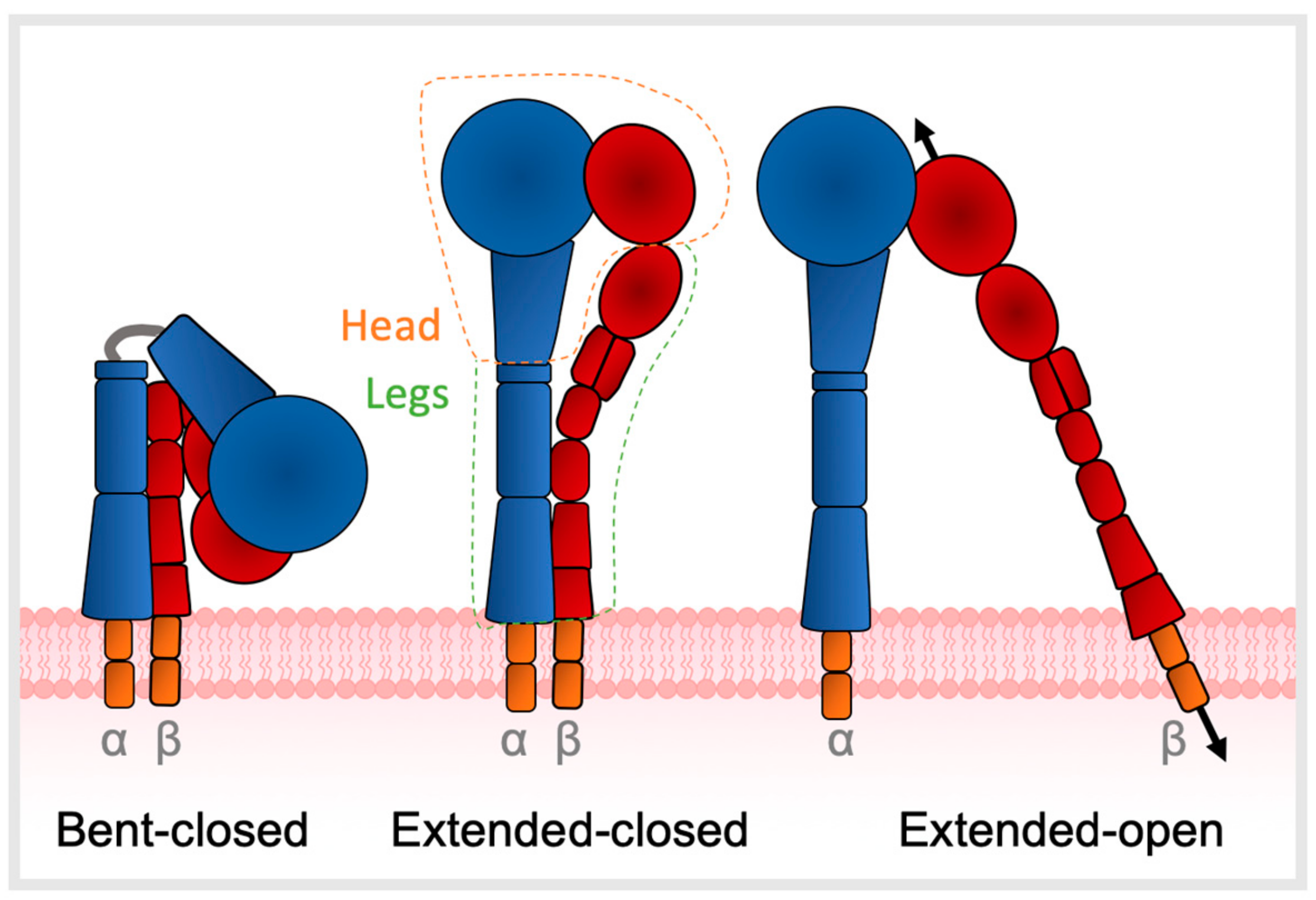
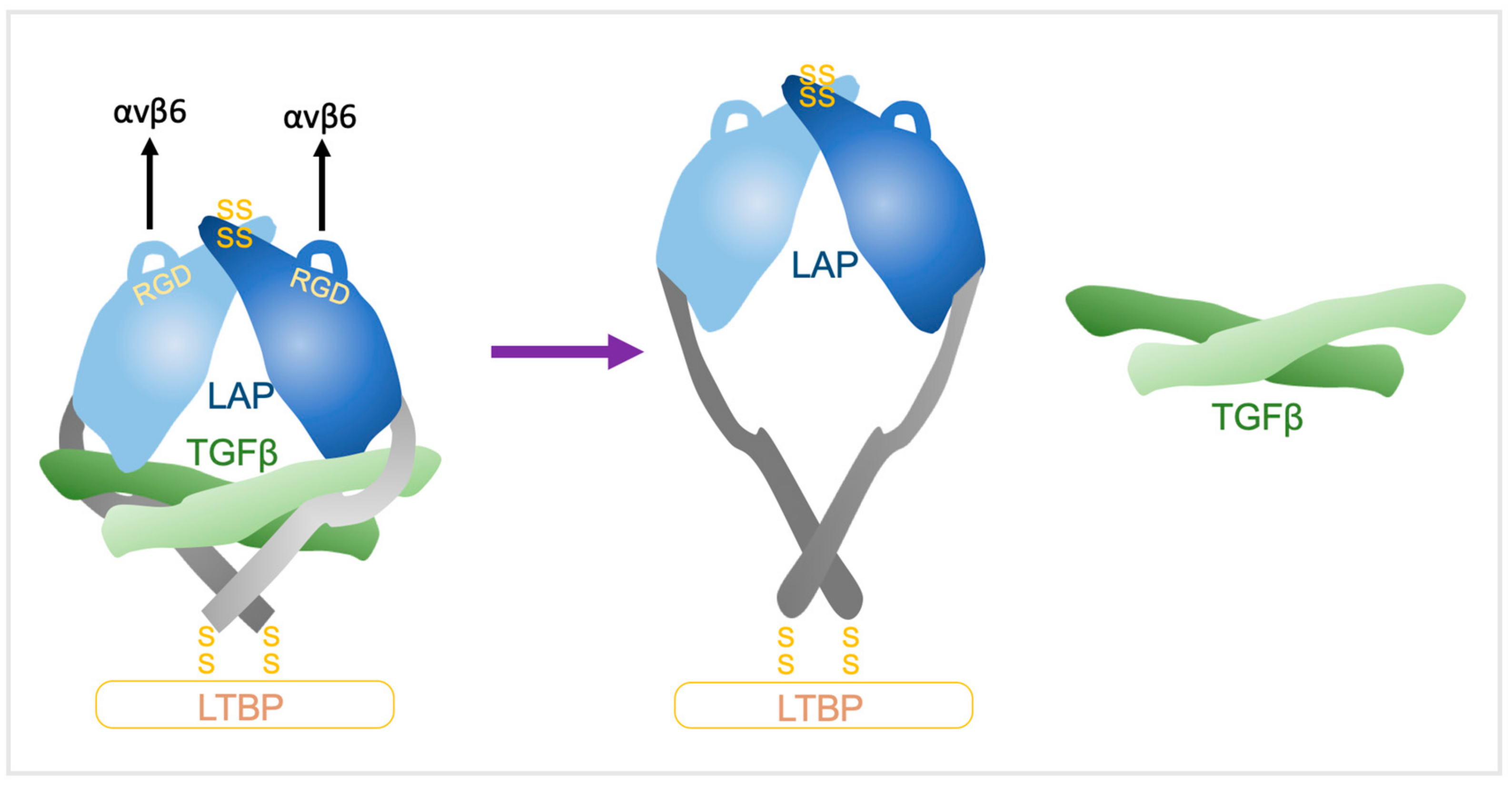
3. TGFβ Activation
3.1. Knockout Mouse Phenotypes
3.2. Ligand Affinity
3.3. Force-Mediated Activation of ECM Bound Latent TGFβ by αvβ6
3.4. Activation of GARP-Bound Latent TGFβ on Treg Cells
3.5. Metalloprotease-Dependent TGFβ Activation
3.6. Other Mediators of TGFβ Activation
4. TGFβ Signalling
5. TGFβ in Cancer
5.1. Proliferation
5.2. EMT & Metastasis
5.3. Angiogenesis
5.4. Stroma
5.5. Immunomodulation
6. Therapeutic Targeting of TGFβ
7. Conclusions
Funding
Conflicts of Interest
References
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of transforming growth factor beta in human disease. N. Engl. J. Med. 2000, 342, 1350–1358. [Google Scholar] [CrossRef]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr. Opin. Genet. Dev. 2011, 21, 93–99. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Calderwood, D.A. Integrin activation. J. Cell Sci. 2004, 117, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Rahmouni, S.; Lindner, A.; Rechenmacher, F.; Neubauer, S.; Sobahi, T.R.; Kessler, H.; Cavalcanti-Adam, E.A.; Spatz, J.P. Hydrogel micropillars with integrin selective peptidomimetic functionalized nanopatterned tops: A new tool for the measurement of cell traction forces transmitted through alphavbeta3-or alpha5beta1-integrins. Adv. Mater. 2013, 25, 5869–5874. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zhao, B.; Iacob, R.E.; Zhu, J.; Koksal, A.C.; Lu, C.; Engen, J.R.; Springer, T.A. Force interacts with macromolecular structure in activation of TGF-beta. Nature 2017, 542, 55–59. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef]
- Taipale, J.; Saharinen, J.; Keski-Oja, J. Extracellular matrix-associated transforming growth factor-beta: Role in cancer cell growth and invasion. Adv. Cancer Res. 1998, 75, 87–134. [Google Scholar]
- Katz, L.H.; Li, Y.; Chen, J.-S.; Muñoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFbeta. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef]
- Lienart, S.; Merceron, R.; Vanderaa, C.; Lambert, F.; Colau, D.; Stockis, J.; van der Woning, B.; De Haard, H.; Saunders, M.; Coulie, P.G.; et al. Structural basis of latent TGF-beta1 presentation and activation by GARP on human regulatory T cells. Science 2018, 362, 952–956. [Google Scholar] [CrossRef]
- Stockis, J.; Dedobbeleer, O.; Lucas, S. Role of GARP in the activation of latent TGF-beta1. Mol. Biosyst. 2017, 13, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Rifkin, D.B. Latent transforming growth factor-beta (TGF-beta) binding proteins: Orchestrators of TGF-beta availability. J. Biol. Chem. 2005, 280, 7409–7412. [Google Scholar] [CrossRef]
- Jenkins, G. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Sakai, T. Biological Significance of Local TGF-β Activation in Liver Diseases. Front. Physiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D. Epithelial-mesenchymal interactions in fibrosis and repair. Transforming growth factor-beta activation by epithelial cells and fibroblasts. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. 1), S21–S23. [Google Scholar] [CrossRef]
