Targeting Ca2+ Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma




Abstract
:Simple Summary
Abstract
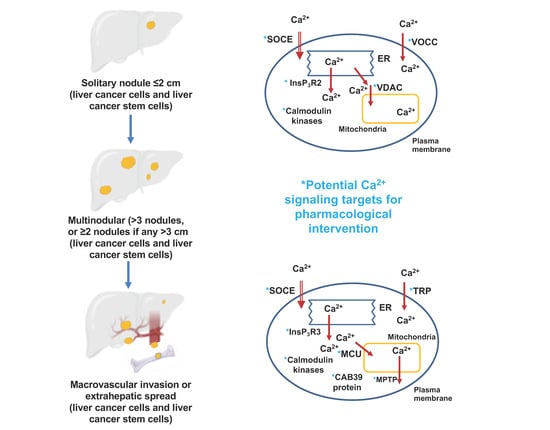
1. Introduction
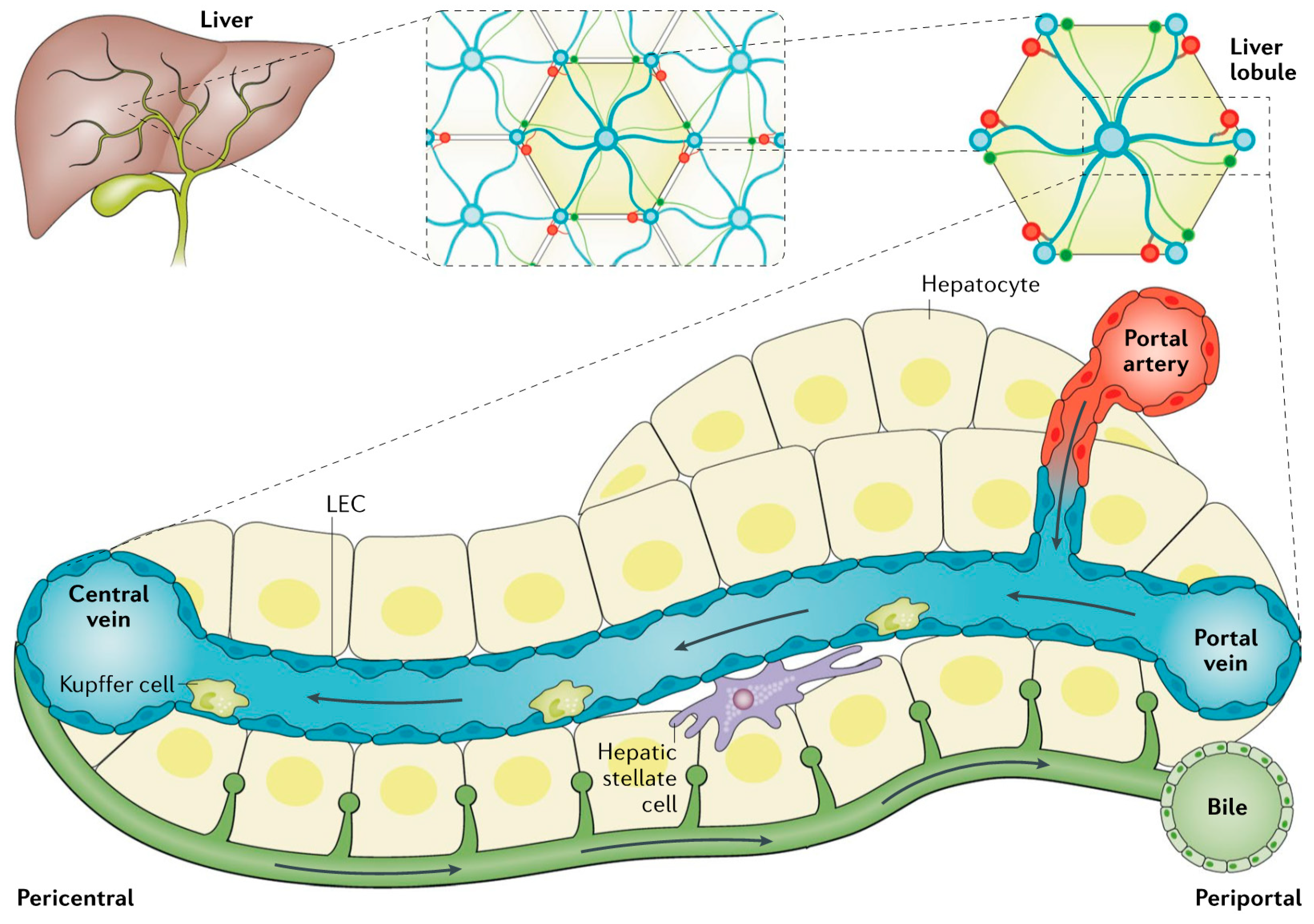
