Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story

Abstract
:1. Introduction
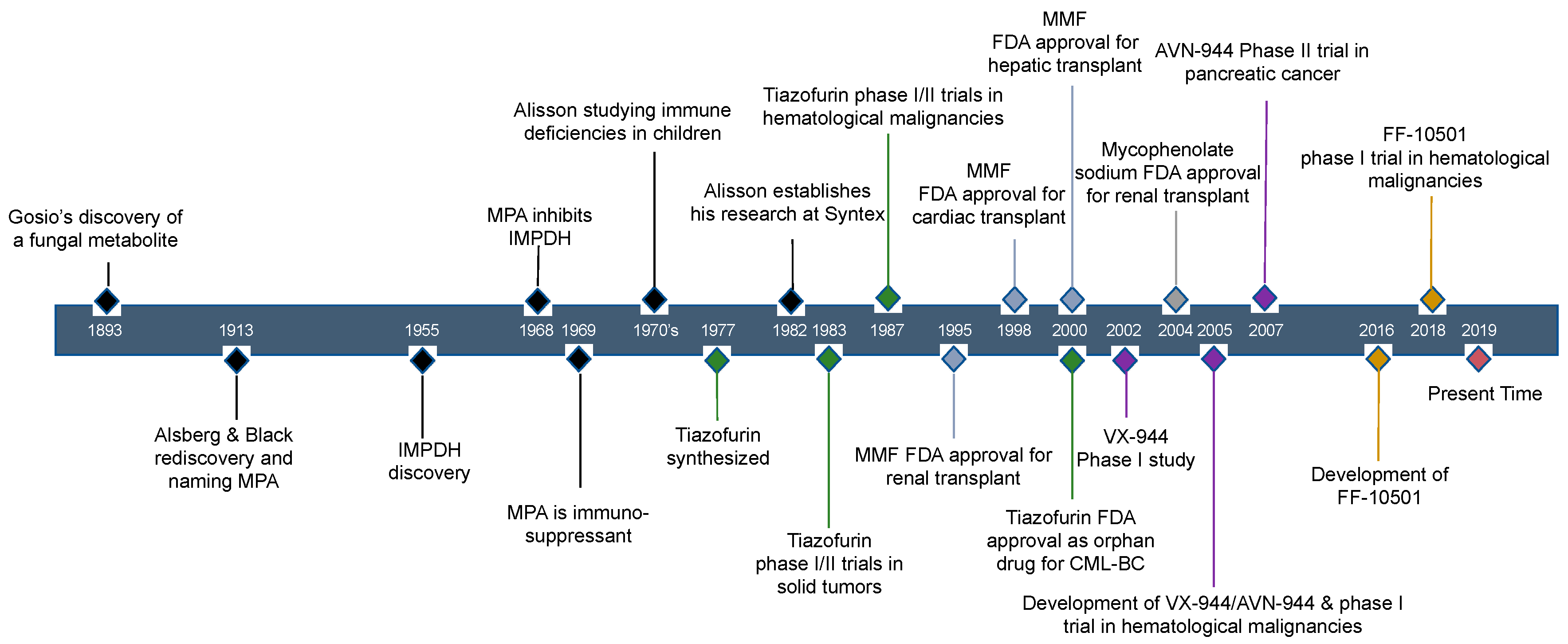
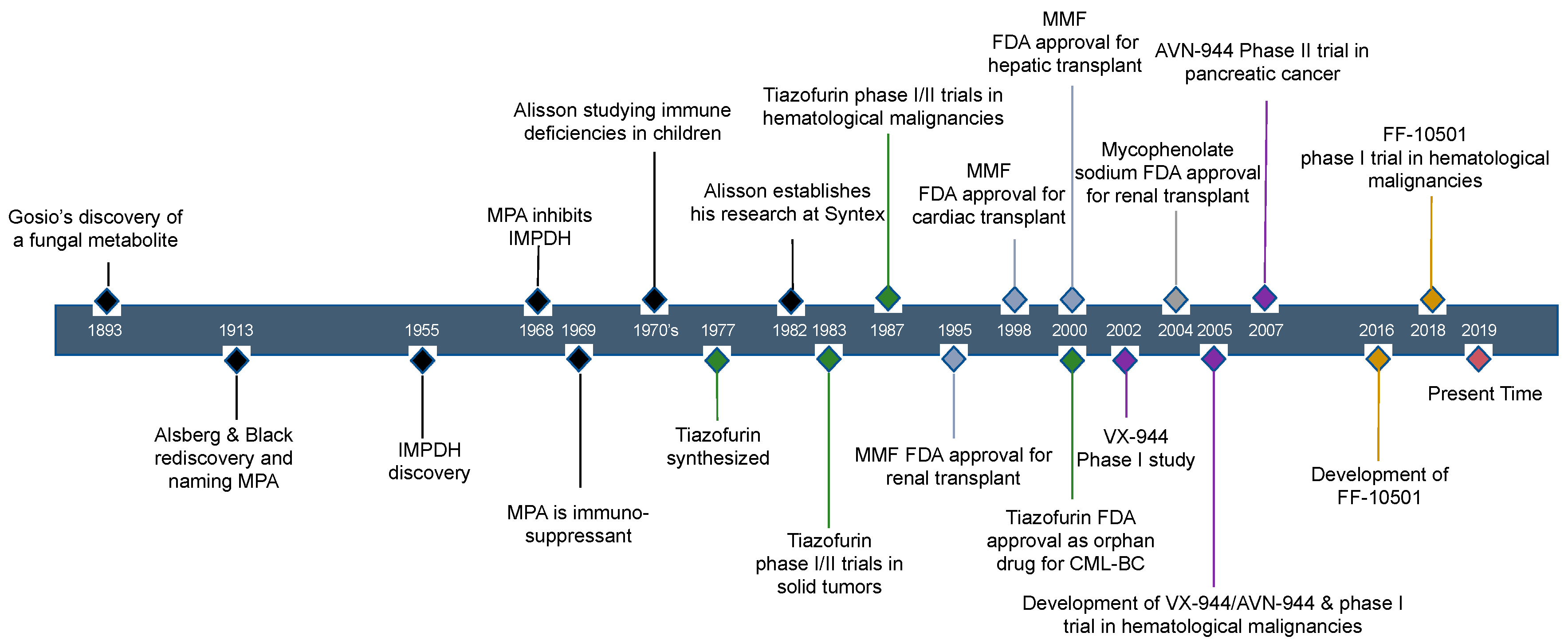
2. Historical Review of IMPDH Inhibitors: The Discovery of Mycophenolic Acid
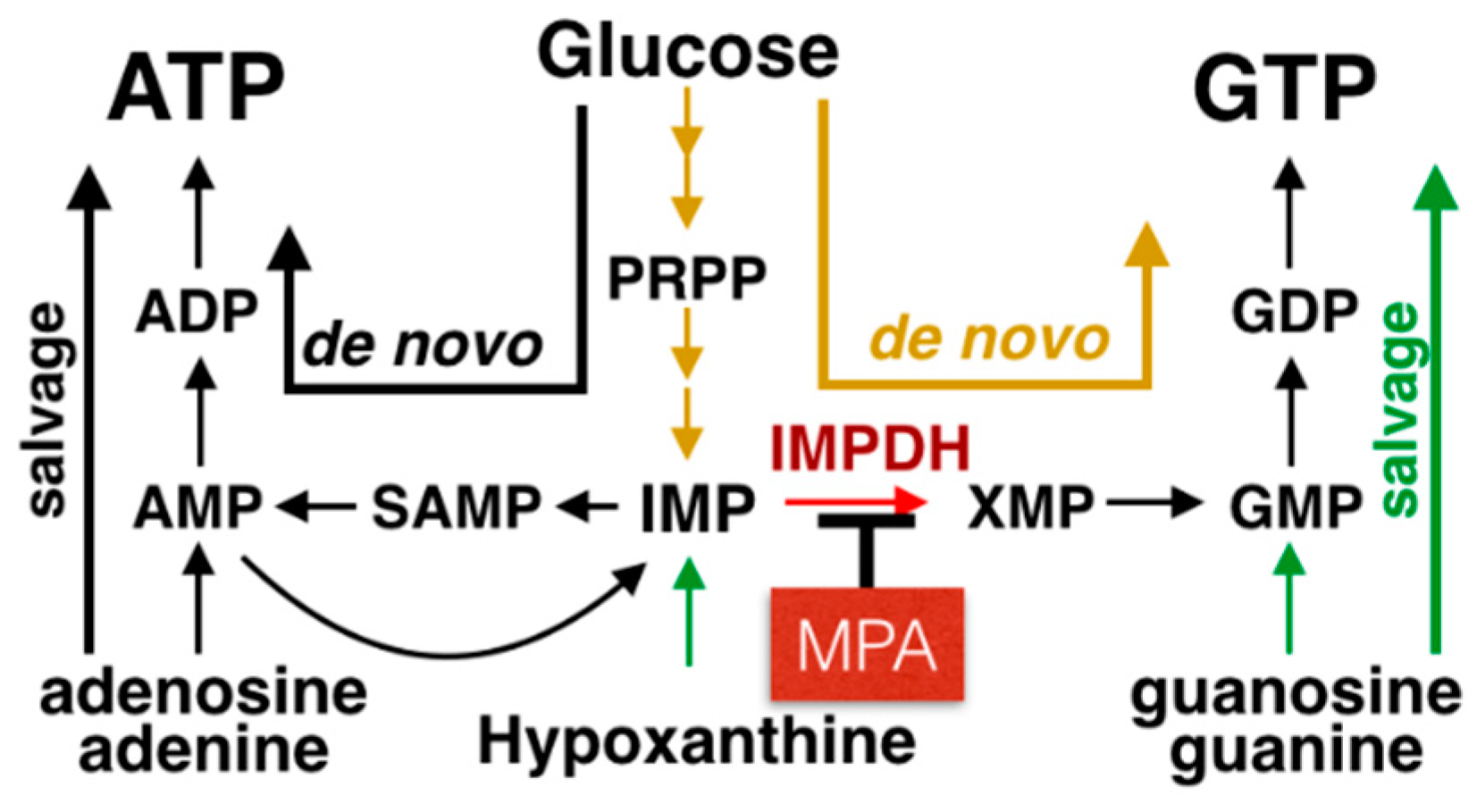
3. MPA Inhibits IMPDH Activity and Possesses an Immunosuppressive Effect
4. Current Use of IMPDH Inhibitors and Their Application in Cancer Therapy
4.1. Evidence of the Antitumor Activity of IMPDH Inhibitors
4.2. Long-Term Treatment Effect of MPA/MMF in Tissue-Transplanted Patients
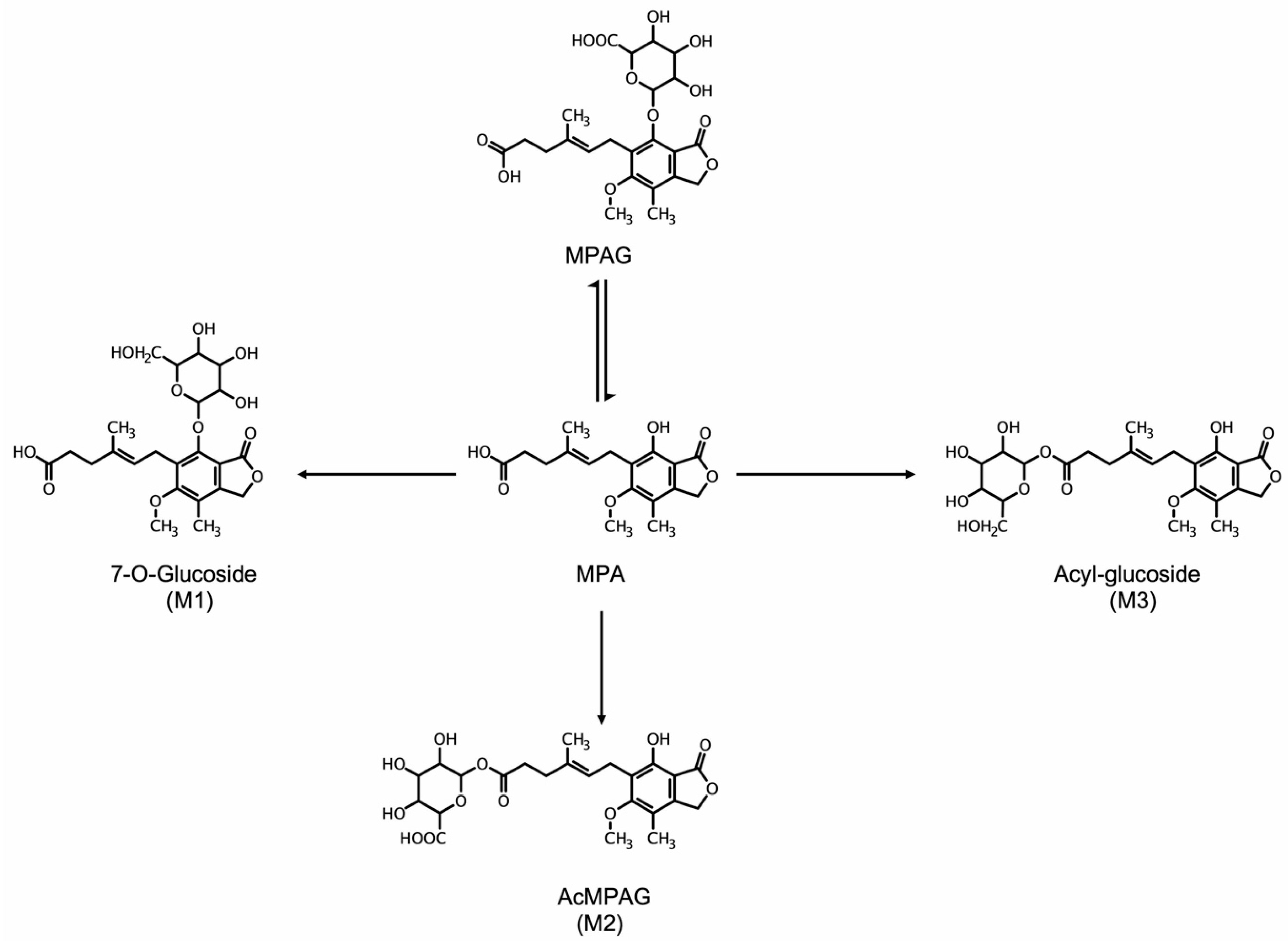
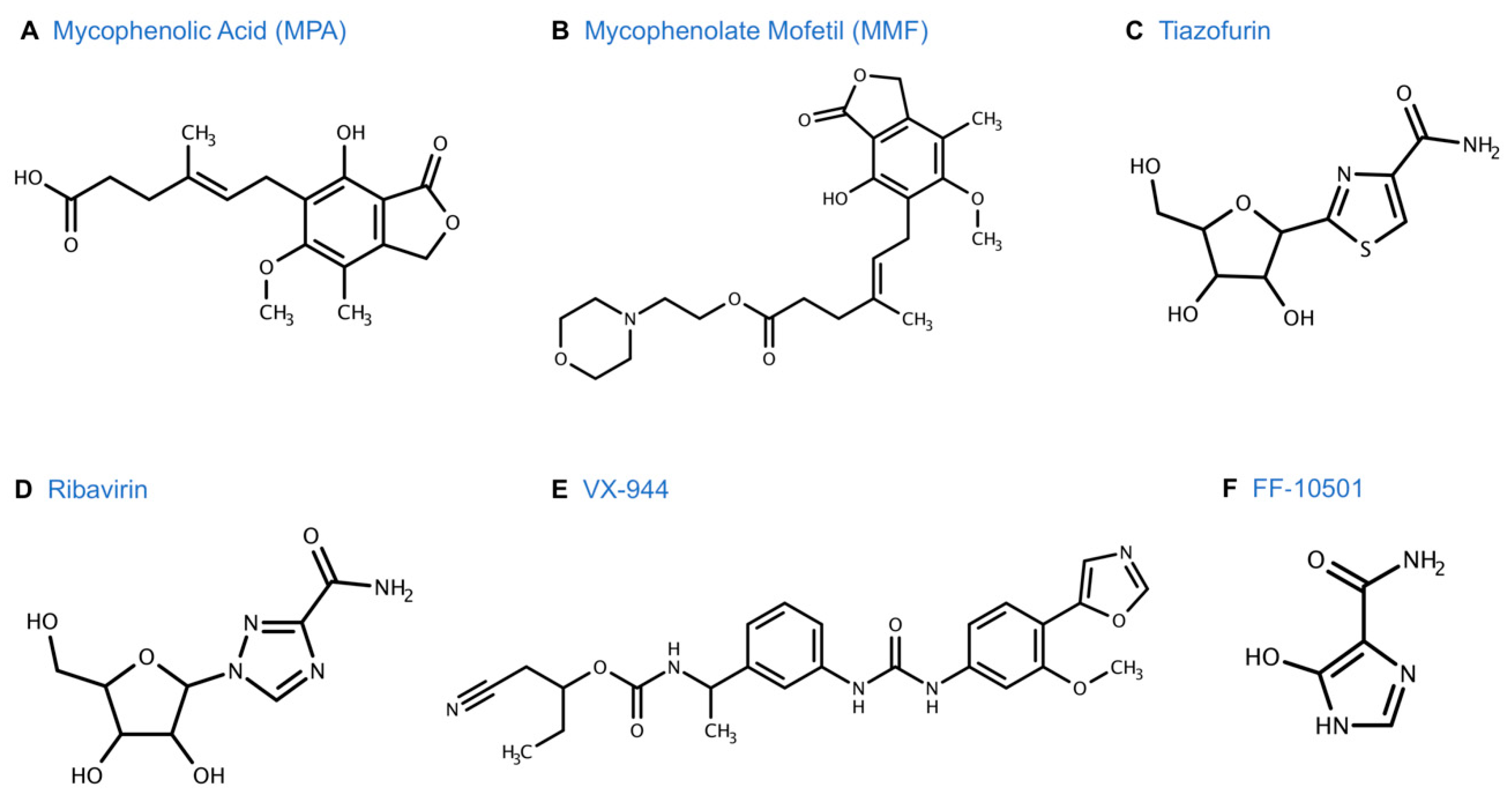
4.3. Metabolism of MPA and Improved Routes of Delivery
5. Other IMPDH Inhibitors
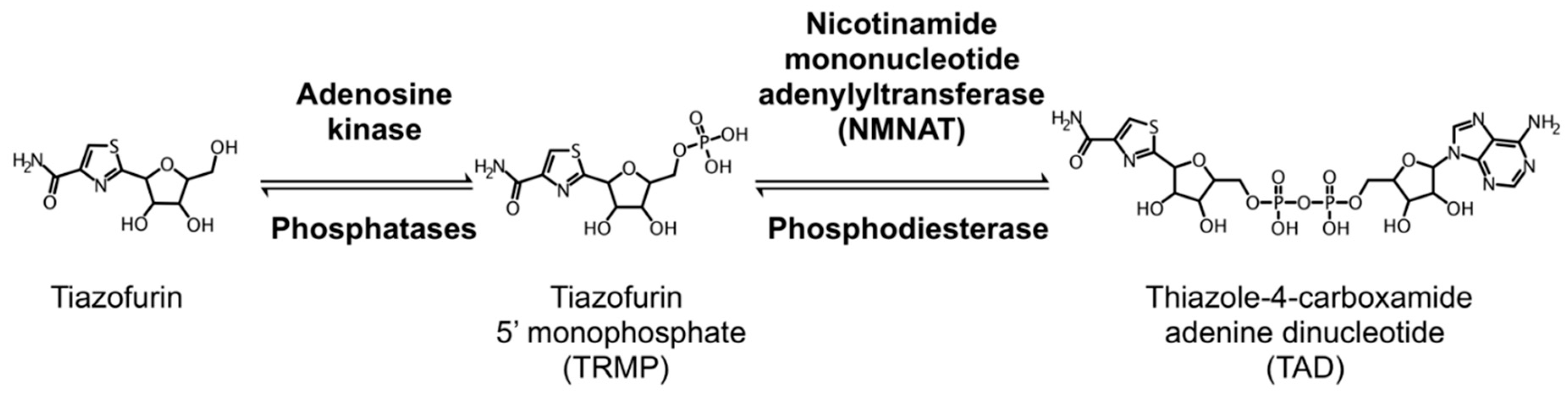
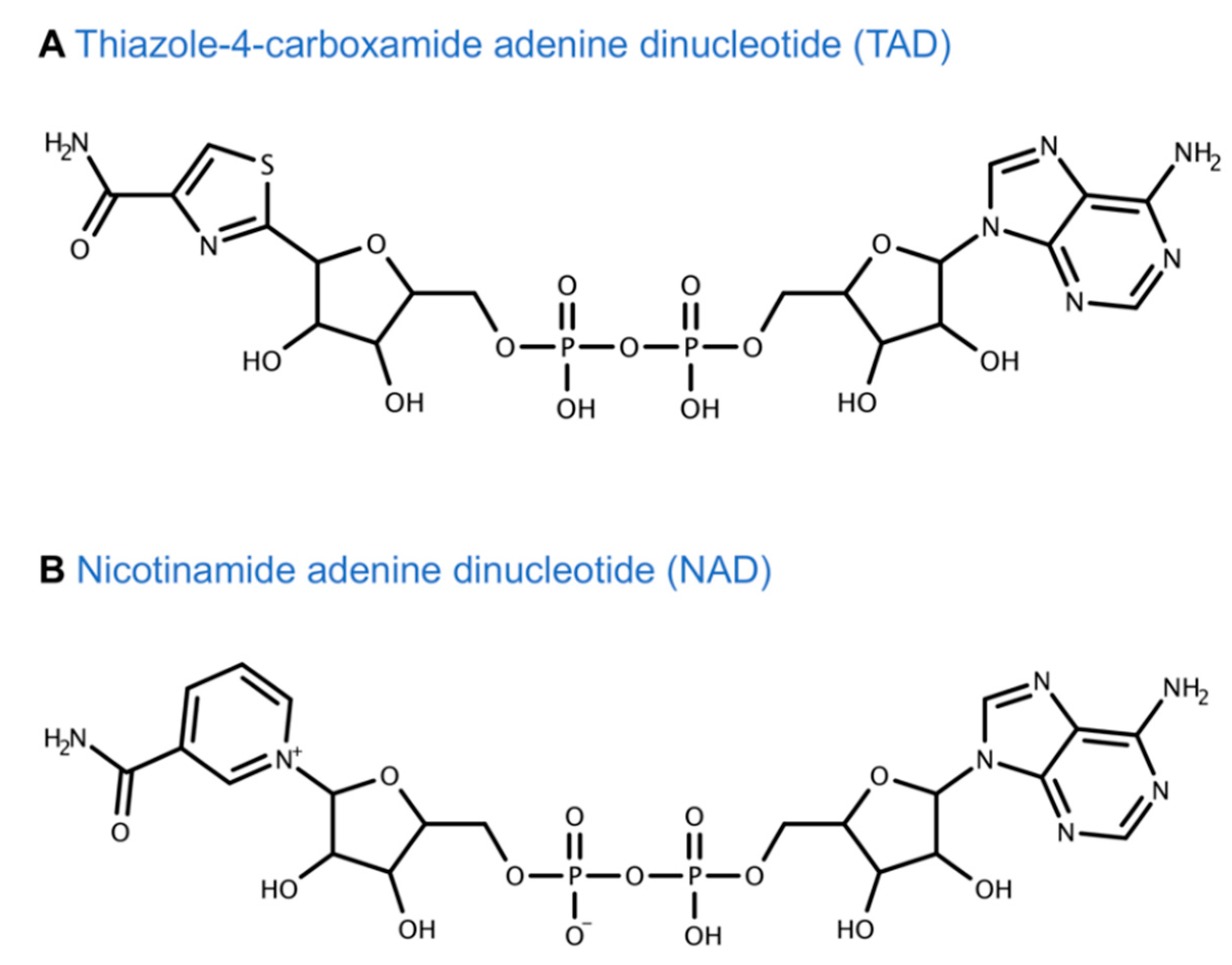
5.1. Tiazofurin Trials for Hematological Malignancy and Solid Tumors
5.2. VX-944/AVN-944 and VX-497, Direct IMPDH Inhibitors
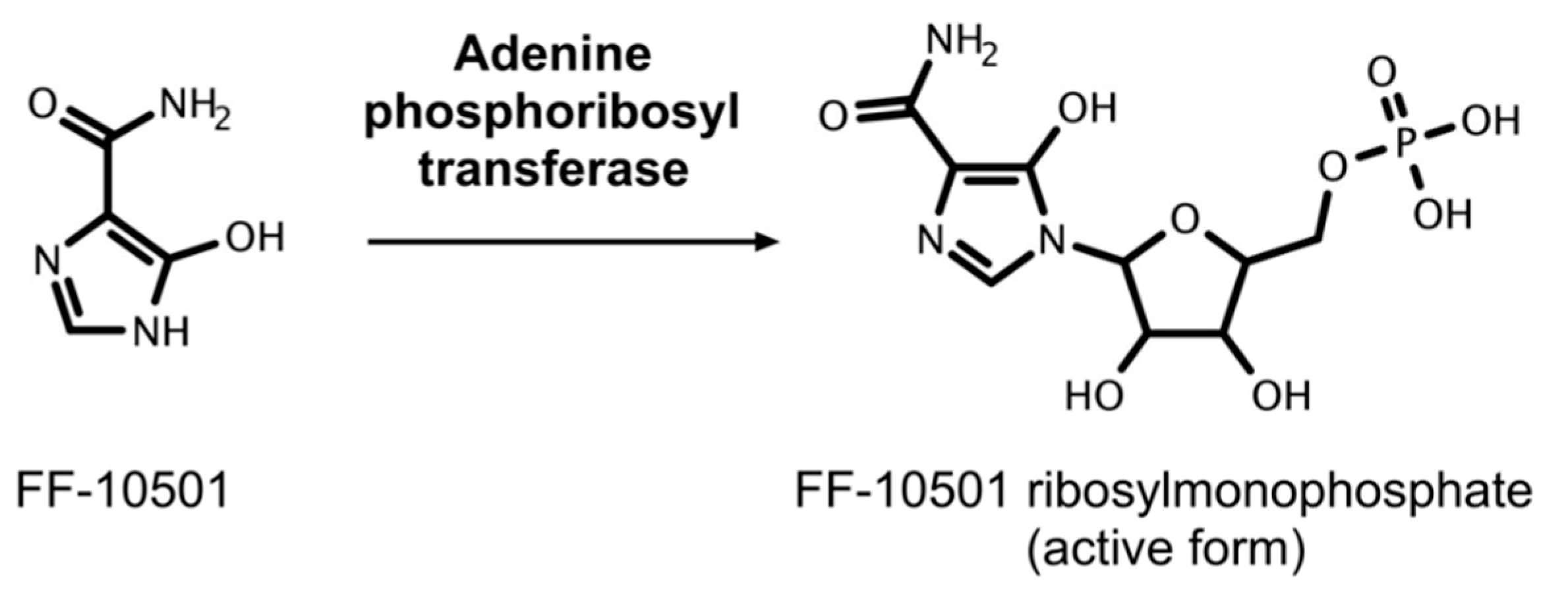
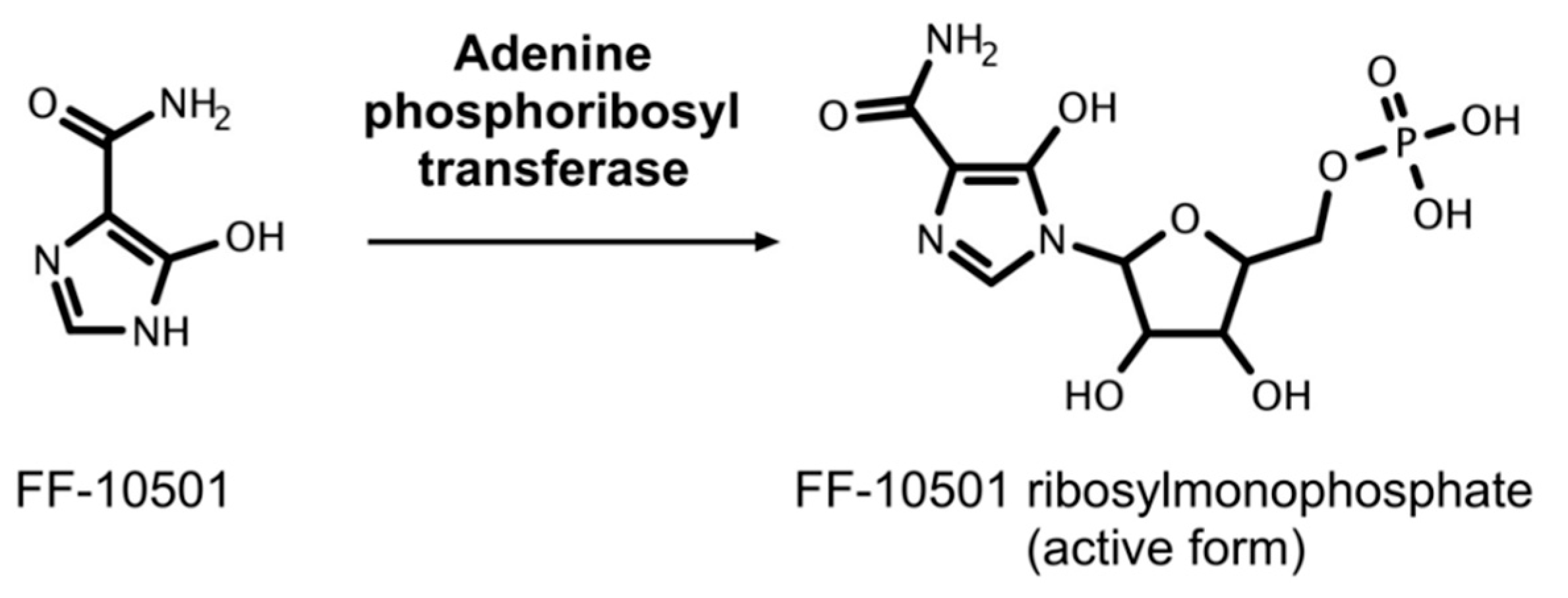
5.3. FF-10501
5.4. Future Directions for IMPDH Inhibitors as Anti-Tumor Drugs
6. Regulation of IMPDH by GTP
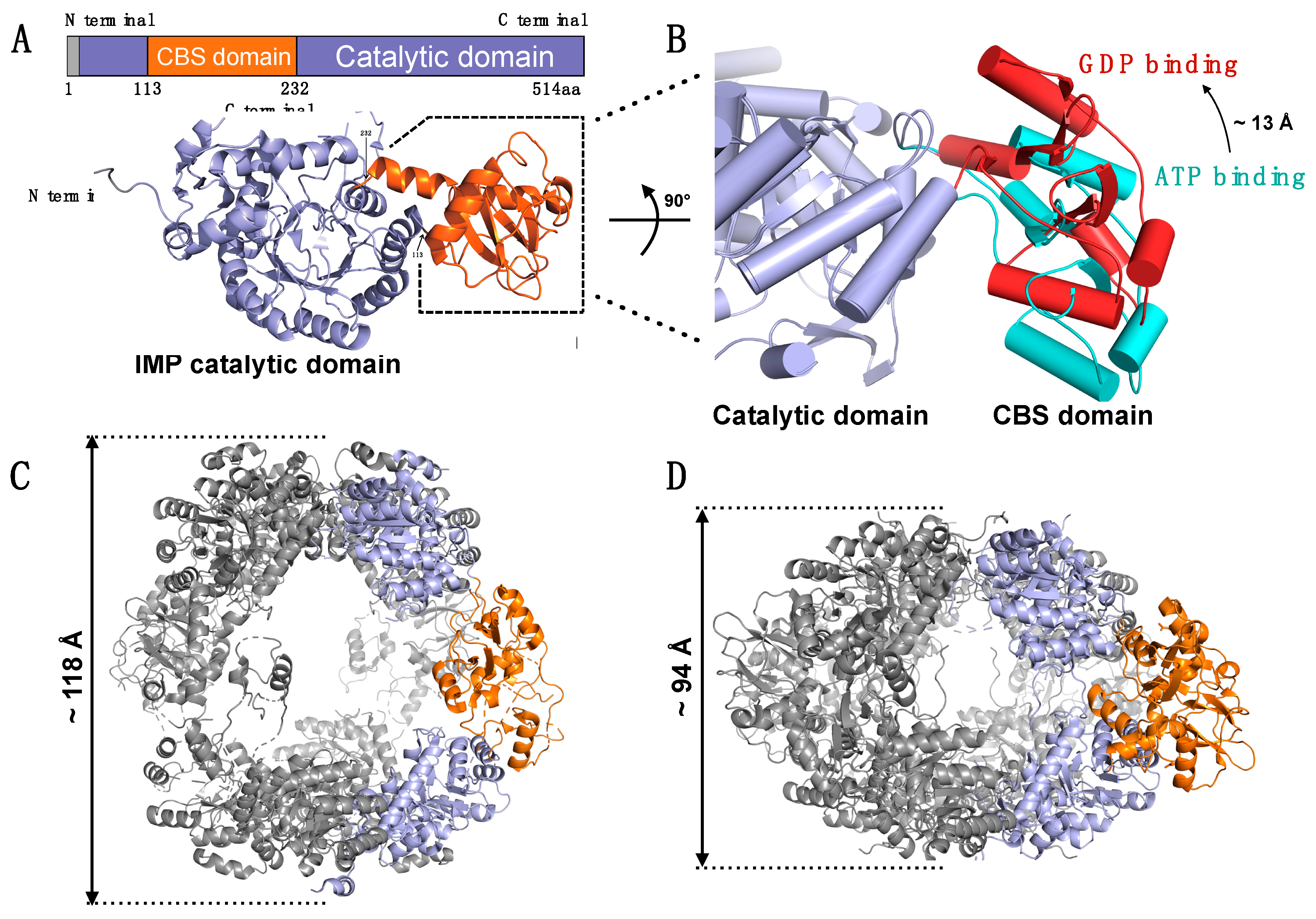
7. Dynamic Feature of IMPDH—Macrostructural Formation
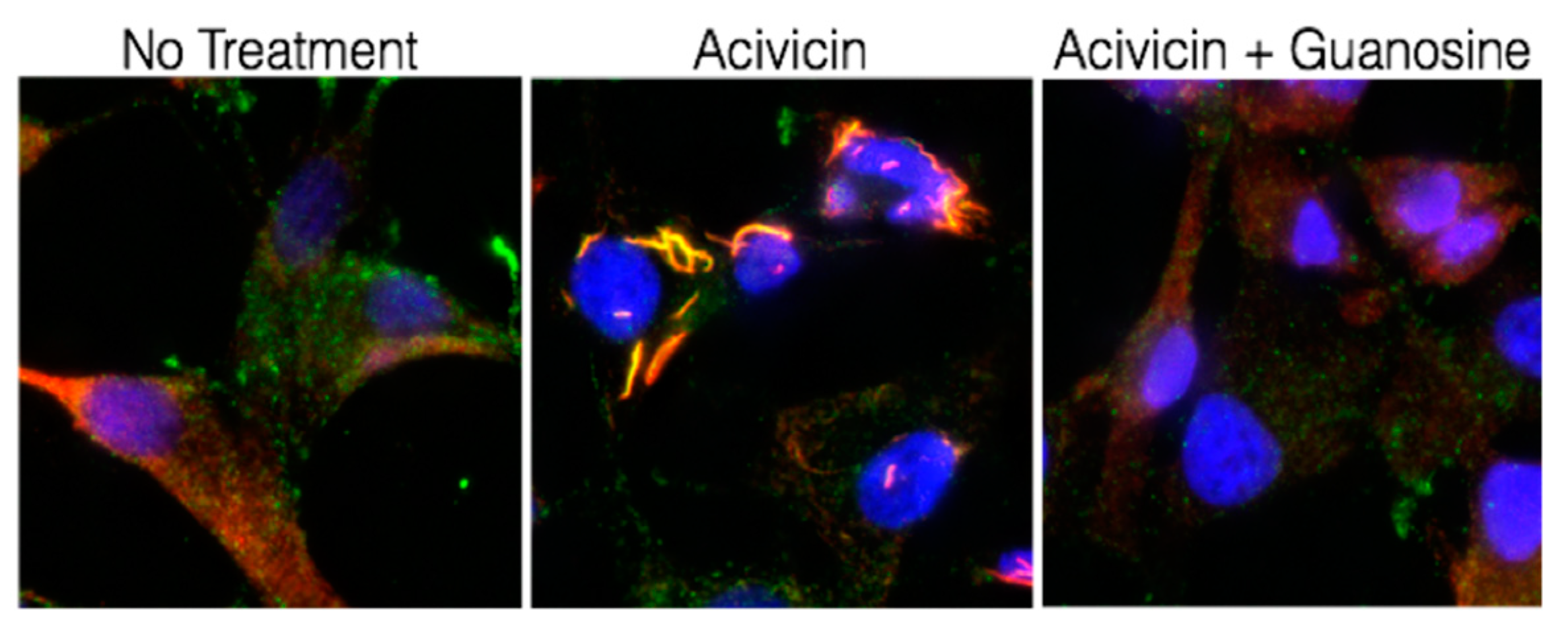
8. IMPDH Immunohistochemical Analysis May Report the Metabolic Status of Tumors
9. Potential Biomarker for the Anti-Tumor Effect of IMPDH Inhibitors in the Target Tumor
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Glesne, D.; Collart, F.; Varkony, T.; Drabkin, H.; Huberman, E. Chromosomal localization and structure of the human type II IMP dehydrogenase gene (IMPDH2). Genomics 1993, 16, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Senda, M.; Natsumeda, Y. Tissue-differential expression of two distinct genes for human IMP dehydrogenase (E.C.1.1.1.205). Life Sci. 1994, 54, 1917–1926. [Google Scholar] [CrossRef]
- Collart, F.R.; Chubb, C.B.; Mirkin, B.L.; Huberman, E. Increased inosine-5′-phosphate dehydrogenase gene expression in solid tumor tissues and tumor cell lines. Cancer Res. 1992, 52, 5826–5828. [Google Scholar] [PubMed]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvezan, A.J.; Turner, M.; Belaid, A.; Lam, H.C.; Miller, S.K.; McNamara, M.C.; Baglini, C.; Housden, B.E.; Perrimon, N.; Kwiatkowski, D.J.; et al. mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 2017, 32, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Ni, M.; Chalishazar, M.D.; Huffman, K.E.; Kim, J.; Cai, L.; Shi, X.; Cai, F.; Zacharias, L.G.; Ireland, A.S.; et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018, 28, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Kimura, T.; Ando, K.; Sawada, M.; Tamura, G. Antitumor activity of mycophenolic acid. J. Antibiot. 1969, 22, 297–302. [Google Scholar] [CrossRef]
- Suzuki, S.; Takaku, S.; Mori, T. Antitumor activity of derivatives of mycophenolic acid. J. Antibiot. 1976, 29, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Tressler, R.J.; Garvin, L.J.; Slate, D.L. Anti-tumor activity of mycophenolate mofetil against human and mouse tumors in vivo. Int. J. Cancer 1994, 57, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Bollet, A.J. Politics and pellagra: The epidemic of pellagra in the U.S. in the early twentieth century. Yale J. Biol. Med. 1992, 65, 211–221. [Google Scholar] [PubMed]
- Gosio, B. Sperimentate su culture pure di bacilli del carbonchio demonstrarato notevole potere antisettica. CR Acad Med. Torino 1893, 61, 484. [Google Scholar]
- Alsberg, C.L.; Black, O.M. Contribution to the study of maize deterioration. U.S. Dept. Agric. Bur. Plant Ind. Bull. 1913, 270, 7–48. [Google Scholar]
- Papageorgiou, C. Enterohepatic recirculation: A powerful incentive for drug discovery in the inosine monophosphate dehydrogenase field. Mini Rev. Med. Chem. 2001, 1, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Abrams, R.; Bentley, M. Biosynthesis of nucleic acid purines. I. Formation of guanine from adenine compounds in bone marrow extracts. Arch. Biochem. Biophys. 1955, 56, 184–195. [Google Scholar] [CrossRef]
- Abrams, R.; Bentley, M. Biosynthesis of nucleic acid purines. II. Role of hypoxanthine and xanthine compounds. Arch. Biochem. Biophys. 1955, 58, 109–118. [Google Scholar] [CrossRef]
- Lagerkvist, U. Enzymic Synthesis of Xanthosine-5-Phosphate and Guanosine-5-Phosphate from Inosine-5-Phosphate. Acta Chem. Scand. 1955, 9, 1028–1029. [Google Scholar] [CrossRef]
- Franklin, T.J.; Cook, J.M. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem. J. 1969, 113, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Franklin, T.J. Pharmaceutical Compositions. UK. Patent Specification GB1203328, 26 August 1970. [Google Scholar]
- Allison, A.C.; Almquist, S.J.; Muller, C.D.; Eugui, E.M. In vitro immunosuppressive effects of mycophenolic acid and an ester pro-drug, RS-61443. Transplant. Proc. 1991, 23, 10–14. [Google Scholar]
- Mitsui, A.; Suzuki, S. Immunosuppressive effect of mycophenolic acid. J. Antibiot. 1969, 22, 358–363. [Google Scholar] [CrossRef]
- Lee, W.A.; Gu, L.; Miksztal, A.R.; Chu, N.; Leung, K.; Nelson, P.H. Bioavailability improvement of mycophenolic acid through amino ester derivatization. Pharm. Res. 1990, 7, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Sollinger, H.W. Mycophenolate mofetil for the prevention of acute rejection in primary cadaveric renal allograft recipients. U.S. Renal Transplant Mycophenolate Mofetil Study Group. Transplantation 1995, 60, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Grinyo, J.G.C.; Pichlmayr, R.; Sadek, S.; Vanrenterghem, Y.; Behrend, M.; Luck, R.; Moreso, F.; Peeters, J.; Rodicio, J.; Morales, J.; et al. Placebo-controlled study of mycophenolate mofetil combined with cyclosporin and corticosteroids for prevention of acute rejection. Lancet 1995, 345, 1321–1325. [Google Scholar]
- Keown, P. A blinded, randomized clinical trial of mycophenolate mofetil for the prevention of acute rejection in cadaveric renal transplantation. The Tricontinental Mycophenolate Mofetil Renal Transplantation Study Group. Transplantation 1996, 61, 1029–1037. [Google Scholar]
- Eckhoff, D.E.; McGuire, B.M.; Frenette, L.R.; Contreras, J.L.; Hudson, S.L.; Bynon, J.S. Tacrolimus (FK506) and mycophenolate mofetil combination therapy versus tacrolimus in adult liver transplantation. Transplantation 1998, 65, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, R.H.; Shorr, J.S.; Steffen, B.J.; Chu, A.H.; Gordon, R.D.; Lake, J.R. Mycophenolate mofetil combination therapy improves long-term outcomes after liver transplantation in patients with and without hepatitis C. Liver Transplant. 2005, 11, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Nashan, B.; Saliba, F.; Durand, F.; Barcena, R.; Herrero, J.I.; Mentha, G.; Neuhaus, P.; Bowles, M.; Patch, D.; Bernardos, A.; et al. Pharmacokinetics, efficacy, and safety of mycophenolate mofetil in combination with standard-dose or reduced-dose tacrolimus in liver transplant recipients. Liver Transplant. 2009, 15, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Eisen, H.J.; Kobashigawa, J.; Keogh, A.; Bourge, R.; Renlund, D.; Mentzer, R.; Alderman, E.; Valantine, H.; Dureau, G.; Mancini, D.; et al. Three-year results of a randomized, double-blind, controlled trial of mycophenolate mofetil versus azathioprine in cardiac transplant recipients. J. Heart Lung Transplant. 2005, 24, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, J.A.; Miller, L.W.; Russell, S.D.; Ewald, G.A.; Zucker, M.J.; Goldberg, L.R.; Eisen, H.J.; Salm, K.; Tolzman, D.; Gao, J.; et al. Tacrolimus with mycophenolate mofetil (MMF) or sirolimus vs. cyclosporine with MMF in cardiac transplant patients: 1-year report. Am. J. Transplant. 2006, 6, 1377–1386. [Google Scholar] [CrossRef]