- Wakefield, L.M.; Winokur, T.S.; Hollands, R.S.; Christopherson, K.; Levinson, A.D.; Sporn, M.B. Recombinant latent transforming growth factor beta 1 has a longer plasma half-life in rats than active transforming growth factor beta 1, and a different tissue distribution. J. Clin. Investig. 1990, 86, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 41, 233–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Wu, J.F.; Cass, D.; Erle, D.J.; Corry, D.; Young, S.G.; Farese, R.V., Jr.; Sheppard, D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J. Cell Biol. 1996, 133, 921–928. [Google Scholar] [CrossRef]
- Kaminski, N.; Allard, J.D.; Pittet, J.F.; Zuo, F.; Griffiths, M.J.; Morris, D.; Huang, X.; Sheppard, D.; Heller, R.A. Global analysis of gene expression in pulmonary fibrosis reveals distinct programs regulating lung inflammation and fibrosis. Proc. Natl. Acad. Sci. USA 2000, 97, 1778–1783. [Google Scholar] [CrossRef]
- Yang, Z.; Mu, Z.; Dabovic, B.; Jurukovski, V.; Yu, D.; Sung, J.; Xiong, X.; Munger, J.S. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J. Cell Biol. 2007, 176, 787–793. [Google Scholar] [CrossRef]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef]
- Aluwihare, P.; Mu, Z.; Zhao, Z.; Yu, D.; Weinreb, P.H.; Horan, G.S.; Violette, S.M.; Munger, J.S. Mice that lack activity of alphavbeta6-and alphavbeta8-integrins reproduce the abnormalities of Tgfb1-and Tgfb3-null mice. J. Cell Sci. 2009, 122, 227–232. [Google Scholar] [CrossRef]
- Wu, B.X.; Li, A.; Lei, L.; Kaneko, S.; Wallace, C.; Li, X.; Li, Z. Glycoprotein A repetitions predominant (GARP) positively regulates transforming growth factor (TGF) beta3 and is essential for mouse palatogenesis. J. Biol. Chem. 2017, 292, 18091–18097. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.T.; Kaartinen, V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 2007, 312, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Klementowicz, J.E.; Travis, M.A. TGFbeta: A sleeping giant awoken by integrins. Trends Biochem. Sci. 2011, 36, 47–54. [Google Scholar] [CrossRef]
- Travis, M.A.; Reizis, B.; Melton, A.C.; Masteller, E.; Tang, Q.; Proctor, J.M.; Wang, Y.; Bernstein, X.; Huang, X.; Reichardt, L.F.; et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007, 449, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Harpel, J.G.; Giancotti, F.G.; Rifkin, D.B. Interactions between growth factors and integrins: Latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol. Biol. Cell 1998, 9, 2627–2638. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, S.B.; Barry, S.T.; Delves, C.J.; Horgan, C.M.T. The integrin alphavbeta3 is a receptor for the latency-associated peptides of transforming growth factors beta1 and beta3. Biochem. J. 2003, 369, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; John, A.E.; Jolly, L.; Habgood, A.; Porte, J.; Brightling, C.; Knox, A.J.; Pang, L.; Sheppard, D.; Huang, X.; et al. Integrin alphavbeta5-mediated TGF-beta activation by airway smooth muscle cells in asthma. J. Immunol. 2011, 187, 6094–6107. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Hudson, N.E.; Lu, C.; Springer, T.A. Structural determinants of integrin beta-subunit specificity for latent TGF-beta. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. [Google Scholar] [CrossRef]
- Annes, J.P.; Rifkin, D.B.; Munger, J.S. The integrin alphaVbeta6 binds and activates latent TGFbeta3. FEBS Lett. 2002, 511, 65–68. [Google Scholar] [CrossRef]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef]