2. Overview of Liver Anatomy, Cell Types, Physiology and Metabolic Pathways
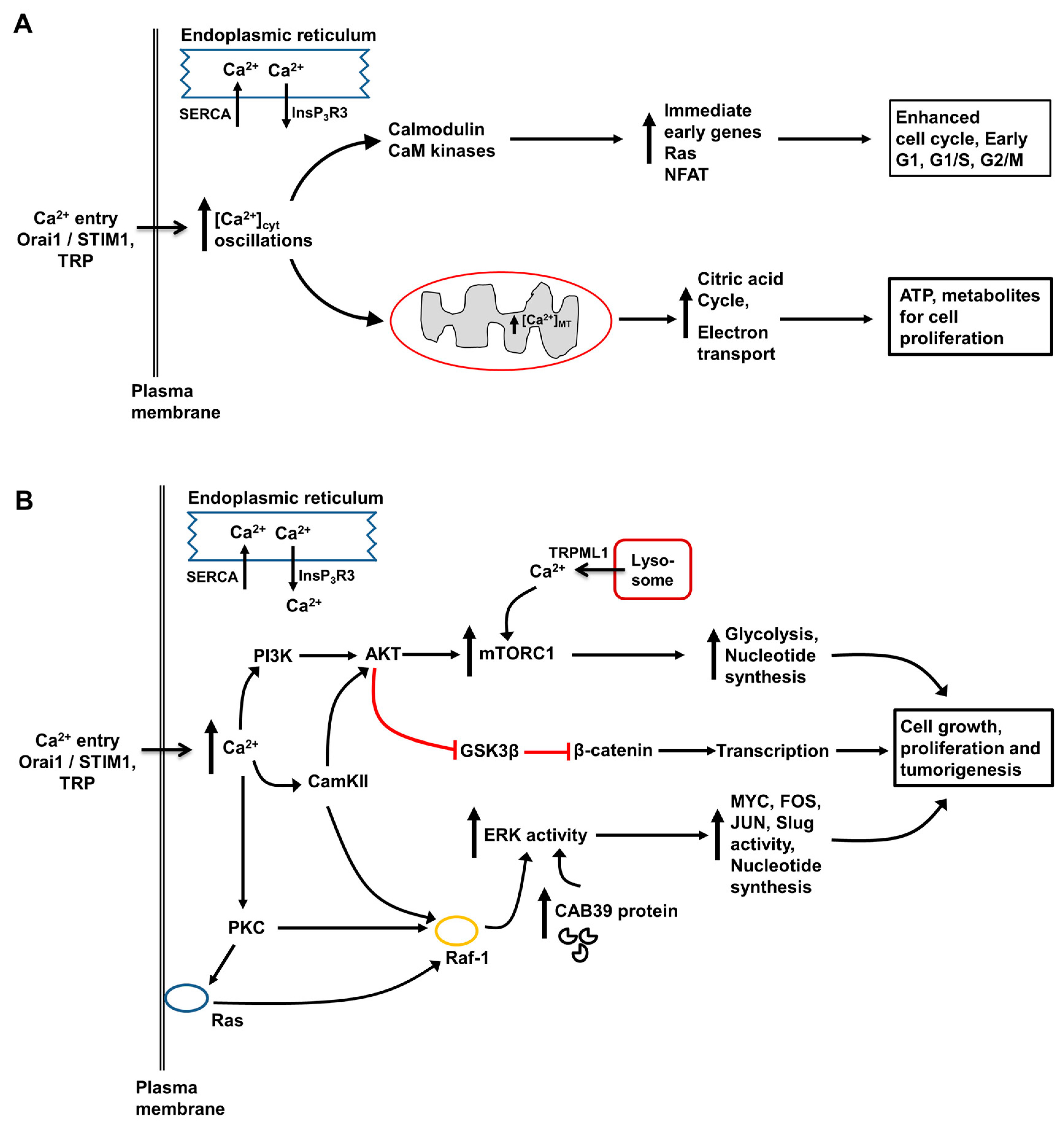
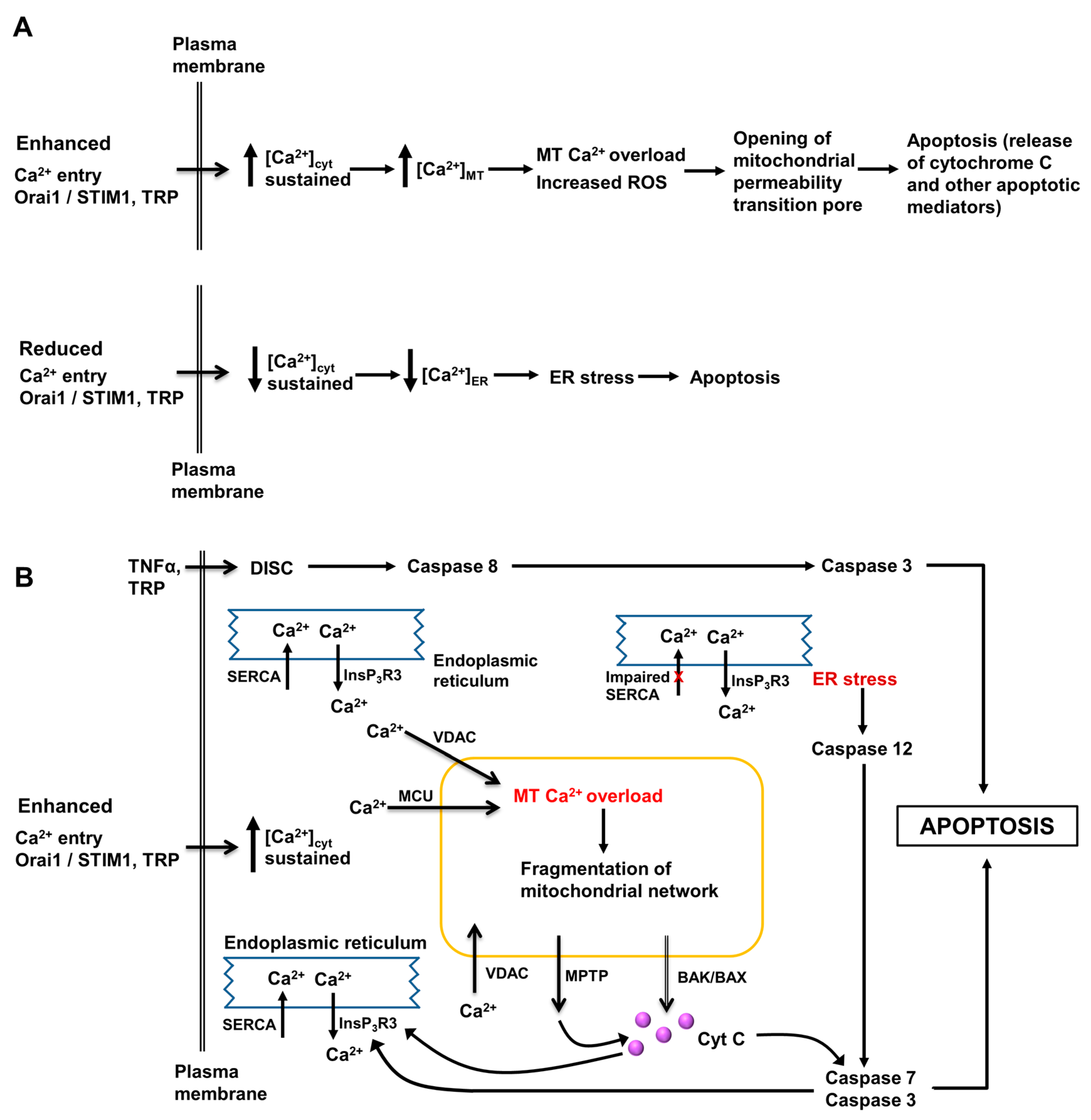