- Kaczmarek, I.; Zaruba, M.M.; Beiras-Fernandez, A.; Reimann, R.; Nickel, T.; Grinninger, C.; Sadoni, S.; Hagl, C.; Meiser, B. Tacrolimus with mycophenolate mofetil or sirolimus compared with calcineurin inhibitor-free immunosuppression (sirolimus/mycophenolate mofetil) after heart transplantation: 5-year results. J. Heart Lung Transplant. 2013, 32, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, A.K.; Andersson, B.; Gustafsson, F.; Eiskjaer, H.; Radegran, G.; Gude, E.; Jansson, K.; Solbu, D.; Sigurdardottir, V.; Arora, S.; et al. Everolimus initiation and early calcineurin inhibitor withdrawal in heart transplant recipients: A randomized trial. Am. J. Transplant. 2014, 14, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Treede, H.; Klepetko, W.; Reichenspurner, H.; Zuckermann, A.; Meiser, B.; Birsan, T.; Wisser, W.; Reichert, B. Tacrolimus versus cyclosporine after lung transplantation: A prospective, open, randomized two-center trial comparing two different immunosuppressive protocols. J. Heart Lung Transplant. 2001, 20, 511–517. [Google Scholar] [CrossRef]
- Zuckermann, A.; Reichenspurner, H.; Birsan, T.; Treede, H.; Deviatko, E.; Reichart, B.; Klepetko, W. Cyclosporine A versus tacrolimus in combination with mycophenolate mofetil and steroids as primary immunosuppression after lung transplantation: One-year results of a 2-center prospective randomized trial. J. Thorac. Cardiovasc. Surg. 2003, 125, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speich, R.; Schneider, S.; Hofer, M.; Irani, S.; Vogt, P.; Weder, W.; Boehler, A. Mycophenolate mofetil reduces alveolar inflammation, acute rejection and graft loss due to bronchiolitis obliterans syndrome after lung transplantation. Pulm. Pharmacol. Ther. 2010, 23, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Ricart, M.J.; Oppenheimer, F.; Andres, A.; Morales, J.M.; Alonso, A.; Fernandez, C. Enteric-coated mycophenolate sodium in de novo and maintenance kidney-pancreas transplant recipients. Clin. Transplant. 2012, 26, 424–431. [Google Scholar] [CrossRef]
- Descourouez, J.L.; Jorgenson, M.R.; Menninga, N.; Leverson, G.; Odorico, J.; Redfield, R. Impact of intensive dosing of mycophenolate on pancreas allograft survival. Clin. Transplant. 2018, 32, e13293. [Google Scholar] [CrossRef] [PubMed]
- Alousi, A.M.; Weisdorf, D.J.; Logan, B.R.; Bolanos-Meade, J.; Carter, S.; Difronzo, N.; Pasquini, M.; Goldstein, S.C.; Ho, V.T.; Hayes-Lattin, B.; et al. Etanercept, mycophenolate, denileukin, or pentostatin plus corticosteroids for acute graft-versus-host disease: A randomized phase 2 trial from the Blood and Marrow Transplant Clinical Trials Network. Blood 2009, 114, 511–517. [Google Scholar] [CrossRef]
- Wolff, D.; Gerbitz, A.; Ayuk, F.; Kiani, A.; Hildebrandt, G.C.; Vogelsang, G.B.; Elad, S.; Lawitschka, A.; Socie, G.; Pavletic, S.Z.; et al. Consensus conference on clinical practice in chronic graft-versus-host disease (GVHD): First-line and topical treatment of chronic GVHD. Biol. Blood Marrow Transplant. 2010, 16, 1611–1628. [Google Scholar] [CrossRef]
- Sabry, W.; Le Blanc, R.; Labbe, A.C.; Sauvageau, G.; Couban, S.; Kiss, T.; Busque, L.; Cohen, S.; Lachance, S.; Roy, D.C.; et al. Graft-versus-host disease prophylaxis with tacrolimus and mycophenolate mofetil in HLA-matched nonmyeloablative transplant recipients is associated with very low incidence of GVHD and nonrelapse mortality. Biol. Blood Marrow Transplant. 2009, 15, 919–929. [Google Scholar] [CrossRef]
- Scheinberg, P.; Nunez, O.; Wu, C.; Young, N.S. Treatment of severe aplastic anaemia with combined immunosuppression: Anti-thymocyte globulin, ciclosporin and mycophenolate mofetil. Br. J. Haematol. 2006, 133, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Zachou, K.; Gatselis, N.K.; Arvaniti, P.; Gabeta, S.; Rigopoulou, E.I.; Koukoulis, G.K.; Dalekos, G.N. A real-world study focused on the long-term efficacy of mycophenolate mofetil as first-line treatment of autoimmune hepatitis. Aliment. Pharmacol. Ther. 2016, 43, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Czaja, A.J.; Gorham, J.D.; Krawitt, E.L.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M. Diagnosis and management of autoimmune hepatitis. Hepatology 2010, 51, 2193–2213. [Google Scholar] [CrossRef] [PubMed]
- Contreras, G.; Pardo, V.; Leclercq, B.; Lenz, O.; Tozman, E.; O’Nan, P.; Roth, D. Sequential therapies for proliferative lupus nephritis. N. Engl. J. Med. 2004, 350, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.M.; Hooi, L.S.; Lim, T.O.; Goh, B.L.; Ahmad, G.; Ghazalli, R.; Teo, S.M.; Wong, H.S.; Tan, S.Y.; Shaariah, W.; et al. Randomized controlled trial of pulse intravenous cyclophosphamide versus mycophenolate mofetil in the induction therapy of proliferative lupus nephritis. Nephrology 2005, 10, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.A.; Jayne, D.; Ginzler, E.M.; Isenberg, D.; Olsen, N.J.; Wofsy, D.; Eitner, F.; Appel, G.B.; Contreras, G.; Lisk, L.; et al. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N. Engl. J. Med. 2011, 365, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; McMahon, M.A.; Wilkinson, A.; Wallace, W.D.; Daikh, D.I.; Fitzgerald, J.D.; Karpouzas, G.A.; Merrill, J.T.; Wallace, D.J.; Yazdany, J.; et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res. 2012, 64, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Meriggioli, M.N.; Ciafaloni, E.; Al-Hayk, K.A.; Rowin, J.; Tucker-Lipscomb, B.; Massey, J.M.; Sanders, D.B. Mycophenolate mofetil for myasthenia gravis: An analysis of efficacy, safety, and tolerability. Neurology 2003, 61, 1438–1440. [Google Scholar] [CrossRef]
- Sanders, D.B.; Hart, I.K.; Mantegazza, R.; Shukla, S.S.; Siddiqi, Z.A.; De Baets, M.H.V.; Melms, A.; Nicolle, M.W.; Solomons, N.; Richman, D.P. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008, 71, 400–406. [Google Scholar] [CrossRef]
- Sieb, J.P. Myasthenia gravis: An update for the clinician. Clin. Exp. Immunol. 2014, 175, 408–418. [Google Scholar] [CrossRef]
- Menter, A.; Korman, N.J.; Elmets, C.A.; Feldman, S.R.; Gelfand, J.M.; Gordon, K.B.; Gottlieb, A.B.; Koo, J.Y.; Lebwohl, M.; Lim, H.W.; et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 4. Guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J. Am. Acad. Dermatol. 2009, 61, 451–485. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, A.J.; Goss, C.H.; Molitor, J.A. Effect of mycophenolate mofetil on pulmonary function in scleroderma-associated interstitial lung disease. Chest 2008, 133, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Derk, C.T.; Grace, E.; Shenin, M.; Naik, M.; Schulz, S.; Xiong, W. A prospective open-label study of mycophenolate mofetil for the treatment of diffuse systemic sclerosis. Rheumatology 2009, 48, 1595–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, E.N.; Wigley, F.M.; Shah, A.A.; Boin, F.; Hummers, L.K. Long-term experience of mycophenolate mofetil for treatment of diffuse cutaneous systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.A.; Nagle, S.J.; Lee, J.B.; Jimenez, S.A. A prospective observational study of mycophenolate mofetil treatment in progressive diffuse cutaneous systemic sclerosis of recent onset. J. Rheumatol. 2012, 39, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Tashkin, D.P.; Roth, M.D.; Clements, P.J.; Furst, D.E.; Khanna, D.; Kleerup, E.C.; Goldin, J.; Arriola, E.; Volkmann, E.R.; Kafaja, S.; et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): A randomised controlled, double-blind, parallel group trial. Lancet Respir. Med. 2016, 4, 708–719. [Google Scholar] [CrossRef]
- Herrick, A.L.; Pan, X.; Peytrignet, S.; Lunt, M.; Hesselstrand, R.; Mouthon, L.; Silman, A.; Brown, E.; Czirjak, L.; Distler, J.H.W.; et al. Treatment outcome in early diffuse cutaneous systemic sclerosis: The European Scleroderma Observational Study (ESOS). Ann. Rheum. Dis. 2017, 76, 1207–1218. [Google Scholar] [CrossRef]
