Cancer Takes a Toll on Skeletal Muscle by Releasing Heat Shock Proteins—An Emerging Mechanism of Cancer-Induced Cachexia


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammatory Signaling Cascades Mediate Skeletal Muscle Protein Degradation in the Cancer Milieu
3. TLR4 Activation in Muscle Cells Causes Muscle Wasting
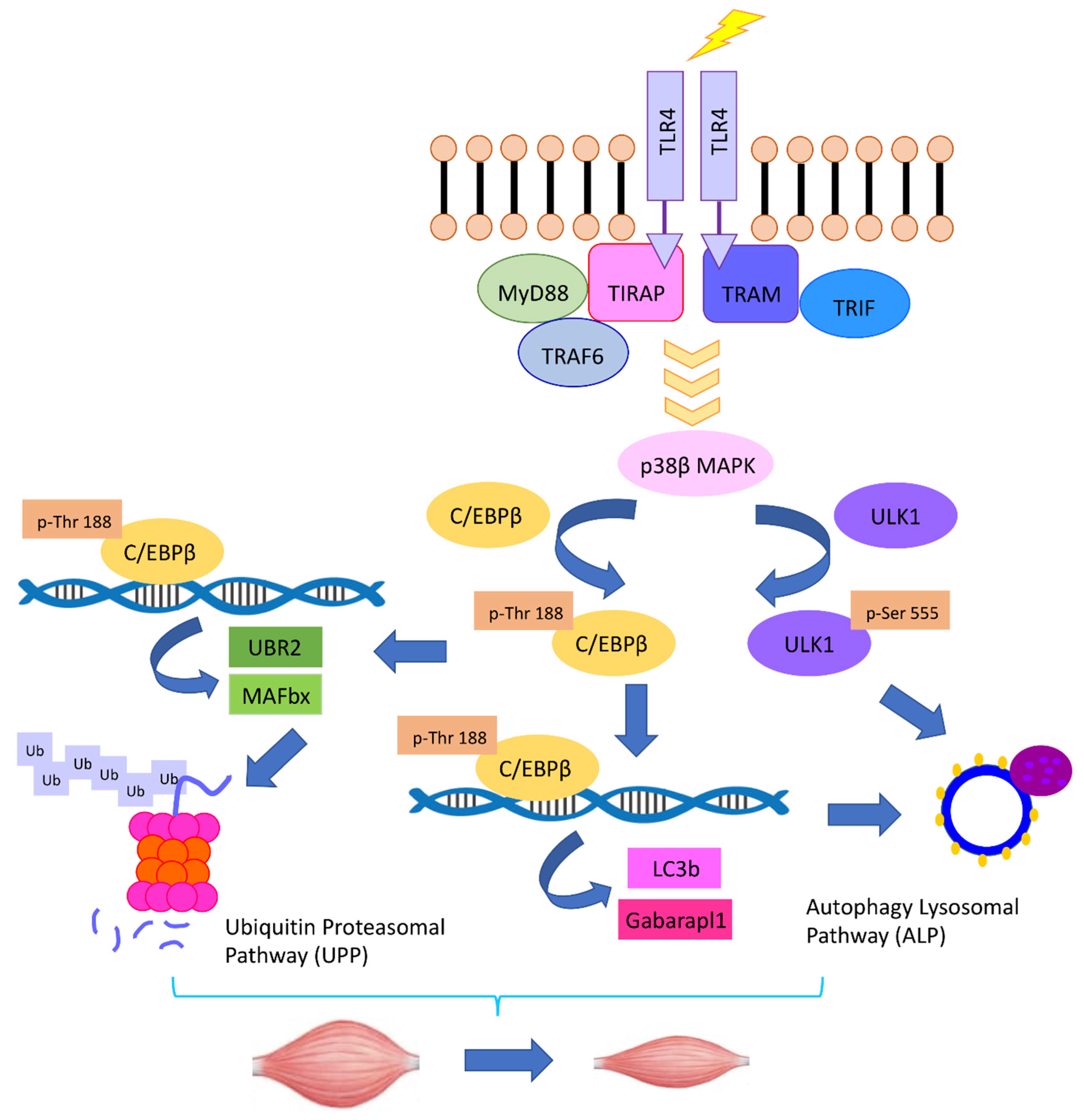
4. Intracellular Signaling Pathways that Mediate TLR4-Induced Muscle Catabolism
5. Cachectic Cancers Induce Muscle Catabolism by Activating TLR4 through Releasing Hsp70 and Hsp90
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; Macdonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Andreyev, H.J.; Norman, A.R.; Oates, J.; Cunningham, D. Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur. J. Cancer 1998, 34, 503–509. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef]
- Polge, C.; Heng, A.E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler-Joseph, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Leibovitch, M.P.; Cornille, K.; Tintignac, L.A.; Leibovitch, S.A. MAFbx/Atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines. J. Biol. Chem. 2009, 284, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Lagirand-Cantaloube, J.; Cornille, K.; Csibi, A.; Batonnet-Pichon, S.; Leibovitch, M.P.; Leibovitch, S.A. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS ONE 2009, 4, e4973. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Bohnert, K.R.; Gallot, Y.S.; Sato, S.; Xiong, G.; Hindi, S.M.; Kumar, A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J. 2016, 30, 3053–3068. [Google Scholar] [CrossRef] [Green Version]
- Talbert, E.E.; Metzger, G.A.; He, W.A.; Guttridge, D.C. Modeling human cancer cachexia in colon 26 tumor-bearing adult mice. J. Cachexia Sarcopenia Muscle 2014, 5, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Liu, Z.; Ding, H.; Miao, H.; Garcia, J.M.; Li, Y.P. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci. Rep. 2017, 7, 2273. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Rossi Fanelli, F.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Skipworth, R.J.; Gallagher, I.J.; Greig, C.A.; Guttridge, D.C.; Ross, J.A.; Fearon, K.C. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 2015, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.; Schols, A.M. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Bonetto, A.; Muscaritoli, M.; Costamagna, D.; Minero, V.G.; Bonelli, G.; Rossi Fanelli, F.; Baccino, F.M.; Costelli, P. Muscle atrophy in experimental cancer cachexia: Is the IGF-1 signaling pathway involved? Int. J. Cancer 2010, 127, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Jin, B.; Li, Y.P. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Lowry, S.F.; Cerami, A. The pathophysiologic role of cachectin/TNF in septic shock and cachexia. Ann. Inst. Pasteur Immunol. 1988, 139, 311–317. [Google Scholar] [CrossRef]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130′s role in Lewis lung carcinoma-induced cachexia. FASEB J. 2014, 28, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bhatnagar, S.; Paul, P.K. TWEAK and TRAF6 regulate skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Muscaritoli, M.; Bonetto, A.; Penna, F.; Reffo, P.; Bossola, M.; Bonelli, G.; Doglietto, G.B.; Baccino, F.M.; Rossi Fanelli, F. Muscle myostatin signalling is enhanced in experimental cancer cachexia. Eur. J. Clin. Investig. 