3. Role of Intracellular Ca2+ in Regulation of Hepatocyte Metabolism, Proliferation, Injury and Death
4. The Pathology of Hepatocellular Carcinoma
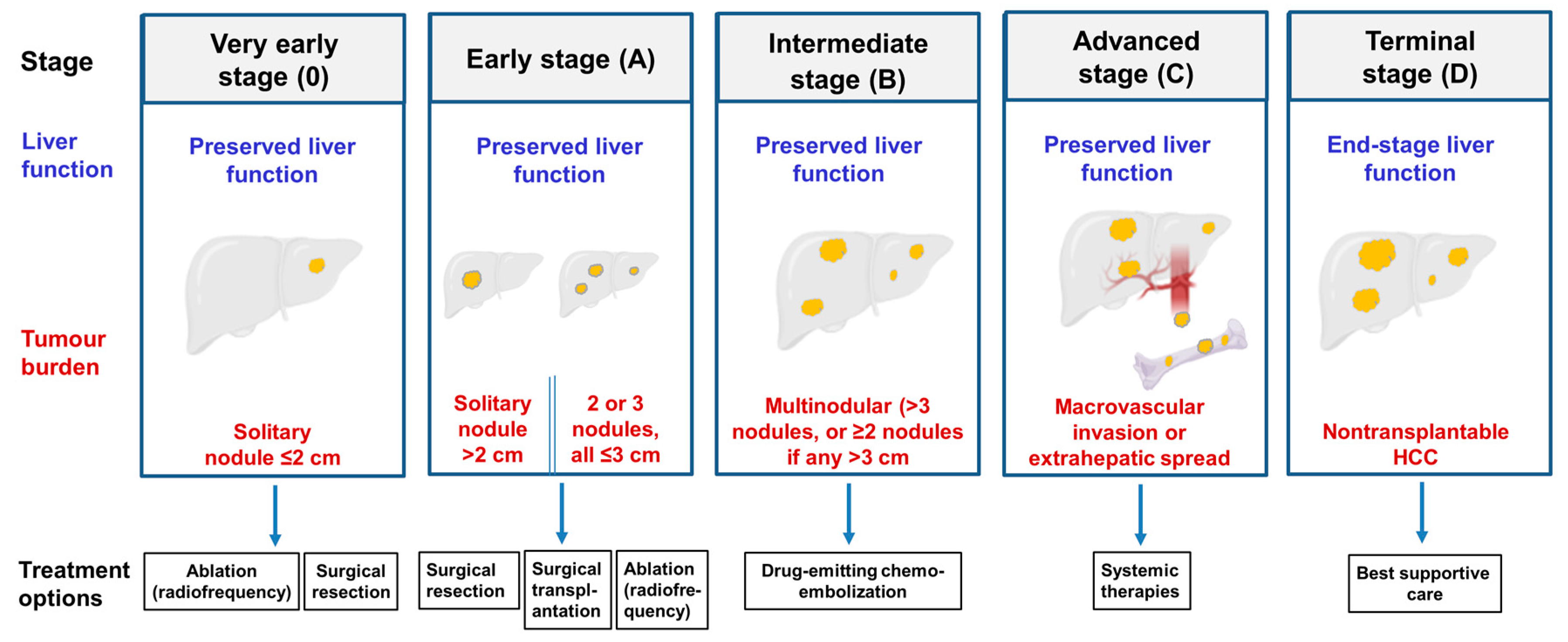
5. Current Treatments for Hepatocellular Carcinoma