- Wipff, P.-J.; Hinz, B. Integrins and the activation of latent transforming growth factor β1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Fontana, L.; Chen, Y.; Prijatelj, P.; Sakai, T.; Fassler, R.; Sakai, L.Y.; Rifkin, D.B. Fibronectin is required for integrin alphavbeta6-mediated activation of latent TGF-beta complexes containing LTBP-1. FASEB J. 2005, 19, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-beta1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Flavell, R.A. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1-and Th17-cell differentiation. Immunity 2007, 26, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Fahlen, L.; Read, S.; Gorelik, L.; Hurst, S.D.; Coffman, R.L.; Flavell, R.A.; Powrie, F. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 2005, 201, 737–746. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitani, A.; Strober, W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 2001, 194, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.Q.; Andersson, J.; Wang, R.; Ramsey, H.; Unutmaz, D.; Shevach, E.M. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13445–13450. [Google Scholar] [CrossRef]
- Cuende, J.; Lienart, S.; Dedobbeleer, O.; van der Woning, B.; De Boeck, G.; Stockis, J.; Huygens, C.; Colau, D.; Somja, J.; Delvenne, P.; et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci. Transl. Med. 2015, 7, 284ra56. [Google Scholar] [CrossRef]
- Edwards, J.P.; Thornton, A.M.; Shevach, E.M. Release of active TGF-β1 from the latent TGF-β1/GARP complex on T regulatory cells is mediated by integrin β8. J. Immunol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Worthington, J.J.; Kelly, A.; Smedley, C.; Bauche, D.; Campbell, S.; Marie, J.C.; Travis, M.A. Integrin alphavbeta8-Mediated TGF-beta Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity 2015, 42, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Stockis, J.; Liénart, S.; Colau, D.; Collignon, A.; Nishimura, S.L.; Sheppard, D.; Coulie, P.G.; Lucas, S. Blocking immunosuppression by human Tregs in vivo with antibodies targeting integrin αVβ8. Proc. Natl. Acad. Sci. USA 2017, 114, E10161–E10168. [Google Scholar] [CrossRef] [PubMed]
- Reszka, A.A.; Hayashi, Y.; Horwitz, A.F. Identification of amino acid sequences in the integrin beta 1 cytoplasmic domain implicated in cytoskeletal association. J. Cell Biol. 1992, 117, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Moyle, M.; Napier, M.A.; McLean, J.W. Cloning and expression of a divergent integrin subunit beta 8. J. Biol. Chem. 1991, 266, 19650–19658. [Google Scholar] [PubMed]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Cambier, S.; Mu, D.Z.; O’Connell, D.; Boylen, K.; Travis, W.; Liu, W.H.; Broaddus, V.C.; Nishimura, S.L. A role for the integrin alphavbeta8 in the negative regulation of epithelial cell growth. Cancer Res. 2000, 60, 7084–7093. [Google Scholar]
- Kelly, A.; Gunaltay, S.; McEntee, C.P.; Shuttleworth, E.E.; Smedley, C.; Houston, S.A.; Fenton, T.M.; Levison, S.; Mann, E.R.; Travis, M.A. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J. Exp. Med. 2018, 215, 2725–2736. [Google Scholar] [CrossRef]
- Reynolds, L.E.; Wyder, L.; Lively, J.C.; Taverna, D.; Robinson, S.D.; Huang, X.; Sheppard, D.; Hynes, R.O.; Hodivala-Dilke, K.M. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 2002, 8, 27–34. [Google Scholar] [CrossRef]
- Rolli, M.; Fransvea, E.; Pilch, J.; Saven, A.; Felding-Habermann, B. Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9482–9487. [Google Scholar] [CrossRef]