2008, 38, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.H.; Tomkotter, L.; Vahle, A.K.; Rokosh, R.; Avanzi, A.; Mahmood, S.K.; Deutsch, M.; Alothman, S.; Alqunaibit, D.; Ochi, A.; et al. TGF-beta Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia. PLoS ONE 2015, 10, e0132786. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.D.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.; Liu, Z.; Lin, R.K.; Li, M.; Li, Y.P. Activin A induces skeletal muscle catabolism via p38beta mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Gao, S.; Durstine, J.L.; Koh, H.J.; Carver, W.E.; Frizzell, N.; Carson, J.A. Acute myotube protein synthesis regulation by IL-6 related cytokines. Am. J. Physiol. Cell Physiol. 2017, 313, C487–C500. [Google Scholar] [CrossRef]
- Li, Y.P.; Schwartz, R.J.; Waddell, I.D.; Holloway, B.R.; Reid, M.B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J. 1998, 12, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Lecker, S.H.; Chen, Y.; Waddell, I.D.; Goldberg, A.L.; Reid, M.B. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J. 2003, 17, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Seto, D.N.; Kandarian, S.C.; Jackman, R.W. A Key Role for Leukemia Inhibitory Factor in C26 Cancer Cachexia. J. Biol. Chem. 2015, 290, 19976–19986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedenmann, B.; Malfertheiner, P.; Friess, H.; Ritch, P.; Arseneau, J.; Mantovani, G.; Caprioni, F.; Van Cutsem, E.; Richel, D.; DeWitte, M.; et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J. Support. Oncol. 2008, 6, 18–25. [Google Scholar] [PubMed]
- Goldberg, R.M.; Loprinzi, C.L.; Mailliard, J.A.; O’Fallon, J.R.; Krook, J.E.; Ghosh, C.; Hestorff, R.D.; Chong, S.F.; Reuter, N.F.; Shanahan, T.G. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 1995, 13, 2856–2859. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Okamoto, M.; Sato, M. Toll-like receptor signaling in anti-cancer immunity. J. Med. Investig. 2003, 50, 9–24. [Google Scholar]
- Cannon, T.Y.; Guttridge, D.; Dahlman, J.; George, J.R.; Lai, V.; Shores, C.; Buzkova, P.; Couch, M.E. The effect of altered Toll-like receptor 4 signaling on cancer cachexia. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 1263–1269. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, A.; Zhang, G.; Abdel Fattah, E.A.; Eissa, N.T.; Li, Y.P. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011, 25, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Puppa, M.J.; White, J.P.; Sato, S.; Cairns, M.; Baynes, J.W.; Carson, J.A. Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim. Biophys. Acta 2011, 1812, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; Goli, P.; Roy, A.; Sharma, A.K.; Xiong, G.; Gallot, Y.S.; Kumar, A. TLR/MyD88/XBP1 signaling axis mediates skeletal muscle wasting during cancer cachexia. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Burfeind, K.G.; Michaelis, K.A.; Braun, T.P.; Olson, B.; Pelz, K.R.; Morgan, T.K.; Marks, D.L. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burfeind, K.G.; Zhu, X.; Levasseur, P.R.; Michaelis, K.A.; Norgard, M.A.; Marks, D.L. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain Behav. Immun. 2018, 73, 364–374. [Google Scholar] [CrossRef]