- Williams, R.H.; Lively, D.H.; DeLong, D.C.; Cline, J.C.; Sweeny, M.J. Mycophenolic acid: Antiviral and antitumor properties. J. Antibiot. 1968, 21, 463–464. [Google Scholar] [CrossRef]
- Florey, H.W.; Jennings, M.A.; Gilliver, K.; Sanders, A.G. Mycophenolic Acid an Antibiotic from Penicillium Brevicompactum Dierckx. Lancet 1946, 247, 46–49. [Google Scholar] [CrossRef]
- Jones, E.L.; Epinette, W.W.; Hackney, V.C.; Menendez, L.; Frost, P. Treatment of psoriasis with oral mycophenolic acid. J. Investig. Dermatol. 1975, 65, 537–542. [Google Scholar] [CrossRef]
- Carter, S.B.; Franklin, T.J.; Jones, D.F.; Leonard, B.J.; Mills, S.D.; Turner, R.W.; Turner, W.B. Mycophenolic acid: An anti-cancer compound with unusual properties. Nature 1969, 223, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.J.; Hoffman, D.H.; Esterman, M.A. Metabolism and biochemistry of mycophenolic acid. Cancer Res. 1972, 32, 1803–1809. [Google Scholar] [PubMed]
- Sweeney, M.J.; Gerzon, K.; Harris, P.N.; Holmes, R.E.; Poore, G.A.; Williams, R.H. Experimental antitumor activity and preclinical toxicology of mycophenolic acid. Cancer Res. 1972, 32, 1795–1802. [Google Scholar] [PubMed]
- Bacus, S.S.; Kiguchi, K.; Chin, D.; King, C.R.; Huberman, E. Differentiation of cultured human breast cancer cells (AU-565 and MCF-7) associated with loss of cell surface HER-2/neu antigen. Mol. Carcinog. 1990, 3, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Floryk, D.; Huberman, E. Mycophenolic acid-induced replication arrest, differentiation markers and cell death of androgen-independent prostate cancer cells DU145. Cancer Lett. 2006, 231, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Kiguchi, K.; Collart, F.R.; Henning-Chubb, C.; Huberman, E. Induction of cell differentiation in melanoma cells by inhibitors of IMP dehydrogenase: Altered patterns of IMP dehydrogenase expression and activity. Cell Growth Differ. 1990, 1, 259–270. [Google Scholar] [PubMed]
- Collart, F.R.; Huberman, E. Expression of IMP dehydrogenase in differentiating HL-60 cells. Blood 1990, 75, 570–576. [Google Scholar] [Green Version]
- Messina, E.; Micheli, V.; Giacomello, A. Guanine nucleotide depletion induces differentiation and aberrant neurite outgrowth in human dopaminergic neuroblastoma lines: A model for basal ganglia dysfunction in Lesch-Nyhan disease. Neurosci. Lett. 2005, 375, 97–100. [Google Scholar] [CrossRef]
- Takebe, N.; Cheng, X.; Bauer, K.; Tricot, G.; Karp, J.; Ross, D. Induction of apoptosis in multiple myeloma (MM) cell lines using mycophenolate mofetil (Cellcept). Blood 2001, 98, 312B. [Google Scholar]
- Takebe, N.; Cheng, X.; Wu, S.; Bauer, K.; Goloubeva, O.G.; Fenton, R.G.; Heyman, M.; Rapoport, A.P.; Badros, A.; Shaughnessy, J.; et al. Phase I clinical trial of the inosine monophosphate dehydrogenase inhibitor mycophenolate mofetil (cellcept) in advanced multiple myeloma patients. Clin. Cancer Res. 2004, 10, 8301–8308. [Google Scholar] [CrossRef]
- Rodriguez-Pascual, J.; Sha, P.; Garcia-Garcia, E.; Rajeshkumar, N.V.; De Vicente, E.; Quijano, Y.; Cubillo, A.; Angulo, B.; Hernando, O.; Hidalgo, M. A preclinical and clinical study of mycophenolate mofetil in pancreatic cancer. Investig. New Drugs 2013, 31, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benlloch, S.; Berenguer, M.; Prieto, M.; Moreno, R.; San Juan, F.; Rayon, M.; Mir, J.; Segura, A.; Berenguer, J. De novo internal neoplasms after liver transplantation: Increased risk and aggressive behavior in recent years? Am. J. Transplant. 2004, 4, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Penn, I. Occurrence of cancers in immunosuppressed organ transplant recipients. Clin. Transplant. 1998, 12, 147–158. [Google Scholar]
- Zafar, S.Y.; Howell, D.N.; Gockerman, J.P. Malignancy after solid organ transplantation: An overview. Oncologist 2008, 13, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Cherikh, W.S.; Kauffman, H.M.; McBride, M.A.; Maghirang, J.; Swinnen, L.J.; Hanto, D.W. Association of the type of induction immunosuppression with posttransplant lymphoproliferative disorder, graft survival, and patient survival after primary kidney transplantation. Transplantation 2003, 76, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Robson, R.; Cecka, J.M.; Opelz, G.; Budde, M.; Sacks, S. Prospective Registry-Based Observational Cohort Study of the Long-Term Risk of Malignancies in Renal Transplant Patients Treated with Mycophenolate Mofetil. Am. J. Transplant. 2005, 5, 2954–2960. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.O.; Edwards, L.B.; Taylor, D.O. Mycophenolate mofetil and risk of developing malignancy after orthotopic heart transplantation: Analysis of the transplant registry of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2006, 25, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- David, K.M.; Morris, J.A.; Steffen, B.J.; Chi-Burris, K.S.; Gotz, V.P.; Gordon, R.D. Mycophenolate mofetil vs. azathioprine is associated with decreased acute rejection, late acute rejection, and risk for cardiovascular death in renal transplant recipients with pre-transplant diabetes. Clin. Transplant. 2005, 19, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Mechanisms of action of mycophenolate mofetil. Lupus 2005, 14 (Suppl. 1), 2–8. [Google Scholar] [CrossRef]
- Behrend, M. Adverse gastrointestinal effects of mycophenolate mofetil: Aetiology, incidence and management. Drug Saf. 2001, 24, 645–663. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.J.; Jacobs, V.N.; Jones, G.; Ple, P. Human colorectal carcinoma cells in vitro as a means to assess the metabolism of analogs of mycophenolic acid. Drug Metab. Dispos. 1997, 25, 367–370. [Google Scholar] [PubMed]
- Lesiak, K.; Watanabe, K.A.; Majumdar, A.; Powell, J.; Seidman, M.; Vanderveen, K.; Goldstein, B.M.; Pankiewicz, K.W. Synthesis of a methylenebis (phosphonate) analogue of mycophenolic adenine dinucleotide: A glucuronidation-resistant MAD analogue of NAD. J. Med. Chem. 1998, 41, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Rejman, D.; Olesiak, M.; Chen, L.; Patterson, S.E.; Wilson, D.; Jayaram, H.N.; Hedstrom, L.; Pankiewicz, K.W. Novel methylenephosphophosphonate analogues of mycophenolic adenine dinucleotide. Inhibition of inosine monophosphate dehydrogenase. J. Med. Chem. 2006, 49, 5018–5022. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Petrelli, R.; Olesiak, M.; Wilson, D.J.; Labello, N.P.; Pankiewicz, K.W. Bis(sulfonamide) isosters of mycophenolic adenine dinucleotide analogues: Inhibition of inosine monophosphate dehydrogenase. Bioorg. Med. Chem. 2008, 16, 7462–7469. [Google Scholar] [CrossRef]
- Han, D.; Sasaki, M.; Yoshino, H.; Kofuji, S.; Sasaki, A.T.; Steckl, A.J. In-vitro evaluation of MPA-loaded electrospun coaxial fiber membranes for local treatment of glioblastoma tumor cells. J. Drug Deliv. Sci. Technol. 2017, 40, 45–50. [Google Scholar] [CrossRef]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef]
- Burger, P.C.; Dubois, P.J.; Schold, S.C., Jr.; Smith, K.R., Jr.; Odom, G.L.; Crafts, D.C.; Giangaspero, F. Computerized tomographic and pathologic studies of the untreated, quiescent, and recurrent glioblastoma multiforme. J. Neurosurg. 1983, 58, 159–169. [Google Scholar] [CrossRef]
- Gaspar, L.E.; Fisher, B.J.; Macdonald, D.R.; LeBer, D.V.; Halperin, E.C.; Schold, S.C., Jr.; Cairncross, J.G. Supratentorial malignant glioma: Patterns of recurrence and implications for external beam local treatment. Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 55–57. [Google Scholar] [CrossRef]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Cooney, D.A.; Grusch, M.; Krupitza, G. Consequences of IMP dehydrogenase inhibition, and its relationship to cancer and apoptosis. Curr. Med. Chem. 1999, 6, 561–574. [Google Scholar] [PubMed]
- Vitale, M.; Zamai, L.; Falcieri, E.; Zauli, G.; Gobbi, P.; Santi, S.; Cinti, C.; Weber, G. IMP dehydrogenase inhibitor, tiazofurin, induces apoptosis in K562 human erythroleukemia cells. Cytometry 1997, 30, 61–66. [Google Scholar] [CrossRef]