- Brooks, P.C.; Stromblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996, 85, 683–693. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Truong, H.H.; Xiong, J.; Ghotra, V.P.; Nirmala, E.; Haazen, L.; Le Devedec, S.E.; Balcioglu, H.E.; He, S.; Snaar-Jagalska, B.E.; Vreugdenhil, E.; et al. Beta1 integrin inhibition elicits a prometastatic switch through the TGFbeta-miR-200-ZEB network in E-cadherin-positive triple-negative breast cancer. Sci. Signal. 2014, 7, ra15. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal transduction by members of the transforming growth factor-beta superfamily. Cytokine Growth Factor Rev. 1996, 7, 327–339. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341. [Google Scholar] [CrossRef]
- Moustakas, A.; Lin, H.Y.; Henis, Y.I.; Plamondon, J.; O’Connor-McCourt, M.D.; Lodish, H.F. The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 1993, 268, 22215–22218. [Google Scholar]
- Chacko, B.M.; Qin, B.; Correia, J.J.; Lam, S.S.; de Caestecker, M.P.; Lin, K. The L3 loop and C-terminal phosphorylation jointly define Smad protein trimerization. Nat. Struct. Biol. 2001, 8, 248–253. [Google Scholar] [CrossRef]
- De Crescenzo, G.; Hinck, C.S.; Shu, Z.; Zuniga, J.; Yang, J.; Tang, Y.; Baardsnes, J.; Mendoza, V.; Sun, L.; Lopez-Casillas, F.; et al. Three key residues underlie the differential affinity of the TGFbeta isoforms for the TGFbeta type II receptor. J. Mol. Biol. 2006, 355, 47–62. [Google Scholar] [CrossRef]
- Qian, S.W.; Burmester, J.K.; Tsang, M.L.; Weatherbee, J.A.; Hinck, A.P.; Ohlsen, D.J.; Sporn, M.B.; Roberts, A.B. Binding affinity of transforming growth factor-beta for its type II receptor is determined by the C-terminal region of the molecule. J. Biol. Chem. 1996, 271, 30656–30662. [Google Scholar] [CrossRef]
- Inman, G.J.; Nicolas, F.J.; Hill, C.S. Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell 2002, 10, 283–294. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Yue, J.; Mulder, K.M. Activation of the mitogen-activated protein kinase pathway by transforming growth factor-beta. Methods Mol. Biol. 2000, 142, 125–131. [Google Scholar] [CrossRef]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-beta targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Garcia, C.; Paules, A.B.; Vicente, M.; Megias, M.; Reyes, G.; de Villalonga, P.; Agell, N.; Lluis, F.; Bachs, O.; et al. Disruption of the antiproliferative TGF-beta signaling pathways in human pancreatic cancer cells. Oncogene 1998, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Myeroff, L.L.; Swinler, S.E.; Rajput, A.; Thiagalingam, S.; Lutterbaugh, J.D.; Neumann, A.; Brattain, M.G.; Chang, J.; Kim, S.J.; et al. Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999, 59, 320–324. [Google Scholar] [PubMed]
- Glick, A.B.; Weinberg, W.C.; Wu, I.-H.; Quan, W.; Yuspa, S.H. Transforming growth factor β1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996, 56, 3645–3650. [Google Scholar] [PubMed]
- Sun, L.; Wu, G.; Willson, J.K.; Zborowska, E.; Yang, J.; Rajkarunanayake, I.; Wang, J.; Gentry, L.E.; Wang, X.F.; Brattain, M.G. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J. Biol. Chem. 1994, 269, 26449–26455. [Google Scholar] [PubMed]