- Braun, T.P.; Grossberg, A.J.; Krasnow, S.M.; Levasseur, P.R.; Szumowski, M.; Zhu, X.X.; Maxson, J.E.; Knoll, J.G.; Barnes, A.P.; Marks, D.L. Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J. 2013, 27, 3572–3582. [Google Scholar] [CrossRef]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef]
- Johns, N.; Stretch, C.; Tan, B.H.; Solheim, T.S.; Sorhaug, S.; Stephens, N.A.; Gioulbasanis, I.; Skipworth, R.J.; Deans, D.A.; Vigano, A.; et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J. Cachexia Sarcopenia Muscle 2017, 8, 122–130. [Google Scholar] [CrossRef]
- Frantz, S.; Kobzik, L.; Kim, Y.D.; Fukazawa, R.; Medzhitov, R.; Lee, R.T.; Kelly, R.A. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J. Clin. Investig. 1999, 104, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birks, E.J.; Felkin, L.E.; Banner, N.R.; Khaghani, A.; Barton, P.J.; Yacoub, M.H. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J. Heart. Lung Transpl. 2004, 23, 228–235. [Google Scholar] [CrossRef]
- Verzola, D.; Bonanni, A.; Sofia, A.; Montecucco, F.; D’Amato, E.; Cademartori, V.; Parodi, E.L.; Viazzi, F.; Venturelli, C.; Brunori, G.; et al. Toll-like receptor 4 signalling mediates inflammation in skeletal muscle of patients with chronic kidney disease. J. Cachexia Sarcopenia Muscle 2017, 8, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Nozaki, R.; Naito, H.; Machida, S. TLR4-defective (C3H/HeJ) mice are not protected from cast immobilization-induced muscle atrophy. Physiol. Rep. 2017, 5, e13255. [Google Scholar] [CrossRef] [PubMed]
- Henriques, F.; Lopes, M.A.; Franco, F.O.; Knobl, P.; Santos, K.B.; Bueno, L.L.; Correa, V.A.; Bedard, A.H.; Guilherme, A.; Birbrair, A.; et al. Toll-Like Receptor-4 Disruption Suppresses Adipose Tissue Remodeling and Increases Survival in Cancer Cachexia Syndrome. Sci. Rep. 2018, 8, 18024. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Atkins, C.M.; Sweatt, J.D.; Reid, M.B. Mitochondria mediate tumor necrosis factor-alpha/NF-kappaB signaling in skeletal muscle myotubes. Antioxid. Redox Signal. 1999, 1, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.; Ruiz-Bonilla, V.; Gresh, L.; Hui, L.; Ballestar, E.; Sousa-Victor, P.; Baeza-Raja, B.; Jardi, M.; Bosch-Comas, A.; Esteller, M.; et al. Genetic analysis of p38 MAP kinases in myogenesis: Fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 2007, 26, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef]
- Gillespie, M.A.; Le Grand, F.; Scime, A.; Kuang, S.; von Maltzahn, J.; Seale, V.; Cuenda, A.; Ranish, J.A.; Rudnicki, M.A. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J. Cell Biol. 2009, 187, 991–1005. [Google Scholar] [CrossRef]