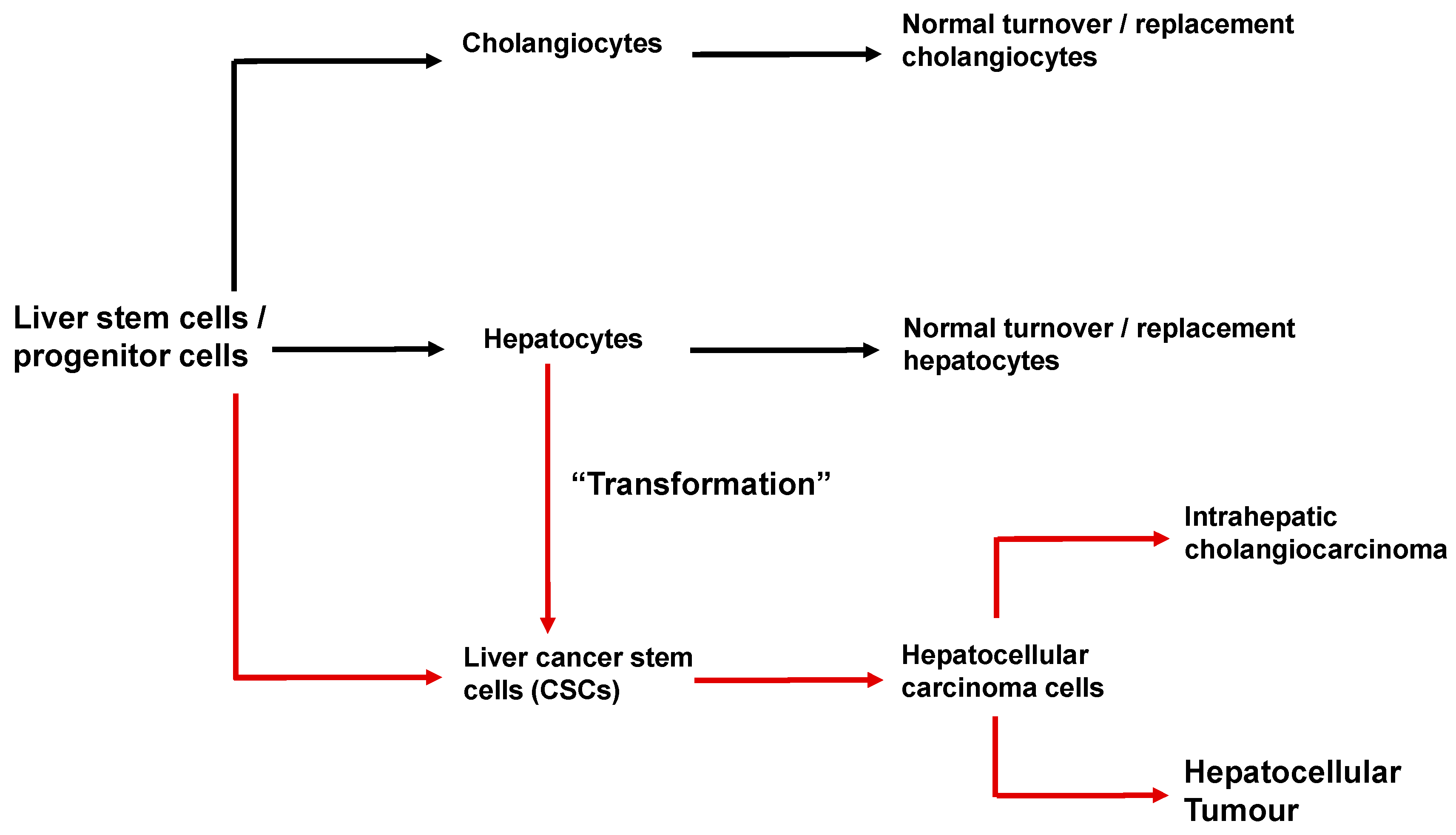
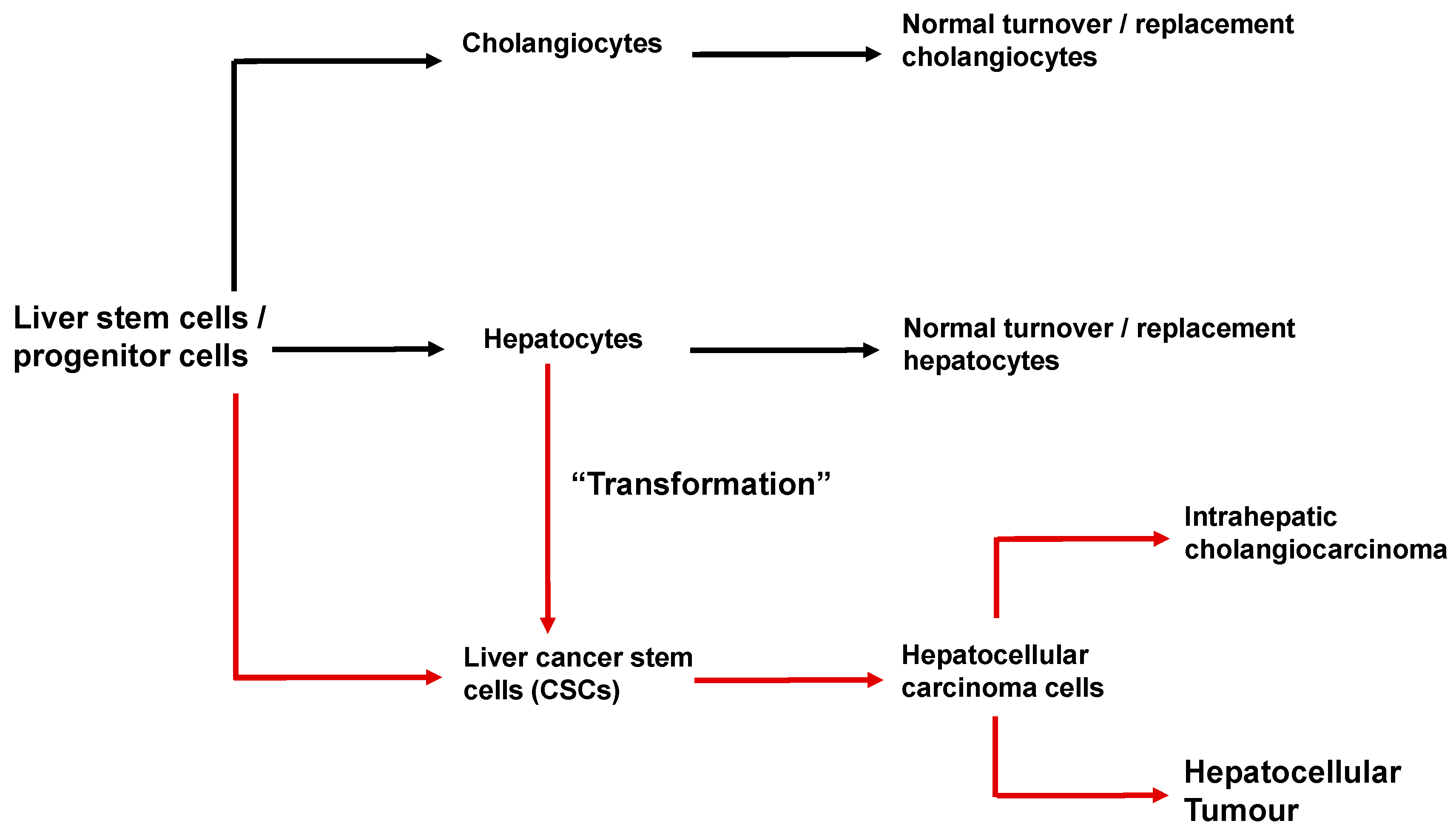
6. Cancer Stem Cells and the Initiation and Progression of Hepatocellular Carcinoma