- Ludlow, A.; Yee, K.O.; Lipman, R.; Bronson, R.; Weinreb, P.; Huang, X.; Sheppard, D.; Lawler, J. Characterization of integrin beta6 and thrombospondin-1 double-null mice. J. Cell. Mol. Med. 2005, 9, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Hezel, A.F.; Deshpande, V.; Zimmerman, S.M.; Contino, G.; Alagesan, B.; O’Dell, M.R.; Rivera, L.B.; Harper, J.; Lonning, S.; Brekken, R.A.; et al. TGF-beta and alphavbeta6 integrin act in a common pathway to suppress pancreatic cancer progression. Cancer Res. 2012, 72, 4840–4845. [Google Scholar] [CrossRef]
- Owens, D.M.; Romero, M.R.; Gardner, C.; Watt, F.M. Suprabasal α6β4 integrin expression in epidermis results in enhanced tumourigenesis and disruption of TGFβ signalling. J. Cell Sci. 2003, 116, 3783–3791. [Google Scholar] [CrossRef]
- Guasch, G.; Schober, M.; Pasolli, H.A.; Conn, E.B.; Polak, L.; Fuchs, E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 2007, 12, 313–327. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Naber, H.P.; Drabsch, Y.; Snaar-Jagalska, B.E.; ten Dijke, P.; van Laar, T. Snail and Slug, key regulators of TGF-beta-induced EMT, are sufficient for the induction of single-cell invasion. Biochem. Biophys. Res. Commun. 2013, 435, 58–63. [Google Scholar] [CrossRef]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef]
- Dumont, N.; Bakin, A.V.; Arteaga, C.L. Autocrine transforming growth factor-beta signaling mediates Smad-independent motility in human cancer cells. J. Biol. Chem. 2003, 278, 3275–3285. [Google Scholar] [CrossRef]
- Feldkoren, B.; Hutchinson, R.; Rapoport, Y.; Mahajan, A.; Margulis, V. Integrin signaling potentiates transforming growth factor-beta 1 (TGF-beta1) dependent down-regulation of E-Cadherin expression—Important implications for epithelial to mesenchymal transition (EMT) in renal cell carcinoma. Exp. Cell Res. 2017, 355, 57–66. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin β1 Signaling Is Necessary for Transforming Growth Factor-β Activation of p38MAPK and Epithelial Plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef]
- Marsh, D.; Dickinson, S.; Neill, G.W.; Marshall, J.F.; Hart, I.R.; Thomas, G.J. Alpha vbeta 6 Integrin promotes the invasion of morphoeic basal cell carcinoma through stromal modulation. Cancer Res. 2008, 68, 3295–3303. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef]
- Moore, K.M.; Thomas, G.J.; Duffy, S.W.; Warwick, J.; Gabe, R.; Chou, P.; Ellis, I.O.; Green, A.R.; Haider, S.; Brouilette, K.; et al. Therapeutic targeting of integrin alphavbeta6 in breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Allen, M.D.; Thomas, G.J.; Clark, S.; Dawoud, M.M.; Vallath, S.; Payne, S.J.; Gomm, J.J.; Dreger, S.A.; Dickinson, S.; Edwards, D.R.; et al. Altered microenvironment promotes progression of preinvasive breast cancer: Myoepithelial expression of alphavbeta6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin. Cancer Res. 2014, 20, 344–357. [Google Scholar] [CrossRef]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Investig. 2005, 115, 339–347. [Google Scholar] [CrossRef]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFbeta and Angiogenesis by Type VII Collagen in Cutaneous SCC. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Fournier, P.G.; Juarez, P.; Jiang, G.; Clines, G.A.; Niewolna, M.; Kim, H.S.; Walton, H.W.; Peng, X.H.; Liu, Y.; Mohammad, K.S.; et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer Cell 2015, 27, 809–821. [Google Scholar] [CrossRef]
- Chiechi, A.; Waning, D.L.; Stayrook, K.R.; Buijs, J.T.; Guise, T.A.; Mohammad, K.S. Role of TGF-β in breast cancer bone metastases. Adv. Biosci. Biotechnol. 2013, 4, 15–30. [Google Scholar] [CrossRef]