- Pogozelski, A.R.; Geng, T.; Li, P.; Yin, X.; Lira, V.A.; Zhang, M.; Chi, J.T.; Yan, Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE 2009, 4, e7934. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.C.; Alcazar, O.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R342–R349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, Y.P. p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet. Muscle 2012, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lin, R.K.; Kwon, Y.T.; Li, Y.P. Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB J. 2013, 27, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.S.; Zhou, X.; Solomon, V.; Baracos, V.E.; Davis, J.; Bannon, A.W.; Boyle, W.J.; Lacey, D.L.; Han, H.Q. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res. 2004, 64, 8193–8198. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.M.; Wu, C.L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Xia, Z.; An, J.Y.; Tasaki, T.; Davydov, I.V.; Seo, J.W.; Sheng, J.; Xie, Y.; Varshavsky, A. Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol. 2003, 23, 8255–8271. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.; Lecker, S.H.; Goldberg, A.L. The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J. Biol. Chem. 1998, 273, 25216–25222. [Google Scholar] [CrossRef] [PubMed]
- Solomon, V.; Baracos, V.; Sarraf, P.; Goldberg, A.L. Rates of ubiquitin conjugation increase when muscles atrophy, largely through activation of the N-end rule pathway. Proc. Natl. Acad. of Sci. USA 1998, 95, 12602–12607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecker, S.H.; Solomon, V.; Price, S.R.; Kwon, Y.T.; Mitch, W.E.; Goldberg, A.L. Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J. Clin. Investig. 1999, 104, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sin, K.W.T.; Ding, H.; Doan, H.A.; Gao, S.; Miao, H.; Wei, Y.; Wang, Y.; Zhang, G.; Li, Y.-P. p38beta MAPK mediates ULK1-dependent induction of autophagy in skeletal muscle of tumor-bearing mice. Cell Stress 2018, 2, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Mansell, A.; Patella, S.; Scott, B.J.; Hedger, M.P.; de Kretser, D.M.; Phillips, D.J. Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia. Proc. Natl. Acad. Sci. USA 2007, 104, 16239–16244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef] [Green Version]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-kB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.A.; Splenser, A.; Guillory, B.; Luo, J.; Mendiratta, M.; Belinova, B.; Halder, T.; Zhang, G.; Li, Y.P.; Garcia, J.M. Ghrelin prevents tumour- and cisplatin-induced muscle wasting: Characterization of multiple mechanisms involved. J. Cachexia Sarcopenia Muscle 2015, 6, 132–143. [Google Scholar] [CrossRef]
- An, J.Y.; Kim, E.; Zakrzewska, A.; Yoo, Y.D.; Jang, J.M.; Han, D.H.; Lee, M.J.; Seo, J.W.; Lee, Y.J.; Kim, T.Y.; et al. UBR2 of the N-end rule pathway is required for chromosome stability via histone ubiquitylation in spermatocytes and somatic cells. PLoS ONE 2012, 7, e37414. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Langen, R.C.; Haegens, A.; Vernooy, J.H.; Wouters, E.F.; de Winther, M.P.; Carlsen, H.; Steele, C.; Shoelson, S.E.; Schols, A.M. NF-kappaB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am. J. Respir. Cell Mol. Biol. 2012, 47, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.; Davis, J.M.; White, J.P.; Carson, J.A. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc (Min/+) mouse. Pflug. Arch. 2009, 457, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, I.J.; Stephens, N.A.; MacDonald, A.J.; Skipworth, R.J.; Husi, H.; Greig, C.A.; Ross, J.A.; Timmons, J.A.; Fearon, K.C. Suppression of skeletal muscle turnover in cancer cachexia: Evidence from the transcriptome in sequential human muscle biopsies. Clin. Cancer Res. 2012, 18, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Gallagher, I.J.; Rooyackers, O.; Skipworth, R.J.; Tan, B.H.; Marstrand, T.; Ross, J.A.; Guttridge, D.C.; Lundell, L.; Fearon, K.C.; et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- D’Orlando, C.; Marzetti, E.; Francois, S.; Lorenzi, M.; Conti, V.; di Stasio, E.; Rosa, F.; Brunelli, S.; Doglietto, G.B.; Pacelli, F.; et al. Gastric cancer does not affect the expression of atrophy-related genes in human skeletal muscle. Muscle Nerve 2013, 49, 528–533. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.P. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J. Cell Biochem. 2007, 100, 960–969. [Google Scholar] [CrossRef]
- Ono, Y.; Sakamoto, K. Lipopolysaccharide inhibits myogenic differentiation of C2C12 myoblasts through the Toll-like receptor 4-nuclear factor-kappaB signaling pathway and myoblast-derived tumor necrosis factor-alpha. PLoS ONE 2017, 12, e0182040. [Google Scholar] [CrossRef]
- Guttridge, D.C.; Mayo, M.W.; Madrid, L.V.; Wang, C.Y.; Baldwin, A.S., Jr. NF-kappaB-induced loss of MyoD messenger RNA: Possible role in muscle decay and cachexia. Science 2000, 289, 2363–2366. [Google Scholar] [CrossRef]
- Wang, H.; Hertlein, E.; Bakkar, N.; Sun, H.; Acharyya, S.; Wang, J.; Carathers, M.; Davuluri, R.; Guttridge, D.C. NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell Biol. 2007, 27, 4374–4387. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed]
- Ramji, D.P.; Foka, P. CCAAT/enhancer-binding proteins: Structure, function and regulation. Biochem. J. 2002, 365, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Nerlov, C. C/EBPs: Recipients of extracellular signals through proteome modulation. Curr. Opin. Cell Biol. 2008, 20, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Zhu, J.Z.; Zhang, G.; Li, Y.P. p300 Mediates Muscle Wasting in Lewis Lung Carcinoma. Cancer Res. 2019, 79, 1331–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Hartl, F.U.; Martin, J. Molecular chaperones in cellular protein folding. Curr. Opin. Struct. Biol. 1995, 5, 92–102. [Google Scholar] [CrossRef]
- Pilon, M.; Schekman, R. Protein translocation: How Hsp70 pulls it off. Cell 1999, 97, 679–682. [Google Scholar] [CrossRef]
- Fisher, E.A.; Zhou, M.; Mitchell, D.M.; Wu, X.; Omura, S.; Wang, H.; Goldberg, A.L.; Ginsberg, H.N. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J. Biol. Chem. 1997, 272, 20427–20434. [Google Scholar] [CrossRef]
- Qian, S.B.; McDonough, H.; Boellmann, F.; Cyr, D.M.; Patterson, C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 2006, 440, 551–555. [Google Scholar] [CrossRef] [Green Version]