- Grifantini, M. Tiazofurine ICN Pharmaceuticals. Curr. Opin. Investig. Drugs 2000, 1, 257–262. [Google Scholar] [PubMed]
- Srivastava, P.C.; Pickering, M.V.; Allen, L.B.; Streeter, D.G.; Campbell, M.T.; Witkowski, J.T.; Sidwell, R.W.; Robins, R.K. Synthesis and antiviral activity of certain thiazole C-nucleosides. J. Med. Chem. 1977, 20, 256–262. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, P.J.; Shoemaker, D.D.; Jayaram, H.N.; Johns, D.G.; Cooney, D.A.; Marsoni, S.; Malspeis, L.; Plowman, J.; Davignon, J.P.; Davis, R.D. Tiazofurin: A new antitumor agent. Investig. New Drugs 1984, 2, 79–84. [Google Scholar] [CrossRef]
- Grem, J.L.; Rubinstein, L.; King, S.A.; Cheson, B.D.; Hawkins, M.J.; Shoemaker, D.D. Clinical toxicity associated with tiazofurin. Investig. New Drugs 1990, 8, 227–238. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Dion, R.L.; Glazer, R.I.; Johns, D.G.; Robins, R.K.; Srivastava, P.C.; Cooney, D.A. Initial studies on the mechanism of action of a new oncolytic thiazole nucleoside, 2-β-d-ribofuranosylthiazole-4-carboxamide NSC 286193. Biochem. Pharmacol. 1982, 31, 2371–2380. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Smith, A.L.; Glazer, R.I.; Johns, D.G.; Cooney, D.A. Studies on the mechanism of action of 2-beta-d-ribofuranosylthiazole-4-carboxamide (NSC 286193)-II. Relationship between dose level and biochemical effects in P388 leukemia in vivo. Biochem. Pharmacol. 1982, 31, 3839–3845. [Google Scholar] [CrossRef]
- Cooney, D.A.; Jayaram, H.N.; Gebeyehu, G.; Betts, C.R.; Kelley, J.A.; Marquez, V.E.; Johns, D.G. The conversion of 2-beta-d-ribofuranosylthiazole-4-carboxamide to an analogue of NAD with potent IMP dehydrogenase-inhibitory properties. Biochem. Pharmacol. 1982, 31, 2133–2136. [Google Scholar] [CrossRef]
- Cooney, D.A.; Jayaram, H.N.; Glazer, R.I.; Kelley, J.A.; Marquez, V.E.; Gebeyehu, G.; Van Cott, A.C.; Zwelling, L.A.; Johns, D.G. Studies on the mechanism of action of tiazofurin metabolism to an analog of NAD with potent IMP dehydrogenase-inhibitory activity. Adv. Enzym. Regul. 1983, 21, 271–303. [Google Scholar] [CrossRef]
- Jayaram, H.N. Biochemical mechanisms of resistance to tiazofurin. Adv. Enzym. Regul. 1985, 24, 67–89. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Zhen, W.; Gharehbaghi, K. Biochemical consequences of resistance to tiazofurin in human myelogenous leukemic K562 cells. Cancer Res. 1993, 53, 2344–2348. [Google Scholar]
- Gharehbaghi, K.; Sreenath, A.; Hao, Z.; Paull, K.D.; Szekeres, T.; Cooney, D.A.; Krohn, K.; Jayaram, H.N. Comparison of biochemical parameters of benzamide riboside, a new inhibitor of IMP dehydrogenase, with tiazofurin and selenazofurin. Biochem. Pharmacol. 1994, 48, 1413–1419. [Google Scholar] [CrossRef]
- Smejkal, R.M.; Page, T.T.; Boyd, V.L.; Nyhan, W.L.; Jacobsen, S.J.; Mangum, J.H.; Robins, R.K. Novel nucleoside inhibitors of guanosine metabolism as antitumor agents. Adv. Enzym. Regul. 1984, 22, 59–68. [Google Scholar] [CrossRef]
- Look, K.Y.; Natsumeda, Y.; Eble, J.N.; Stehman, F.B.; Clarence, E.E.; Olah, E.; Prajda, N.; Bosze, P.; Eckhardt, S.; Weber, G. Inhibition by tiazofurin of inosine 5′-phosphate dehydrogenase (IMP DH) activity in extracts of ovarian carcinomas. Gynecol. Oncol. 1992, 47, 66–70. [Google Scholar] [CrossRef]
- Zhen, W.; Jayaram, H.N.; Weber, G. Antitumor activity of tiazofurin in human colon carcinoma HT-29. Cancer Investig. 1992, 10, 505–511. [Google Scholar] [CrossRef]
- Tóvári, J.; Bocsi, J.; Ladányi, A.; Lapis, K.; Timár, J. The antitumor effect of Tiazofurin (TR) consists of anti-proliferative and anti-invasive elements. Anticancer Res. 1996, 16, 3307–3312. [Google Scholar]
- Sidi, Y.; Beery, E.; Panet, C.; Wasserman, L.; Novogrodsky, A.; Nordenberg, J. Growth inhibition and induction of phenotypic alterations by tiazofurin: Differential effects on MCF-7 breast cancer and HBL-100 breast cell lines. Eur. J. Cancer Clin. Oncol. 1989, 25, 883–885. [Google Scholar] [CrossRef]
- Sokoloski, J.A.; Blair, O.C.; Sartorelli, A.C. Alterations in glycoprotein synthesis and guanosine triphosphate levels associated with the differentiation of HL-60 leukemia cells produced by inhibitors of inosine 5′-phosphate dehydrogenase. Cancer Res. 1986, 46, 2314–2319. [Google Scholar]
- Olah, E.; Natsumeda, Y.; Ikegami, T.; Kote, Z.; Horanyi, M.; Szelenyi, J.; Paulik, E.; Kremmer, T.; Hollan, S.R.; Sugar, J.; et al. Induction of erythroid differentiation and modulation of gene expression by tiazofurin in K-562 leukemia cells. Proc. Natl. Acad. Sci. USA 1988, 85, 6533–6537. [Google Scholar] [CrossRef]
- Robins, R.K.; Srivastava, P.C.; Narayanan, V.L.; Plowman, J.; Paull, K.D. 2-beta-d-Ribofuranosylthiazole-4-carboxamide, a novel potential antitumor agent for lung tumors and metastases. J. Med. Chem. 1982, 25, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, G.S.; Jayaram, H.N.; Plowman, J.P.; Cooney, D.A.; Johns, D.G. Studies on the mechanism of action of 2-beta-d-ribofuranosylthiazole-4-carboxamide-V. Factors governing the response of murine tumors to tiazofurin. Biochem. Pharmacol. 1984, 33, 1195–1203. [Google Scholar] [CrossRef]
- Lui, M.S.; Faderan, M.A.; Liepnieks, J.J.; Natsumeda, Y.; Olah, E.; Jayaram, H.N.; Weber, G. Modulation of IMP dehydrogenase activity and guanylate metabolism by tiazofurin (2-beta-d-ribofuranosylthiazole-4-carboxamide). J. Biol. Chem. 1984, 259, 5078–5082. [Google Scholar] [PubMed]
- Yamada, Y.; Natsumeda, Y.; Yamaji, Y.; Jayaram, H.N.; Tricot, G.J.; Hoffman, R.; Weber, G. IMP dehydrogenase: Inhibition by the anti-leukemic drug, tiazofurin. Leuk. Res. 1989, 13, 179–184. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Pillwein, K.; Nichols, C.R.; Hoffman, R.; Weber, G. Selective sensitivity to tiazofurin of human leukemic cells. Biochem. Pharmacol. 1986, 35, 2029–2032. [Google Scholar] [CrossRef]
- Tricot, G.J.; Jayaram, H.N.; Lapis, E.; Natsumeda, Y.; Nichols, C.R.; Kneebone, P.; Heerema, N.; Weber, G.; Hoffman, R. Biochemically directed therapy of leukemia with tiazofurin, a selective blocker of inosine 5′-phosphate dehydrogenase activity. Cancer Res. 1989, 49, 3696–3701. [Google Scholar]
- Tricot, G.J.; Jayaram, H.N.; Nichols, C.R.; Pennington, K.; Lapis, E.; Weber, G.; Hoffman, R. Hematological and biochemical action of tiazofurin (NSC 286193) in a case of refractory acute myeloid leukemia. Cancer Res. 1987, 47, 4988–4991. [Google Scholar]
- Tricot, G.; Weber, G. Biochemically targeted therapy of refractory leukemia and myeloid blast crisis of chronic granulocytic leukemia with Tiazofurin, a selective blocker of inosine 5′-phosphate dehydrogenase activity. Anticancer Res. 1996, 16, 3341–3347. [Google Scholar]
- Malek, K.; Boosalis, M.S.; Waraska, K.; Mitchell, B.S.; Wright, D.G. Effects of the IMP-dehydrogenase inhibitor, Tiazofurin, in bcr-abl positive acute myelogenous leukemia. Part I. In vivo studies. Leuk. Res. 2004, 28, 1125–1136. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Lapis, E.; Tricot, G.; Kneebone, P.; Paulik, E.; Zhen, W.; Engeler, G.P.; Hoffman, R.; Weber, G. Clinical pharmacokinetic study of tiazofurin administered as a 1-hour infusion. Int. J. Cancer 1992, 51, 182–188. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Klecker, R.W., Jr.; Jayaram, H.N.; Jenkins, J.F.; Grygiel, J.; Ihde, D.C.; Eddy, J.L.; Fine, R.L.; Kerr, I.G.; Collins, J.M. Phase I and pharmacokinetic study of tiazofurin (TCAR, NSC 286193) administered by continuous infusion. Investig. New Drugs 1985, 3, 349–355. [Google Scholar] [CrossRef]
- Melink, T.J.; Von Hoff, D.; Kuhn, J.G.; Hersh, M.R.; Sternson, L.A.; Patton, T.F.; Siegler, R.; Boldt, D.H.; Clark, G. Phase I Evaluation and Pharmacokinetics of Tiazofurin (2-β-d-Ribofuranosylthiazole-4-carboxamide, NSC 286193). Cancer Res. 1985, 45, 2859–2865. [Google Scholar] [PubMed]