- Dutta, A.; Li, J.; Lu, H.; Akech, J.; Pratap, J.; Wang, T.; Zerlanko, B.J.; FitzGerald, T.J.; Jiang, Z.; Birbe, R.; et al. Integrin alphavbeta6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res. 2014, 74, 1598–1608. [Google Scholar] [CrossRef]
- Dutta, A.; Li, J.; Fedele, C.; Sayeed, A.; Singh, A.; Violette, S.M.; Manes, T.D.; Languino, L.R. αvβ6 integrin is required for TGFβ1-mediated matrix metalloproteinase2 expression. Biochem. J. 2015, 466, 525–536. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Schniewind, B.; Groth, S.; Sebens Muerkoster, S.; Sipos, B.; Schafer, H.; Kalthoff, H.; Fandrich, F.; Ungefroren, H. Dissecting the role of TGF-beta type I receptor/ALK5 in pancreatic ductal adenocarcinoma: Smad activation is crucial for both the tumor suppressive and prometastatic function. Oncogene 2007, 26, 4850–4862. [Google Scholar] [CrossRef]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar]
- Liu, Z.; Kobayashi, K.; van Dinther, M.; van Heiningen, S.H.; Valdimarsdottir, G.; van Laar, T.; Scharpfenecker, M.; Lowik, C.W.; Goumans, M.J.; Ten Dijke, P.; et al. VEGF and inhibitors of TGFbeta type-I receptor kinase synergistically promote blood-vessel formation by inducing alpha5-integrin expression. J. Cell Sci. 2009, 122, 3294–3302. [Google Scholar] [CrossRef]
- Elayadi, A.N.; Samli, K.N.; Prudkin, L.; Liu, Y.H.; Bian, A.; Xie, X.J.; Wistuba, I.I.; Roth, J.A.; McGuire, M.J.; Brown, K.C. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007, 67, 5889–5895. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. αV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef]
- Silva, R.; D’Amico, G.; Hodivala-Dilke, K.M.; Reynolds, L.E. Integrins: The keys to unlocking angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1703–1713. [Google Scholar] [CrossRef]
- Zhu, J.; Motejlek, K.; Wang, D.; Zang, K.; Schmidt, A.; Reichardt, L.F. Beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 2002, 129, 2891–2903. [Google Scholar]
- Tchaicha, J.H.; Mobley, A.K.; Hossain, M.G.; Aldape, K.D.; McCarty, J.H. A mosaic mouse model of astrocytoma identifies alphavbeta8 integrin as a negative regulator of tumor angiogenesis. Oncogene 2010, 29, 4460–4472. [Google Scholar] [CrossRef]
- Massague, J. The Transforming Growth Factor-beta Family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef]
- Hayashida, T.; Wu, M.H.; Pierce, A.; Poncelet, A.C.; Varga, J.; Schnaper, H.W. MAP-kinase activity necessary for TGFbeta1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J. Cell Sci. 2007, 120, 4230–4240. [Google Scholar] [CrossRef]
- Lamar, J.M.; Iyer, V.; DiPersio, C.M. Integrin alpha3beta1 potentiates TGFbeta-mediated induction of MMP-9 in immortalized keratinocytes. J. Investig. Dermatol. 2008, 128, 575–586. [Google Scholar] [CrossRef]
- Bacman, D.; Merkel, S.; Croner, R.; Papadopoulos, T.; Brueckl, W.; Dimmler, A. TGF-beta receptor 2 downregulation in tumour-associated stroma worsens prognosis and high-grade tumours show more tumour-associated macrophages and lower TGF-beta1 expression in colon carcinoma: A retrospective study. BMC Cancer 2007, 7, 156. [Google Scholar] [CrossRef]
- Van Aarsen, L.A.; Leone, D.R.; Ho, S.; Dolinski, B.M.; McCoon, P.E.; LePage, D.J.; Kelly, R.; Heaney, G.; Rayhorn, P.; Reid, C.; et al. Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res. 2008, 68, 561–570. [Google Scholar] [CrossRef]