- McArdle, A.; Dillmann, W.H.; Mestril, R.; Faulkner, J.A.; Jackson, M.J. Overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age-related muscle dysfunction. FASEB J. 2004, 18, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Senf, S.M.; Dodd, S.L.; McClung, J.M.; Judge, A.R. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008, 22, 3836–3845. [Google Scholar] [CrossRef] [PubMed]
- Senf, S.M.; Dodd, S.L.; Judge, A.R. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am. J. Physiol. Cell Physiol. 2010, 298, C38–C45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smuder, A.J.; Morton, A.B.; Hall, S.E.; Wiggs, M.P.; Ahn, B.; Wawrzyniak, N.R.; Sollanek, K.J.; Min, K.; Kwon, O.S.; Nelson, W.B.; et al. Effects of exercise preconditioning and HSP72 on diaphragm muscle function during mechanical ventilation. J. Cachexia Sarcopenia Muscle 2019. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Botzler, C.; Issels, R.; Multhoff, G. Heat-shock protein 72 cell-surface expression on human lung carcinoma cells in associated with an increased sensitivity to lysis mediated by adherent natural killer cells. Cancer Immunol. Immunother. 1996, 43, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Botzler, C.; Schmidt, J.; Luz, A.; Jennen, L.; Issels, R.; Multhoff, G. Differential Hsp70 plasma-membrane expression on primary human tumors and metastases in mice with severe combined immunodeficiency. Int. J. Cancer 1998, 77, 942–948. [Google Scholar] [CrossRef]
- Multhoff, G.; Botzler, C.; Jennen, L.; Schmidt, J.; Ellwart, J.; Issels, R. Heat shock protein 72 on tumor cells: A recognition structure for natural killer cells. J. Immunol. 1997, 158, 4341–4350. [Google Scholar] [PubMed]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef]
- Becker, B.; Multhoff, G.; Farkas, B.; Wild, P.J.; Landthaler, M.; Stolz, W.; Vogt, T. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp. Dermatol. 2004, 13, 27–32. [Google Scholar] [CrossRef]
- O’Gorman, P.; McMillan, D.C.; McArdle, C.S. Longitudinal study of weight, appetite, performance status, and inflammation in advanced gastrointestinal cancer. Nutr. Cancer 1999, 35, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Takeuchi, S.; Kubota, K.; Kobayashi, Y.; Kozakai, S.; Ukai, I.; Shichiku, A.; Okubo, M.; Numasaki, M.; Kanemitsu, Y.; et al. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKK-IRF3 axis activation. J. Biol. Chem. 2018, 293, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice with Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles from Cancer Cells. Gastroenterology 2019, 156, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ito, Y.; Wakai, K.; Kawado, M.; Hashimoto, S.; Seki, N.; Ando, M.; Nishino, Y.; Kondo, T.; Watanabe, Y.; et al. Serum heat shock protein 70 levels and lung cancer risk: A case-control study nested in a large cohort study. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, X.; Lou, J.; Han, X.; Zhang, L.; Wang, Q.; Li, B.; Dong, M.; Zhang, Y. Plasma levels of heat shock protein 90 alpha associated with lung cancer development and treatment responses. Clin. Cancer Res. 2014, 20, 6016–6022. [Google Scholar] [CrossRef]