- Raghavan, D.; Bishop, J.; Sampson, D.; Grygiel, J.; Woods, R.; Coates, A.; Fox, R. Phase I and pharmacokinetic study of tiazofurin (NSC 286193) administered by 5-day continuous infusion. Cancer Chemother. Pharmacol. 1986, 16, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Trump, D.L.; Tutsch, K.D.; Koeller, J.M.; Tormey, D.C. Phase I Clinical Study with Pharmacokinetic Analysis of 2-β-d-Ribofuranosylthiazole-4-carboxamide (NSC 286193) Administered as a Five-Day Infusion. Cancer Res. 1985, 45, 2853–2858. [Google Scholar] [PubMed]
- Roberts, J.D.; Stewart, J.A.; McCormack, J.J.; Krakoff, I.R.; Culham, C.A.; Hartshorn, J.N.; Newman, R.A.; Haugh, L.D.; Young, J.A. Phase I trial of tiazofurin administered by i.v. bolus daily for 5 days, with pharmacokinetic evaluation. Cancer Treat. Rep. 1987, 71, 141–149. [Google Scholar] [PubMed]
- Maroun, J.A.; Stewart, D.J. Phase I study of tiazofurin (2-β-d-ribofuranosylthiazole-4-carboxamide, NSC 286193). Investig. New Drugs 1990, 8, S39. [Google Scholar] [CrossRef]
- Stewart, D.J.; Eisenhauer, E.; Macdonald, D.R.; Cairncross, J.G.; Langleben, A. Phase II study of tiazofurin in gliomas in adults. A National Cancer Institute of Canada study. J. Neurooncol. 1993, 15, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Holoye, P.Y.; Carr, D.T.; Dhingra, H.M.; Glisson, B.S.; Lee, J.S.; Murphy, W.K.; Umsawasdi, T.; Jeffries, D. Phase II study of tiazofurin (NSC 286193) in the treatment of advanced small cell bronchogenic carcinoma. Investig. New Drugs 1988, 6, 217–218. [Google Scholar] [CrossRef]
- Maroun, J.A.; Eisenhauer, E.; Cripps, C.; Maksymiuk, A. Phase II study of tiazofurin in colorectal carcinoma: A National Cancer Institute of Canada Study. Cancer Treat. Rep. 1987, 71, 1297–1298. [Google Scholar]
- Dimery, I.W.; Neidhart, J.A.; McCarthy, K.; Krakoff, I.H.; Hong, W.K. Phase II trial of tiazofurin in recurrent squamous cell carcinoma of the head and neck. Cancer Treat. Rep. 1987, 71, 425–426. [Google Scholar] [PubMed]
- Cuny, G.D.; Suebsuwong, C.; Ray, S.S. Inosine-5′-monophosphate dehydrogenase (IMPDH) inhibitors: A patent and scientific literature review (2002–2016). Expert Opin. Ther. Pat. 2017, 27, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Almquist, S.J.; Shlyakhter, D.; Harding, M.W. VX-497: A novel, selective IMPDH inhibitor and immunosuppressive agent. J. Pharm. Sci. 2001, 90, 625–637. [Google Scholar] [CrossRef]
- Markland, W.; McQuaid, T.J.; Jain, J.; Kwong, A.D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: A comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 2000, 44, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Smith, J.; Bukreyeva, N.; Koma, T.; Manning, J.T.; Kalkeri, R.; Kwong, A.D.; Paessler, S. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antivir. Res. 2018, 149, 34–40. [Google Scholar] [CrossRef]
- Tossing, G. Merimepodib (Vertex). IDrugs 2003, 6, 372–376. [Google Scholar] [PubMed]
- McHutchison, J.G.; Shiffman, M.L.; Cheung, R.C.; Gordon, S.C.; Wright, T.L.; Pottage, J.C., Jr.; McNair, L.; Ette, E.; Moseley, S.; Alam, J. A randomized, double-blind, placebo-controlled dose-escalation trial of merimepodib (VX-497) and interferon-alpha in previously untreated patients with chronic hepatitis C. Antivir. Ther. 2005, 10, 635–643. [Google Scholar]
- Jain, J.; Ma, J.G.; Furey, B.; Recher, C.; Demur, C.; Poondru, S.; Zhang, J.B.; Li, S.L.; Firestone, B.; Olson, K.; et al. The IMPDH inhibitor VX-944 demonstrates in vivo efficacy in an aggressive leukemia model. Blood 2004, 104, 693a–694a. [Google Scholar]
- Ishitsuka, K.; Hideshima, T.; Hamasaki, M.; Raje, N.; Kumar, S.; Podar, K.; Gouill, S.L.; Shiraishi, N.; Yasui, H.; Roccaro, A.M.; et al. Novel inosine monophosphate dehydrogenase inhibitor VX-944 induces apoptosis in multiple myeloma cells primarily via caspase-independent AIF/Endo G pathway. Oncogene 2005, 24, 5888–5896. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Harding, M.; Genna, T.; Bol, D. A Phase I Dose-Ranging Study of the Pharmacokinetics, Pharmacodynamics, Safety, and Tolerability of AVN944, an IMPDH Inhibitor, in Healthy Male Volunteers. J. Clin. Pharmacol. 2009, 49, 30–38. [Google Scholar] [CrossRef]
- Jain, J.; Almquist, S.; Hoover, R.; Shlyakhter, D.; Ford, P.; Markland, W.; Dauffenbach, L.; Kerfoot, C.; Mosher, R. 488 VX-944: An inosine monophosphate dehydrogenase inhibitor with unique anti-cancer activity. Eur. J. Cancer Suppl. 2004, 2, 149. [Google Scholar] [CrossRef]
- Cappellacci, L.; Petrelli, R. Novel Inhibitors of Inosine Monophosphate Dehydrogenase in Patent Literature of the Last Decade. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 103–125. [Google Scholar]
- Klisovic, R.B.; Tricot, G.; Coutre, S.; Kovacsovics, T.; Giles, F.; Genna, T.; Bol, D.K.; Strovel, J.W.; Hamilton, J.M.; Mitchell, B. A phase I trial of AVN944 in patients with advanced hematologic malignancies. J. Clin. Oncol. 2007, 25, 14026. [Google Scholar]
- Yang, H.; Fang, Z.; Wei, Y.; Bohannan, Z.S.; Ganan-Gomez, I.; Pierola, A.A.; Paradiso, L.J.; Iwamura, H.; Garcia-Manero, G. Preclinical activity of FF-10501-01, a novel inosine-5′-monophosphate dehydrogenase inhibitor, in acute myeloid leukemia. Leuk. Res. 2017, 59, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Kiyohara, T.; Fukui, M.; Atsumi, T.; Ogino, S.; Inaba, M.; Tsukagoshi, S.; Sakurai, Y. Antitumor activities of newly synthesized 5-carbamoyl-1 H-imidazol-4yl 1-adamantanecarboxylate and 5-carbamoyl-1H-imidazol-4yl piperonylate. Cancer Res. 1980, 40, 3810–3814. [Google Scholar] [PubMed]
- Kimura, K.; Suzuoki, Y.; Ogawa, M.; Miyazaki, T.; Uzuka, Y.; Sakai, Y.; Ohno, R.; Yamada, K.; Ota, K.; Yoshida, T. Phase I study and early phase II of SM-108 (4-carbamoylimidazolium-5-olate) in lung cancer. SM-108 Study Group. Gan Kagaku Ryoho. Cancer Chemother. 1989, 16, 113–121. [Google Scholar]
- Kimura, K.; Yamada, K.; Uzuka, Y.; Masaoka, T.; Hirano, M.; Ohno, R.; Ogawa, M. Phase II study of SM-108 (4-carbamoylimidazolium-5-olate) in hematological malignancies. Gan Kagaku Ryoho. Cancer Chemother. 1989, 16, 123–130. [Google Scholar]
- Uzuka, Y.; Saito, Y. Treatment of myelodysplastic syndrome using SM-108 in relation to stem cell kinetics. Gan Kagaku Ryoho. Cancer Chemother. 1988, 15, 1215–1222. [Google Scholar]
- Nishikawa, M.; Morita, K.; Sekine, T.; Takeda, M.; Tamaki, S.; Katayama, N.; Kobayashi, T.; Mukai, K.; Tanaka, H.; Minami, N. [Clinical trial of SM-108 in myeloproliferative disorders]. Gan Kagaku Ryoho. Cancer Chemother. 1987, 14, 3078–3082. [Google Scholar]
- Murase, M.; Iwamura, H.; Komatsu, K.; Saito, M.; Maekawa, T.; Nakamura, T.; Yokokawa, T.; Shimada, Y. Lack of cross-resistance to FF-10501, an inhibitor of inosine-5′-monophosphate dehydrogenase, in azacitidine-resistant cell lines selected from SKM-1 and MOLM-13 leukemia cell lines. Pharmacol. Res. Perspect. 2016, 4, e00206. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Kurman, M.R.; DiNardo, C.D.; Pemmaraju, N.; Alvarado, Y.; Naqvi, K.; Ravandi, F.; Jabbour, E.J.; Delumpa, R.; Madden, T.; et al. Results of a Phase 1, Dose-Escalation Study of FF-10501-01 in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML) or Hypomethylating Agent (HMA)-Resistant Myelodysplastic Syndrome (MDS). Blood 2018, 132, 1438. [Google Scholar]
- Garcia-Manero, G.; Yang, H.; Fang, Z.; Kantarjian, H.M.; DiNardo, C.D.; Jabbour, E.J.; Pemmaraju, N.; Daver, N.; Delumpa, R.; Loiselle, C.; et al. Phase 1 Results of FF-10501-01, a Novel Inosine 5′-Monophosphate Dehydrogenase Inhibitor, in Advanced Acute Myeloid Leukemia (AML) and Myelodysplastic Syndromes (MDS), Including Hypomethylating Agent (HMA) Failures. Blood 2016, 128, 1640. [Google Scholar]
- A Study of FF-10501-01 in Combination with Azacitidine in Patients with Myelodysplastic Syndrome. Available online: https://ClinicalTrials.gov/show/NCT03486353 (accessed on 22 July 2019).