- Eberlein, C.; Kendrew, J.; McDaid, K.; Alfred, A.; Kang, J.S.; Jacobs, V.N.; Ross, S.J.; Rooney, C.; Smith, N.R.; Rinkenberger, J.; et al. A human monoclonal antibody 264RAD targeting αvβ6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 2012, 32, 4406. [Google Scholar] [CrossRef]
- Eberlein, C.; Rooney, C.; Ross, S.J.; Farren, M.; Weir, H.M.; Barry, S.T. E-Cadherin and EpCAM expression by NSCLC tumour cells associate with normal fibroblast activation through a pathway initiated by integrin alphavbeta6 and maintained through TGFbeta signalling. Oncogene 2015, 34, 704–716. [Google Scholar] [CrossRef]
- Reader, C.S.; Vallath, S.; Steele, C.W.; Haider, S.; Brentnall, A.; Desai, A.; Moore, K.M.; Jamieson, N.B.; Chang, D.; Bailey, P.; et al. The integrin αvβ6 drives pancreatic cancer through diverse mechanisms and represents an effective target for therapy. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter-and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Park, E.J.; Prajuabjinda, O.; Soe, Z.Y.; Darkwah, S.; Appiah, M.G.; Kawamoto, E.; Momose, F.; Shiku, H.; Shimaoka, M. Exosomal regulation of lymphocyte homing to the gut. Blood Adv. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Shimaoka, M.; Kawamoto, E.; Gaowa, A.; Okamoto, T.; Park, E.J. Connexins and Integrins in Exosomes. Cancers 2019, 11, 106. [Google Scholar] [CrossRef]
- Singh, A.; Fedele, C.; Lu, H.; Nevalainen, M.T.; Keen, J.H.; Languino, L.R. Exosome-mediated Transfer of αvβ3 Integrin from Tumorigenic to Nontumorigenic Cells Promotes a Migratory Phenotype. J. Mol. Cancer Res. 2016, 14, 1136–1146. [Google Scholar] [CrossRef]
- Chen, X.; Song, C.-H.; Feng, B.-S.; Li, T.-L.; Li, P.; Zheng, P.-Y.; Chen, X.-M.; Xing, Z.; Yang, P.-C. Intestinal epithelial cell-derived integrin αβ6 plays an important role in the induction of regulatory T cells and inhibits an antigen-specific Th2 response. J. Leukoc. Biol. 2011, 90, 751–759. [Google Scholar] [CrossRef]
- Fedele, C.; Singh, A.; Zerlanko, B.J.; Iozzo, R.V.; Languino, L.R. The alphavbeta6 integrin is transferred intercellularly via exosomes. J. Biol. Chem. 2015, 290, 4545–4551. [Google Scholar] [CrossRef]
- Languino, L.R.; Singh, A.; Prisco, M.; Inman, G.J.; Luginbuhl, A.; Curry, J.M.; South, A.P. Exosome-mediated transfer from the tumor microenvironment increases TGFβ signaling in squamous cell carcinoma. Am. J. Transl. Res. 2016, 8, 2432–2437. [Google Scholar]
- Li, M.O.; Flavell, R.A. TGF-beta: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Gong, D.; Shi, W.; Yi, S.-j.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef]
- Lu, H.; Bowler, N.; Harshyne, L.A.; Craig Hooper, D.; Krishn, S.R.; Kurtoglu, S.; Fedele, C.; Liu, Q.; Tang, H.Y.; Kossenkov, A.V.; et al. Exosomal alphavbeta6 integrin is required for monocyte M2 polarization in prostate cancer. Matrix Biol. 2018, 70, 20–35. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef]
- Marcoe, J.P.; Lim, J.R.; Schaubert, K.L.; Fodil-Cornu, N.; Matka, M.; McCubbrey, A.L.; Farr, A.R.; Vidal, S.M.; Laouar, Y. TGF-beta is responsible for NK cell immaturity during ontogeny and increased susceptibility to infection during mouse infancy. Nat. Immunol. 2012, 13, 843–850. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef]
- Kim, B.G.; Li, C.; Qiao, W.; Mamura, M.; Kasprzak, B.; Anver, M.; Wolfraim, L.; Hong, S.; Mushinski, E.; Potter, M.; et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006, 441, 1015–1019. [Google Scholar] [CrossRef]