- Rong, B.; Zhao, C.; Liu, H.; Ming, Z.; Cai, X.; Gao, W.; Yang, S. Identification and verification of Hsp90-beta as a potential serum biomarker for lung cancer. Am. J. Cancer Res. 2014, 4, 874–885. [Google Scholar]
- Gunther, S.; Ostheimer, C.; Stangl, S.; Specht, H.M.; Mozes, P.; Jesinghaus, M.; Vordermark, D.; Combs, S.E.; Peltz, F.; Jung, M.P.; et al. Correlation of Hsp70 Serum Levels with Gross Tumor Volume and Composition of Lymphocyte Subpopulations in Patients with Squamous Cell and Adeno Non-Small Cell Lung Cancer. Front. Immunol. 2015, 6, 556. [Google Scholar] [CrossRef] [Green Version]
- Balazs, M.; Zsolt, H.; Laszlo, G.; Gabriella, G.; Lilla, T.; Gyula, O.; Balazs, D.; Eva, M.; Zoltan, B.; Zoltan, P.; et al. Serum Heat Shock Protein 70, as a Potential Biomarker for Small Cell Lung Cancer. Pathol. Oncol. Res. 2016, 23, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Luo, S.; Xu, F.; Zou, G.; Xu, G.; He, J.; Huang, Y.; Zhu, H.; Li, Y. The expression of DAMP proteins HSP70 and cancer-testis antigen SPAG9 in peripheral blood of patients with HCC and lung cancer. Cell Stress Chaperones 2017, 22, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, J.; Madaras, B.; Toth, E.K.; Fust, G.; Prohaszka, Z. Serum level of soluble 70-kD heat shock protein is associated with high mortality in patients with colorectal cancer without distant metastasis. Cell Stress Chaperones 2010, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.K.; Girotra, M.; Singla, M.; Dutta, A.; Otis Stephen, F.; Nair, P.P.; Merchant, N.B. Serum HSP70: A novel biomarker for early detection of pancreatic cancer. Pancreas 2012, 41, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Garcia, V.; Rodriguez, M.; Compte, M.; Cisneros, E.; Veguillas, P.; Garcia, J.M.; Dominguez, G.; Campos-Martin, Y.; Cuevas, J.; et al. Analysis of exosome release and its prognostic value in human colorectal cancer. Genes Chromosomes Cancer 2012, 51, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ni, Q.; Wang, X.; Zhu, H.; Wang, Z.; Huang, J. High expression of RAB27A and TP53 in pancreatic cancer predicts poor survival. Med. Oncol. 2015, 32, 372. [Google Scholar] [CrossRef] [PubMed]
- Rabinowits, G.; Gercel-Taylor, C.; Day, J.M.; Taylor, D.D.; Kloecker, G.H. Exosomal microRNA: A diagnostic marker for lung cancer. Clin. Lung Cancer 2009, 10, 42–46. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sin, T.K.; Zhang, G.; Zhang, Z.; Gao, S.; Li, M.; Li, Y.-P. Cancer Takes a Toll on Skeletal Muscle by Releasing Heat Shock Proteins—An Emerging Mechanism of Cancer-Induced Cachexia. Cancers 2019, 11, 1272. https://doi.org/10.3390/cancers11091272
Sin TK, Zhang G, Zhang Z, Gao S, Li M, Li Y-P. Cancer Takes a Toll on Skeletal Muscle by Releasing Heat Shock Proteins—An Emerging Mechanism of Cancer-Induced Cachexia. Cancers. 2019; 11(9):1272. https://doi.org/10.3390/cancers11091272
Chicago/Turabian StyleSin, Thomas K, Guohua Zhang, Zicheng Zhang, Song Gao, Min Li, and Yi-Ping Li. 2019. "Cancer Takes a Toll on Skeletal Muscle by Releasing Heat Shock Proteins—An Emerging Mechanism of Cancer-Induced Cachexia" Cancers 11, no. 9: 1272. https://doi.org/10.3390/cancers11091272