- Jayaram, H.N.; Gharehbaghi, K.; Jayaram, N.H.; Rieser, J.; Krohn, K.; Paull, K.D. Cytotoxicity of a new IMP dehydrogenase inhibitor, benzamide riboside, to human myelogenous leukemia K562 cells. Biochem. Biophys. Res. Commun. 1992, 186, 1600–1606. [Google Scholar] [CrossRef]
- Gharehbaghi, K.; Paull, K.D.; Kelley, J.A.; Barchi, J.J., Jr.; Marquez, V.E.; Cooney, D.A.; Monks, A.; Scudiero, D.; Krohn, K.; Jayaram, H.N. Cytotoxicity and characterization of an active metabolite of benzamide riboside, a novel inhibitor of IMP dehydrogenase. Int. J. Cancer 1994, 56, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Gharehbaghi, K.; Grunberger, W.; Jayaram, H.N. Studies on the mechanism of action of benzamide riboside: A novel inhibitor of IMP dehydrogenase. Curr. Med. Chem. 2002, 9, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Roussel, B.; Johnson-Farley, N.; Kerrigan, J.E.; Scotto, K.W.; Banerjee, D.; Felczak, K.; Pankiewicz, K.W.; Gounder, M.; Lin, H.; Abali, E.E.; et al. A second target of benzamide riboside: Dihydrofolate reductase. Cancer Biol. Ther. 2012, 13, 1290–1298. [Google Scholar] [CrossRef]
- Grusch, M.; Rosenberger, G.; Fuhrmann, G.; Braun, K.; Titscher, B.; Szekeres, T.; Fritzer-Skekeres, M.; Oberhuber, G.; Krohn, K.; Hengstschlaeger, M.; et al. Benzamide riboside induces apoptosis independent of Cdc25A expression in human ovarian carcinoma N.1 cells. Cell Death Differ. 1999, 6, 736–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunakova, L.; Bies, J.; Sedlak, J.; Duraj, J.; Jakubikova, J.; Takacsova, X.; Novotny, L.; Chorvath, B. Differential sensitivity of ovarian carcinoma cell lines to apoptosis induced by the IMPDH inhibitor benzamide riboside. Neoplasma 2000, 47, 274–279. [Google Scholar]
- Rauko, P.; Novotny, L.; Dovinova, I.; Hunakova, L.; Szekeres, T.; Jayaram, H.N. Antitumor activity of benzamide riboside and its combination with cisplatin and staurosporine. Eur. J. Pharm. Sci. 2001, 12, 387–394. [Google Scholar] [CrossRef]
- Jayaram, H.N.; Yalowitz, J.A.; Arguello, F.; Greene, J.F., Jr. Toxicity and efficacy of benzamide riboside in cancer chemotherapy models. Curr. Med. Chem. 2002, 9, 787–792. [Google Scholar] [CrossRef] [PubMed]
- McLennan, G.; Cressman, E.N.; Xu, Y.; Zhang, D.; Jagtap, M.R.; Jayaram, H.N. The effect of benzamide riboside on the VX2 model of liver cancer in rabbits. J. Vasc. Interv. Radiol. 2005, 16, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Tsujino, M.; Yoshizawa, M.; Mizuno, K.; Hayano, K. Action of bredinin on mammalian cells. Cancer Res. 1975, 35, 1643–1648. [Google Scholar] [PubMed]
- Kusumi, T.; Tsuda, M.; Katsunuma, T.; Yamamura, M. Dual inhibitory effect of bredinin. Cell Biochem. Funct. 1989, 7, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Tsuji, M. Genetic and biochemical studies on the activation and cytotoxic mechanism of bredinin, a potent inhibitor of purine biosynthesis in mammalian cells. Biochem. Pharmacol. 1983, 32, 3547–3553. [Google Scholar] [CrossRef]
- Mizuno, K.; Tsujino, M.; Takada, M.; Hayashi, M.; Atsumi, K. Studies on bredinin. I. Isolation, characterization and biological properties. J. Antibiot. 1974, 27, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ihara, H.; Takahara, S.; Takada, K.; Shrestha, G.R.; Ishibashi, M.; Arima, M.; Sagawa, S.; Sonoda, T. The immunosuppressive mode of action of mizoribine. Transplantation 1984, 38, 262–267. [Google Scholar] [CrossRef]
- Kamata, K.; Okubo, M.; Ishigamori, E.; Masaki, Y.; Uchida, H.; Watanabe, K.; Kashiwagi, N. Immunosuppressive effect of bredinin on cell-mediated and humoral immune reactions in experimental animals. Transplantation 1983, 35, 144–149. [Google Scholar] [CrossRef]
- Tzoneva, G.; Dieck, C.L.; Oshima, K.; Ambesi-Impiombato, A.; Sanchez-Martin, M.; Madubata, C.J.; Khiabanian, H.; Yu, J.; Waanders, E.; Iacobucci, I.; et al. Clonal evolution mechanisms in NT5C2 mutant-relapsed acute lymphoblastic leukaemia. Nature 2018, 553, 511–514. [Google Scholar] [CrossRef]
- Kawasaki, Y. Mizoribine: A new approach in the treatment of renal disease. Clin. Dev. Immunol. 2009, 2009, 681482. [Google Scholar] [CrossRef]
- Xia, Z.K.; Gao, Y.F.; Rong, L.P.; Dang, X.Q.; Shen, Q.; Jiang, X.Y.; Yi, Z.W.; Xu, H. Usefulness of mizoribine administration in children with frequently relapsing nephrotic syndrome, and the relationship between pharmacokinetic parameters and efficacy: A multicenter prospective cohort study in China. World J. Pediatr. 2019, 15, 262–269. [Google Scholar] [CrossRef]
- Hashimoto, T.; Kawakami, T.; Koga, H.; Ohyama, B.; Hamada, T.; Dainichi, T.; Nakama, T.; Yasumoto, S.; Tsuruta, D.; Ishii, N. Therapeutic effect of mizoribine on pemphigus vulgaris and pemphigus foliaceus. Dermatol. Ther. 2012, 25, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, K.; Suda, M.; Rokutanda, R.; Kishimoto, M.; Okada, M. Mizoribine is as effective as methotrexate for the treatment of polymyalgia rheumatica: a retrospective case series analysis. Arch. Rheumatol. 2018, 33, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Streeter, D.G.; Witkowski, J.T.; Khare, G.P.; Sidwell, R.W.; Bauer, R.J.; Robins, R.K.; Simon, L.N. Mechanism of Action of 1-d-Ribofuranosyl-1,2,4-Triazole-3-Carboxamide (Virazole), A New Broad-Spectrum Antiviral Agent. Proc. Natl. Acad. Sci. USA 1973, 70, 1174–1178. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 2005, 436, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, Y.; Sano, E.; Okamoto, Y.; Yoshimura, S.; Makita, K.; Yamamuro, S.; Ohta, T.; Ogino, A.; Tadakuma, H.; Ueda, T.; et al. Efficacy of ribavirin against malignant glioma cell lines: Follow-up study. Oncol. Rep. 2018, 39, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L.; Culjkovic-Kraljacic, B. Ribavirin as an anti-cancer therapy: Acute Myeloid Leukemia and beyond? Leuk. Lymphoma 2010, 51, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Assouline, S.; Culjkovic, B.; Cocolakis, E.; Rousseau, C.; Beslu, N.; Amri, A.; Caplan, S.; Leber, B.; Roy, D.C.; Miller, W.H.; et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood 2009, 114, 257–260. [Google Scholar] [CrossRef]
- Urtishak, K.A.; Wang, L.-S.; Culjkovic-Kraljacic, B.; Davenport, J.W.; Porazzi, P.; Vincent, T.L.; Teachey, D.T.; Tasian, S.K.; Moore, J.S.; Seif, A.E.; et al. Targeting EIF4E signaling with ribavirin in infant acute lymphoblastic leukemia. Oncogene 2019, 38, 2241–2262. [Google Scholar] [CrossRef]
- Kai, J.; Wang, Y.; Xiong, F.; Wang, S. Genetic and pharmacological inhibition of eIF4E effectively targets esophageal cancer cells and augments 5-FU’s efficacy. J. Thorac. Dis. 2018, 10, 3983–3991. [Google Scholar] [CrossRef]
- Xi, C.; Wang, L.; Yu, J.; Ye, H.; Cao, L.; Gong, Z. Inhibition of eukaryotic translation initiation factor 4E is effective against chemo-resistance in colon and cervical cancer. Biochem. Biophys. Res. Commun. 2018, 503, 2286–2292. [Google Scholar] [CrossRef]
- Pettersson, F.; Yau, C.; Dobocan, M.C.; Culjkovic-Kraljacic, B.; Retrouvay, H.; Puckett, R.; Flores, L.M.; Krop, I.E.; Rousseau, C.; Cocolakis, E.; et al. Ribavirin Treatment Effects on Breast Cancers. Clin. Cancer Res. 1995, 17, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, T.; Shinojima, T.; Kikuchi, K.; Hagiwara, S.; Kojima, S.-i.; Hongo, H.; Ueda, K.; Miyake, S.; Oya, M. A phase 1/2a trial of docetaxel plus ribavirin for reprogramming efficacy in patients with progressive metastatic castration resistant prostate cancer who have previously received docetaxel alone: DRREEM trial. J. Clin. Oncol. 2018, 36, 329. [Google Scholar] [CrossRef]
- Ribavirin for Patients with Recurrent/Metastatic (R/M) Human Papillomavirus (HPV)-Related Malignancies. Available online: https://clinicaltrials.gov/ct2/show/NCT02308241?cond=ribavirin&age=12&rank=9 (accessed on 18 July 2019).
- A Pilot Investigator-Initiated Study of Ribavirin in Indolent Follicular Lymphoma and Mantle Cell Lymphoma. Available online: https://ClinicalTrials.gov/show/NCT03585725 (accessed on 22 July 2019).
- Peginterferon Plus Ribavirin for Hepatitis C Patients Concomitant with Hepatocellular Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT00834860?cond=ribavirin&draw=3&rank=39 (accessed on 18 July 2019).
- Ribavirin aerosol approved for severe cases of RSV in infants and young children. FDA Drug Bull 1986, 16, 7.