- Trapani, J.A. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell 2005, 8, 349–350. [Google Scholar] [CrossRef]
- Shang, B.; Liu, Y.; Jiang, S.-j.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef]
- Melton, A.C.; Bailey-Bucktrout, S.L.; Travis, M.A.; Fife, B.T.; Bluestone, J.A.; Sheppard, D. Expression of alphavbeta8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Investig. 2010, 120, 4436–4444. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Budhu, S.; Schaer, D.A.; Li, Y.; Toledo-Crow, R.; Panageas, K.; Yang, X.; Zhong, H.; Houghton, A.N.; Silverstein, S.C.; Merghoub, T.; et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Hazelbag, S.; Kenter, G.G.; Gorter, A.; Dreef, E.J.; Koopman, L.A.; Violette, S.M.; Weinreb, P.H.; Fleuren, G.J. Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J. Pathol. 2007, 212, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Sipos, B.; Hahn, D.; Carceller, A.; Piulats, J.; Hedderich, J.; Kalthoff, H.; Goodman, S.L.; Kosmahl, M.; Klöppel, G. Immunohistochemical screening for β6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 2004, 45, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.A.; Khazaeli, M.B.; DelGrosso, A.; Saleh, M.N.; Lin, C.Y.; Huse, W.; LoBuglio, A.F. A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother. Radiopharm. 2001, 16, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Hersey, P.; Sosman, J.; O’Day, S.; Richards, J.; Bedikian, A.; Gonzalez, R.; Sharfman, W.; Weber, R.; Logan, T.; Buzoianu, M.; et al. A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or − dacarbazine in patients with stage IV metastatic melanoma. Cancer 2010, 116, 1526–1534. [Google Scholar] [CrossRef]
- Elez, E.; Kocakova, I.; Hohler, T.; Martens, U.M.; Bokemeyer, C.; Van Cutsem, E.; Melichar, B.; Smakal, M.; Csoszi, T.; Topuzov, E.; et al. Abituzumab combined with cetuximab plus irinotecan versus cetuximab plus irinotecan alone for patients with KRAS wild-type metastatic colorectal cancer: The randomised phase I/II POSEIDON trial. Ann. Oncol. 2015, 26, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncology 2015, 17, 708–717. [Google Scholar] [CrossRef]
- Weller, M.; Nabors, L.B.; Gorlia, T.; Leske, H.; Rushing, E.; Bady, P.; Hicking, C.; Perry, J.; Hong, Y.-K.; Roth, P.; et al. Cilengitide in newly diagnosed glioblastoma: Biomarker expression and outcome. Oncotarget 2016, 7, 15018–15032. [Google Scholar] [CrossRef] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, A.; Kostine, M.; Barnetche, T.; Truchetet, M.E.; Schaeverbeke, T. Immune related adverse events associated with anti-CTLA-4 antibodies: Systematic review and meta-analysis. BMC Med. 2015, 13, 211. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, N.F.; Marshall, J.F. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers 2019, 11, 1221. https://doi.org/10.3390/cancers11091221
Brown NF, Marshall JF. Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers. 2019; 11(9):1221. https://doi.org/10.3390/cancers11091221
Chicago/Turabian StyleBrown, Nicholas F., and John F. Marshall. 2019. "Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment" Cancers 11, no. 9: 1221. https://doi.org/10.3390/cancers11091221
APA StyleBrown, N. F., & Marshall, J. F. (2019). Integrin-Mediated TGFβ Activation Modulates the Tumour Microenvironment. Cancers, 11(9), 1221. https://doi.org/10.3390/cancers11091221