- Davis, G.L.; Esteban-Mur, R.; Rustgi, V.; Hoefs, J.; Gordon, S.C.; Trepo, C.; Shiffman, M.L.; Zeuzem, S.; Craxi, A.; Ling, M.H.; et al. Interferon alfa-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N. Engl. J. Med. 1998, 339, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Karlsson, A.; Wang, L.; Bohman, C.; Horská, K.; Votruba, I.; Fridland, A.; Van Aerschot, A.; Herdewijn, P.; De Clercq, E. Eicar (5-ethynyl-1-beta-d-ribofuranosylimidazole-4-carboxamide). A novel potent inhibitor of inosinate dehydrogenase activity and guanylate biosynthesis. J. Biol. Chem. 1993, 268, 24591–24598. [Google Scholar] [PubMed]
- De Clercq, E.; Cools, M.; Balzarini, J.; Snoeck, R.; Andrei, G.; Hosoya, M.; Shigeta, S.; Ueda, T.; Minakawa, N.; Matsuda, A. Antiviral activities of 5-ethynyl-1-beta-d-ribofuranosylimidazole-4-carboxamide and related compounds. Antimicrob. Agents Chemother. 1991, 35, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, N.; Takeda, T.; Sasaki, T.; Matsuda, A.; Ueda, T. Nucleosides and nucleotides. 96. Synthesis and antitumor activity of 5-ethynyl-1-beta-d-ribofuranosylimidazole-4-carboxamide (EICAR) and its derivatives. J. Med. Chem. 1991, 34, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.C.; Robins, R.K. Synthesis and antitumor activity of 2-beta-d-ribofuranosylselenazole-4-carboxamide and related derivatives. J. Med. Chem. 1983, 26, 445–448. [Google Scholar] [CrossRef]
- Streeter, D.G.; Robins, R.K. Comparative in vitro studies of Tiazofurin and a selenazole analog. Biochem. Biophys. Res. Commun. 1983, 115, 544–550. [Google Scholar] [CrossRef]
- Kirsi, J.J.; North, J.A.; McKernan, P.A.; Murray, B.K.; Canonico, P.G.; Huggins, J.W.; Srivastava, P.C.; Robins, R.K. Broad-spectrum antiviral activity of 2-beta-d-ribofuranosylselenazole-4-carboxamide, a new antiviral agent. Antimicrob. Agents Chemother. 1983, 24, 353–361. [Google Scholar] [CrossRef]
- Kirsi, J.J.; McKernan, P.A.; Burns, N.J., 3rd; North, J.A.; Murray, B.K.; Robins, R.K. Broad-spectrum synergistic antiviral activity of selenazofurin and ribavirin. Antimicrob. Agents Chemother. 1984, 26, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchetti, P.; Cappellacci, L.; Grifantini, M.; Barzi, A.; Nocentini, G.; Yang, H.; O’Connor, A.; Jayaram, H.N.; Carrell, C.; Goldstein, B.M. Furanfurin and thiophenfurin: Two novel tiazofurin analogues. Synthesis, structure, antitumor activity, and interactions with inosine monophosphate dehydrogenase. J. Med. Chem. 1995, 38, 3829–3837. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Bitencourt, S.; Laufer, S.; Ines Goettert, M. Myricetin inhibits panel of kinases implicated in tumorigenesis. Basic Clin. Pharmacol. Toxicol. 2019, 125, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Hua, Q.; Wang, J.; Liu, Z.; Wu, D.; Lu, W.; Huang, J. Myricetin is a novel inhibitor of human inosine 5′-monophosphate dehydrogenase with anti-leukemia activity. Biochem. Biophys. Res. Commun. 2016, 477, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, J.; Yu, X.; Xu, S.; Li, D.; Zheng, Q.; Yin, Y. Myricetin Suppresses the Propagation of Hepatocellular Carcinoma via Down-Regulating Expression of YAP. Cells 2019, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Choi, Y.J.; Park, S.H.; Nam, M.J. Potential role of nucleoside diphosphate kinase in myricetin-induced selective apoptosis in colon cancer HCT-15cells. Food Chem. Toxicol. 2018, 116, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.W.; Chen, Y.Q.; Zhao, L.Q.; Feng, J.G. Myricetin induces apoptosis and enhances chemosensitivity in ovarian cancer cells. Oncol. Lett. 2017, 13, 4974–4978. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhang, C.; Huang, H.; Yang, B.; Xiao, G.; Kong, D.; Tian, Q.; Song, Q.; Song, Y.; Tan, H.; et al. The Natural Compound Myricetin Effectively Represses the Malignant Progression of Prostate Cancer by Inhibiting PIM1 and Disrupting the PIM1/CXCR4 Interaction. Cell. Physiol. Biochem. 2018, 48, 1230–1244. [Google Scholar] [CrossRef]
- Knickle, A.; Fernando, W.; Greenshields, A.L.; Rupasinghe, H.P.V.; Hoskin, D.W. Myricetin-induced apoptosis of triple-negative breast cancer cells is mediated by the iron-dependent generation of reactive oxygen species from hydrogen peroxide. Food Chem. Toxicol. 2018, 118, 154–167. [Google Scholar] [CrossRef]
- Ha, T.K.; Jung, I.; Kim, M.E.; Bae, S.K.; Lee, J.S. Anti-cancer activity of myricetin against human papillary thyroid cancer cells involves mitochondrial dysfunction-mediated apoptosis. Biomed. Pharmacother. 2017, 91, 378–384. [Google Scholar] [CrossRef]
- Yazdanparast, R.; Sadeghi, H. Nucleic acid synthesis in cancerous cells under the effect of gnidilatimonoein from Daphne mucronata. Life Sci. 2004, 74, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.; Yazdanparast, R. Gnidilatimonoein from Daphne mucronata induces differentiation and apoptosis in leukemia cell lines. Arch. Pharm. Res. 2007, 30, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Almquist, S.J.; Heiser, A.D.; Shlyakhter, D.; Leon, E.; Memmott, C.; Moody, C.S.; Nimmesgern, E.; Decker, C. Characterization of pharmacological efficacy of VX-148, a new, potent immunosuppressive inosine 5′-monophosphate dehydrogenase inhibitor. J. Pharmacol. Exp. Ther. 2002, 302, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.-U.; Zaman, K. Topics in Anti-Cancer Research; Bentham Science Publishers: Sharjah, UAE, 2014; Volume 3, p. 46. [Google Scholar]
- SEC Filing. Vertex Pharmaceuticals. Available online: https://investors.vrtx.com/node/14376/html (accessed on 5 September 2019).
- Watterson, S.H.; Chen, P.; Zhao, Y.; Gu, H.H.; Dhar, T.G.; Xiao, Z.; Ballentine, S.K.; Shen, Z.; Fleener, C.A.; Rouleau, K.A.; et al. Acridone-based inhibitors of inosine 5′-monophosphate dehydrogenase: Discovery and SAR leading to the identification of N-(2-(6-(4-ethylpiperazin-1-yl)pyridin-3-yl)propan-2-yl)-2-fluoro-9-oxo-9,10-dihydroacridine-3-carboxamide (BMS-566419). J. Med. Chem. 2007, 50, 3730–3742. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Morokata, T.; Kubo, K.; Umeno, H.; Eikyu, Y.; Kozuki, Y.; Seki, N. Effect of the inosine 5′-monophosphate dehydrogenase inhibitor BMS-566419 on rat cardiac allograft rejection. Int. Immunopharmacol. 2010, 10, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Dhar, T.G.M.; Guo, J.; Shen, Z.; Pitts, W.J.; Gu, H.H.; Chen, B.-C.; Zhao, R.; Bednarz, M.S.; Iwanowicz, E.J. A modified approach to 2-(N-aryl)-1,3-oxazoles: Application to the synthesis of the IMPDH inhibitor BMS-337197 and analogues. Org. Lett. 2002, 4, 2091–2093. [Google Scholar] [CrossRef] [PubMed]
- Dhar, T.G.M.; Shen, Z.; Guo, J.; Liu, C.; Watterson, S.H.; Gu, H.H.; Pitts, W.J.; Fleener, C.A.; Rouleau, K.A.; Sherbina, N.Z.; et al. Discovery of N-[2-[2-[[3-methoxy-4-(5-oxazolyl)phenyl]amino]-5-oxazolyl]phenyl]-N-methyl-4-morpholineacetamide as a novel and potent inhibitor of inosine monophosphate dehydrogenase with excellent in vivo activity. J. Med. Chem. 2002, 45, 2127–2130. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kozuki, Y.; Eikyu, Y.; Kubo, K.; Kawato, Y.; Marui, T.; Seki, N.; Masunaga, T.; Tamura, K.; Morokata, T. In vitro and in vivo characterization of AS2643361, a novel and highly potent inosine 5′-monophosphate dehydrogenase inhibitor. Eur. J. Pharmacol. 2012, 674, 58–63. [Google Scholar] [CrossRef]
- Carr, S.F.; Papp, E.; Wu, J.C.; Natsumeda, Y. Characterization of human type I and type II IMP dehydrogenases. J. Biol. Chem. 1993, 268, 27286–27290. [Google Scholar]
- Sintchak, M.D.; Fleming, M.A.; Futer, O.; Raybuck, S.A.; Chambers, S.P.; Caron, P.R.; Murcko, M.A.; Wilson, K.P. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell 1996, 85, 921–930. [Google Scholar] [CrossRef]
- Colby, T.D.; Vanderveen, K.; Strickler, M.D.; Markham, G.D.; Goldstein, B.M. Crystal structure of human type II inosine monophosphate dehydrogenase: Implications for ligand binding and drug design. Proc. Natl. Acad. Sci. USA 1999, 96, 3531–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Ignoul, S.; Eggermont, J. CBS domains: Structure, function, and pathology in human proteins. Am. J. Physiol. Cell Physiol. 2005, 289, C1369–C1378. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhee, K.H.; McPhie, P.; Miles, E.W. Domain architecture of the heme-independent yeast cystathionine beta-synthase provides insights into mechanisms of catalysis and regulation. Biochemistry 2000, 39, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Nimmesgern, E.; Black, J.; Futer, O.; Fulghum, J.R.; Chambers, S.P.; Brummel, C.L.; Raybuck, S.A.; Sintchak, M.D. Biochemical analysis of the modular enzyme inosine 5′-monophosphate dehydrogenase. Protein Expr. Purif. 1999, 17, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Buey, R.M.; Ledesma-Amaro, R.; Velazquez-Campoy, A.; Balsera, M.; Chagoyen, M.; de Pereda, J.M.; Revuelta, J.L. Guanine nucleotide binding to the Bateman domain mediates the allosteric inhibition of eukaryotic IMP dehydrogenases. Nat. Commun. 2015, 6, 8923. [Google Scholar] [CrossRef] [PubMed]
- Buey, R.M.; Fernandez-Justel, D.; Marcos-Alcalde, I.; Winter, G.; Gomez-Puertas, P.; de Pereda, J.M.; Luis Revuelta, J. A nucleotide-controlled conformational switch modulates the activity of eukaryotic IMP dehydrogenases. Sci. Rep. 2017, 7, 2648. [Google Scholar] [CrossRef] [PubMed]
- Chitrakar, I.; Kim-Holzapfel, D.M.; Zhou, W.; French, J.B. Higher order structures in purine and pyrimidine metabolism. J. Struct. Biol. 2017, 197, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chiaro, C.R.; Zhang, L.; Smith, P.B.; Chan, C.Y.; Pedley, A.M.; Pugh, R.J.; French, J.B.; Patterson, A.D.; Benkovic, S.J. Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 2015, 290, 6705–6713. [Google Scholar] [CrossRef]
- O’Connell, J.D.; Zhao, A.; Ellington, A.D.; Marcotte, E.M. Dynamic reorganization of metabolic enzymes into intracellular bodies. Annu. Rev. Cell Dev. Biol. 2012, 28, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Carcamo, W.C.; Calise, S.J.; von Muhlen, C.A.; Satoh, M.; Chan, E.K. Molecular cell biology and immunobiology of mammalian rod/ring structures. Int. Rev. Cell Mol. Biol. 2014, 308, 35–74. [Google Scholar] [PubMed]
- Aughey, G.N.; Liu, J.L. Metabolic regulation via enzyme filamentation. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Petrovska, I.; Nuske, E.; Munder, M.C.; Kulasegaran, G.; Malinovska, L.; Kroschwald, S.; Richter, D.; Fahmy, K.; Gibson, K.; Verbavatz, J.M.; et al. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. Elife 2014, 3, e02409. [Google Scholar] [CrossRef] [PubMed]
- Willingham, M.C.; Richert, N.D.; Rutherford, A.V. A novel fibrillar structure in cultured cells detected by a monoclonal antibody. Exp. Cell Res. 1987, 171, 284–295. [Google Scholar] [CrossRef]
- Juda, P.; Smigova, J.; Kovacik, L.; Bartova, E.; Raska, I. Ultrastructure of cytoplasmic and nuclear inosine-5′-monophosphate dehydrogenase 2 “rods and rings” inclusions. J. Histochem. Cytochem. 2014, 62, 739–750. [Google Scholar] [CrossRef]
- Carcamo, W.C.; Satoh, M.; Kasahara, H.; Terada, N.; Hamazaki, T.; Chan, J.Y.; Yao, B.; Tamayo, S.; Covini, G.; von Muhlen, C.A.; et al. Induction of cytoplasmic rods and rings structures by inhibition of the CTP and GTP synthetic pathway in mammalian cells. PLoS ONE 2011, 6, e29690. [Google Scholar] [CrossRef] [PubMed]
- Keppeke, G.D.; Chang, C.C.; Peng, M.; Chen, L.Y.; Lin, W.C.; Pai, L.M.; Andrade, L.E.C.; Sung, L.Y.; Liu, J.L. IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div. 2018, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calise, S.J.; Purich, D.L.; Nguyen, T.; Saleem, D.A.; Krueger, C.; Yin, J.D.; Chan, E.K. ‘Rod and ring’ formation from IMP dehydrogenase is regulated through the one-carbon metabolic pathway. J. Cell Sci. 2016, 129, 3042–3052. [Google Scholar] [CrossRef]
- Calise, S.J.; Carcamo, W.C.; Krueger, C.; Yin, J.D.; Purich, D.L.; Chan, E.K. Glutamine deprivation initiates reversible assembly of mammalian rods and rings. Cell. Mol. Life. Sci. 2014, 71, 2963–2973. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, W.C.; Pai, L.M.; Lee, H.S.; Wu, S.C.; Ding, S.T.; Liu, J.L.; Sung, L.Y. Cytoophidium assembly reflects upregulation of IMPDH activity. J. Cell Sci. 2015, 128, 3550–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Gu, J.; Makhov, A.M.; Griffith, J.D.; Mitchell, B.S. Regulation of the interaction of inosine monophosphate dehydrogenase with mycophenolic Acid by GTP. J. Biol. Chem. 2006, 281, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Justel, D.; Nunez, R.; Martin-Benito, J.; Jimeno, D.; Gonzalez-Lopez, A.; Soriano, E.M.; Revuelta, J.L.; Buey, R.M. A Nucleotide-Dependent Conformational Switch Controls the Polymerization of Human IMP Dehydrogenases to Modulate their Catalytic Activity. J. Mol. Biol. 2019, 431, 956–969. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Ji, Y.; Itahana, K.; Zhang, Y.; Mitchell, B. Guanine nucleotide depletion inhibits pre-ribosomal RNA synthesis and causes nucleolar disruption. Leuk. Res. 2008, 32, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indication | Studies |
|---|---|
| Labeled (FDA Approved) Indications | Studies that led to FDA approvals |
| Renal transplant | Sollinger 1995 [24], Grinyo 1995 [25], Keown 1996 [26] |
| Liver transplant | Eckhoff 1998 [27], Wiesner 2005 [28], Nashan 2009 [29] |
| Cardiac transplant | Eisen 2005 [30], Kobashigawa 2006 [31], Kaczarek 2013 [32], Andreassen 2014 [33] |
| Off-Label Use | Studies supporting off-label use |
| Lung transplant | Treede 2001 [34], Zuckermann 2003 [35], Speich 2010 [36] |
| Pancreatic transplant | Ricart 2012 [37], Descourouez 2018 [38] |
| Refractory acute graft-versus-host disease | Alousi 2009 [39] |
| Refractory chronic graft-versus-host disease | Wolff 2010 [40] |
| Prevention of graft-versus-host disease | Sabry 2009 [41] |
| Aplastic anemia | Scheinberg 2006 [42] |
| Autoimmune hepatitis, first line | Zachou 2016 [43] |
| Refractory autoimmune hepatitis | Manns 2010 [44] |
| Lupus nephritis | Contreras 2004 [45], Ong 2005 [46], Dooley 2011 [47], Hahn 2012 [48] |
| Myasthenia gravis | Meriggiolo 2003 [49], Sanders 2016 [50], Sieb 2014 [51] |
| Psoriasis | Menter 2009 [52] |
| Systemic sclerosis | Gerbino 2008 [53], Derk 2009 [54], Le 2011 [55], Mendoza 2012 [56], Tashkin 2016 [57], Herrick 2017 [58] |
| Phase | Study Population | Dose | Clinical Response | References |
|---|---|---|---|---|
| I/II | Relapsed/refractory AML, CML-BC, and MDS. n = 27 | Biochemically directed protocol. Starting dose 2200 mg/m2 daily, dose escalated based on IMPDH and GTP levels in the leukemic cells | Complete response (CR) 20% Objective response rate (ORR) 48% | [117,118,119] |
| II | CML-BC n = 6 | Started at 2200 mg/m2 daily for 10 days and escalated based on hematological and biochemical response. | Objective response rate (ORR) 100% but no complete response (CR) | [120] |
| Phase | Study Population | Dose | Clinical Response | References |
|---|---|---|---|---|
| I | Advanced solid malignancies | Maximum tolerated dose varied between studies | Response reported with only one trial Maroun et al. reported 12 of 25 patients had stable disease, including one patient with anaplastic astrocytoma who was in remission for 50 months. | [123,124,125,126,127,128] |
| II | Glioma | 1100 to 1375 mg/m2 IV daily for five days | Five of 16 patients had stable disease for a median of 75 days, but no responses seen. | [129,130,131,132] |
| IMPDH Inhibitor | Mechanism of Action | Study Results |
|---|---|---|
| VX-497 (Merimepodib) (S)-N-3-[3-(3-methoxy-4-oxazol-5-yl-phenyl)-ureido]-benzyl-carbamic acid tetrahydrofuran-3-yl-ester (Figure 7L) | VX-497 is a non-nucleoside, orally bioavailable, selective, reversible, uncompetitive inhibitor of IMPDH, which was developed by Vertex Pharmaceuticals [134]. | Clinical studies VX-497 was efficacious as monotherapy for Hepatitis C in combination with interferon-alpha in treatment naïve patients [138]. However, phase II trials in patients with genotype 1 chronic hepatitis C, who were non responders to standard treatment, showed mixed results. |
| Phase | Study Population | Dose | Clinical Response | Toxicity | References |
|---|---|---|---|---|---|
| I | Relapsed/refractory AML and MDS n = 37 | Escalating doses from 50–500 mg/m2. Recommended phase II dose 400 mg/m2 for 21 days every 28-day cycle. | Response observed in 4 of 37 patients | Well tolerated, frequently Grade 1–2 | [152,153] |
| IMPDH Inhibitor | Mechanism of Action | Study Results |
|---|---|---|
| Reversible nucleoside inhibitors | ||
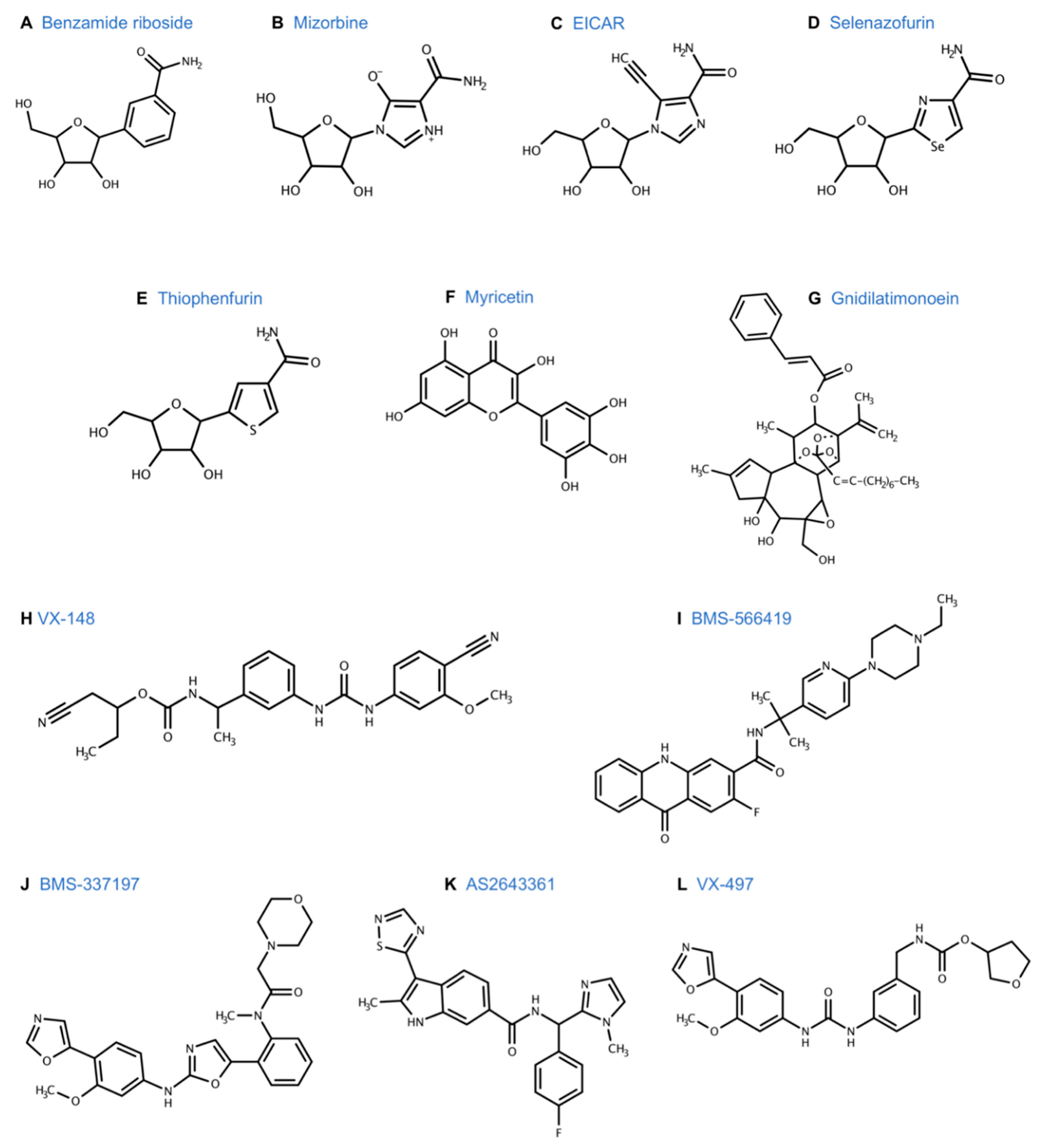
| Benzamide riboside (BR) 3-(1-Deoxyribofuranosyl) benzamide (Figure 7A) | Benzamide riboside (BR) was first synthesized in 1992 [155]. Similar to tiazofurin, BR, is converted to its active metabolite, BAD (benzamide adenine dinucleotide) intracellularly via NMNAT. BAD is proposed as a dual inhibitor of IMPDH and NAD kinase. IMPDH inhibition leads to depletion of guanine nucleotides and halts DNA/RNA synthesis [156,157]. NAD kinase inhibition leads to decreased levels of NADPH. Low NADP+ and NADPH levels lead to instability and lower levels of dihydrofolate reductase [158]. | BR was more cytotoxic than tiazofurin in a broad panel of human cancer cell lines, including leukemia, lung, colon, CNS, melanoma, ovarian, and renal cell carcinoma [104,159,160]. CNS cell lines showed selective sensitivity to BR. BR was 3-10 times more cytotoxic than tiazofurin against leukemia [104]. In vivo, BR prolonged survival of a mouse model with murine leukemia L1210 [161] but caused significant skeletal muscle toxicity [162]. BR induced apoptosis in the VX2 model of liver cancer in rabbits via hepatic artery infusion [163]. Mouse model of LX-1 human small cell lung carcinoma was relatively refractory to treatment with BR in vivo and the high doses required for anti-tumor effect lead to significant morbidity and mortality [162]. The clinical application of BR was limited by its toxicity profile. |
| Mizoribine (MZR) (INN, trade name Bredinin) 5-hydroxy-1-β-D-ribofuranosyl-1H-imidazole-4-carboxamide (Figure 7B) | An imidazole nucleoside isolated from Eupenicillium brefeldianum, mizoribine (MZR) is metabolized to MZR-5’-monophosphate (MZRP) by adenosine kinase. MZRP, the active metabolite, inhibits IMPDH and guanosine monophosphate synthetase, which are sequential enzymes in the de novo pathway. Therefore, MZR completely inhibits the synthesis of guanine nucleotides [164,165,166]. MZR selectively inhibits lymphocyte proliferation, thereby inhibiting both humoral and cellular immunity [167,168]. | MZR was originally isolated as an antibiotic with activity against Candida albicans [167] but was subsequently found to have potent immunosuppressive activity [169]. Preclinical Early pre-clinical studies reported that MZR was not active against mice inoculated with Ehrlich and P388 tumor cells and had a minimal life prolonging effect on mice inoculated with L1210 leukemia cells [167]. However, more recently, MZR was found to produce a marked anti-leukemic response and increased survival in mice inoculated with resistant acute lymphoblastic leukemia with NT5C2+/R367Q mutation [170]. The expression status of adenosine kinase dramatically affects the efficacy of MZR [157]. Further preclinical studies are needed to better define the role of MZR in leukemia. Clinical MZR is currently used as an immunosuppressive drug. It has a favorable adverse effect profile and is usually used in combination with other drugs. It has been approved in Japan to prevent rejection after renal transplantation (1984), lupus nephritis (1990), rheumatoid arthritis (1992), and nephritic syndrome (1995) [171]. The use of MZR is being investigated in other nephropathies [172], pemphigus vulgaris [173], and polymyalgia rheumatica [174]. |
| Ribavirin 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide (Figure 4D) | Ribavirin is a guanosine analogue that is phosphorylated intracellularly to ribavirin-5-monophosphate, which inhibits IMPDH [175]. Ribavirin has broad spectrum antiviral activity [176]. It exerts antitumor activity through inhibition of IMPDH, eukaryotic translation initiation factor 4E (eIF4E), and histone methyltransferase, Enhancer of Zeste Homolog 2 (EZH2) [177,178]. | Pre-clinical studies have shown that ribavirin inhibits the proliferation of several tumor types including malignant glioma [177], acute myeloid leukemia [179], acute lymphoblastic leukemia [180], esophageal [181], colon, cervical [182], breast [183], and prostate cancer [184]. Clinical studies Phase I/II trials are underway for assessing the use of ribavirin in various cancers including head and neck cancer, mantle cell, and follicular lymphoma [185,186,187]. Ribavirin has been approved by the FDA as an inhaled agent for respiratory syncytial virus [188] and in combination with interferon-alpha for the treatment of chronic hepatitis C [189]. |
| EICAR 5-ethynyl-1- β -D-ribofuranosylimidazole-4-carboxamide (Figure 7C) | Imidazole derivative of ribavirin, EICAR is metabolized intracellularly via adenosine kinase to EICAR 5’-monophosphate, which inhibits IMPDH [190]. | EICAR had broad antiviral activity, which was 10-100 fold greater than ribavirin [191]. It was cytotoxic to several human cancer cell lines in vitro and murine leukemia L1210 and P388 in vivo [192]. |
| Selenazofurin 2- β -D-ribofuranosylselenazole-4-carboxamide (Figure 7D) | Selenium analogue of tiazofurin, selenazofurin is converted to its active metabolite, selenazole-4-carboxamide adenine dinucleotide (SAD) intracellularly, via NMNAT. SAD is a NAD analogue and inhibits IMPDH [193,194]. | As an antitumor agent, selenazofurin was found to be 5–10 fold more potent compared to tiazofurin in several in vitro studies [194]. It had broad antiviral activity [195] and was synergistic in combination with ribavirin [196]. |
| Thiophenfurin 5-β-D-ribofuranosylthiophene-3-carboxamide (Figure 7E) | Thiophene analogue of tiazofurin, it is converted intracellularly to thiophene-3-carboxamide adenine dinucleotide (TFAD), a NAD analogue, which inhibits IMPDH [197]. | In vitro studies showed that thiophenfurine was cytotoxic toward several cancer cell lines, including human promyelocytic leukemia HL-60, human colon adenocarcinoma LoVo, and B16 melanoma at similar concentrations as tiazofurin [197]. |
| Flavonoids | ||
| Myricetin 3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)-4-chromenone (Figure 7F) | Myricetin is a dietary flavonoid found in berries and vegetables. It causes cell cycle arrest and apoptosis through various mechanisms, including inhibition of tumorigenic kinases [198], which increases mitochondrial apoptotic pathways, reactive oxygen species, and IMPDH inhibition [199]. | Myricetin has extensive biological activity, including anti-viral, anti-inflammatory, and anti-cancer [200]. In vitro studies have shown that myricetin has anti-leukemia effect on K562 cell lines through IMPDH inhibition [199]. It is cytotoxic to several other human cancer cell lines like colon [201], ovarian [202], prostate [203], breast [204], and thyroid [205] cancer cell lines by targeting various pathways. |
| Diterpene ester | ||
| Gnidilatimonoein (Gn) (Figure 7G) | Diterpene ester isolated from the leaves of Daphne mucronata, Gn exerts anti-neoplastic activity through inhibition of IMPDH [206]. | In vitro studies have shown that Gn has antiproliferative activity against several human cancer cell lines and induced differentiation in the HL-60 human leukemia cell line [207]. |
| IMPDH Inhibitor | Mechanism of Action | Study Results |
|---|---|---|
| Mizoribine | See Table 5. | |
| VX-148 1-cyanobutan-2-yl N-[(1S)-1-[3-[(4-cyano-3-methoxyphenyl) carbamoyl amino] phenyl] ethyl] carbamate (Figure 7H) | VX-148 noncompetitively inhibits IMPDH by binding to the NAD cofactor binding site. It is an orally bioavailable small molecule that was developed by structural modification of VX-497 by Vertex Pharmaceuticals [208]. | VX-148 was found to have in vivo and in vitro immunosuppressive activity similar to MPA but with less cytotoxicity [208]. Vertex Pharmaceuticals selected it as its lead drug development candidate for autoimmune diseases [134]. VX-148 has been evaluated in a Phase II trial in moderate to severe psoriasis in 2004. It was well tolerated. The most frequent adverse events were diarrhea and itching. It showed a statistically significant clinical activity with a response rate of 18% compared to a placebo [209,210]. |
| BMS-566419 N-(1-(6-(4-Ethyl-1-piperazinyl)-3-pyridinyl)-1-methylethyl)-2-fluoro-9,10-dihydro-9-oxo-3-acridinecarboxamide (Figure 7I) | Acridone based derivative of VX-497, BMS-566419 is an orally bioavailable IMPDH inhibitor developed in 2007 [211]. | In vitro studies demonstrated the anti-proliferative activity of BMS-566419 on immune cells. Preclinical studies showed that it was efficacious in the murine model of rheumatoid arthritis and prevented cardiac allograft rejection with less GI toxicity compared to MMF [211,212]. |
| BMS-337197 N-[2-[2-(3-methoxy-4-oxazol-5-yl-anilino) oxazol-5-yl] phenyl]-N-methyl-2-morpholino-acetamide (Figure 7J) | 2-aminooxazole derivative of VX-497, BMS-337197 is an orally bioavailable, uncompetitive inhibitor of IMPDH [213]. | Preclinical studies showed that BMS-337197 had potent immunosuppressive activity. It inhibited antibody production in mice and was efficacious as an anti-arthritis drug in a murine model of rheumatoid arthritis [214]. |
| AS2643361 N-((4-fluorophenyl) (1-methyl-1H-imidazol-2-yl) methyl)- 2-methyl-3-(1,2,4-thiadiazol-5-yl)-1H-indole-6-carboxamide (Figure 7K) | An indole derivative of MMF developed from the Astellas compound library, AS2643361 is an orally bioavailable IMPDH inhibitor [215]. In vitro, it has similar inhibitory activity as mycophenolate to inhibit IMPDH. | AS2643361 had lower serum protein binding activity. In vivo, it showed higher potency and less toxicity than MMF as an immunosuppressant. It prevented cardiac allograft rejection in a murine model [215]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naffouje, R.; Grover, P.; Yu, H.; Sendilnathan, A.; Wolfe, K.; Majd, N.; Smith, E.P.; Takeuchi, K.; Senda, T.; Kofuji, S.; et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 2019, 11, 1346. https://doi.org/10.3390/cancers11091346
Naffouje R, Grover P, Yu H, Sendilnathan A, Wolfe K, Majd N, Smith EP, Takeuchi K, Senda T, Kofuji S, et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers. 2019; 11(9):1346. https://doi.org/10.3390/cancers11091346
Chicago/Turabian StyleNaffouje, Rand, Punita Grover, Hongyang Yu, Arun Sendilnathan, Kara Wolfe, Nazanin Majd, Eric P. Smith, Koh Takeuchi, Toshiya Senda, Satoshi Kofuji, and et al. 2019. "Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story" Cancers 11, no. 9: 1346. https://doi.org/10.3390/cancers11091346
APA StyleNaffouje, R., Grover, P., Yu, H., Sendilnathan, A., Wolfe, K., Majd, N., Smith, E. P., Takeuchi, K., Senda, T., Kofuji, S., & Sasaki, A. T. (2019). Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers, 11(9), 1346. https://doi.org/10.3390/cancers11091346