7. Methodology and Terminology for Hepatocellular Carcinoma Liver Samples and Hepatocellular Carcinoma Cell Lines
8. Mutations and Altered Expression of Ca2+-Signaling Proteins in Hepatocellular Carcinoma
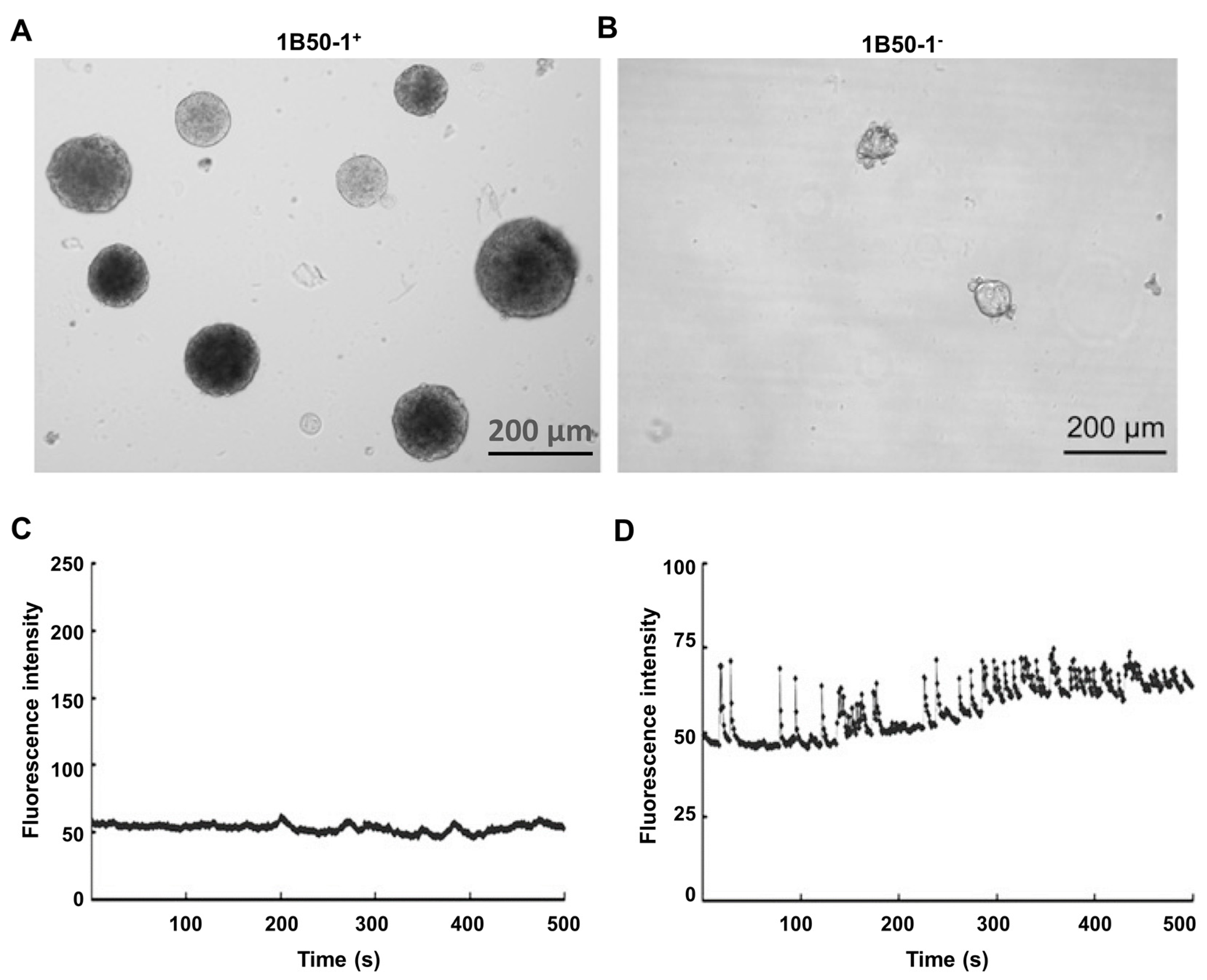
9. Voltage-Operated Ca2+ Channels, InsP3 Receptors and TRPV2 Channels in Liver Cancer Stem Cells
10. Mitochondrial Ca2+ and Store-Operated Ca2+ Entry in HBV and HCV Infection
11. Store-Operated Ca2+ Entry, SERCA2b and Ca2+/Calmodulin-Dependent Protein Kinases in Initiation and Progression of Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease
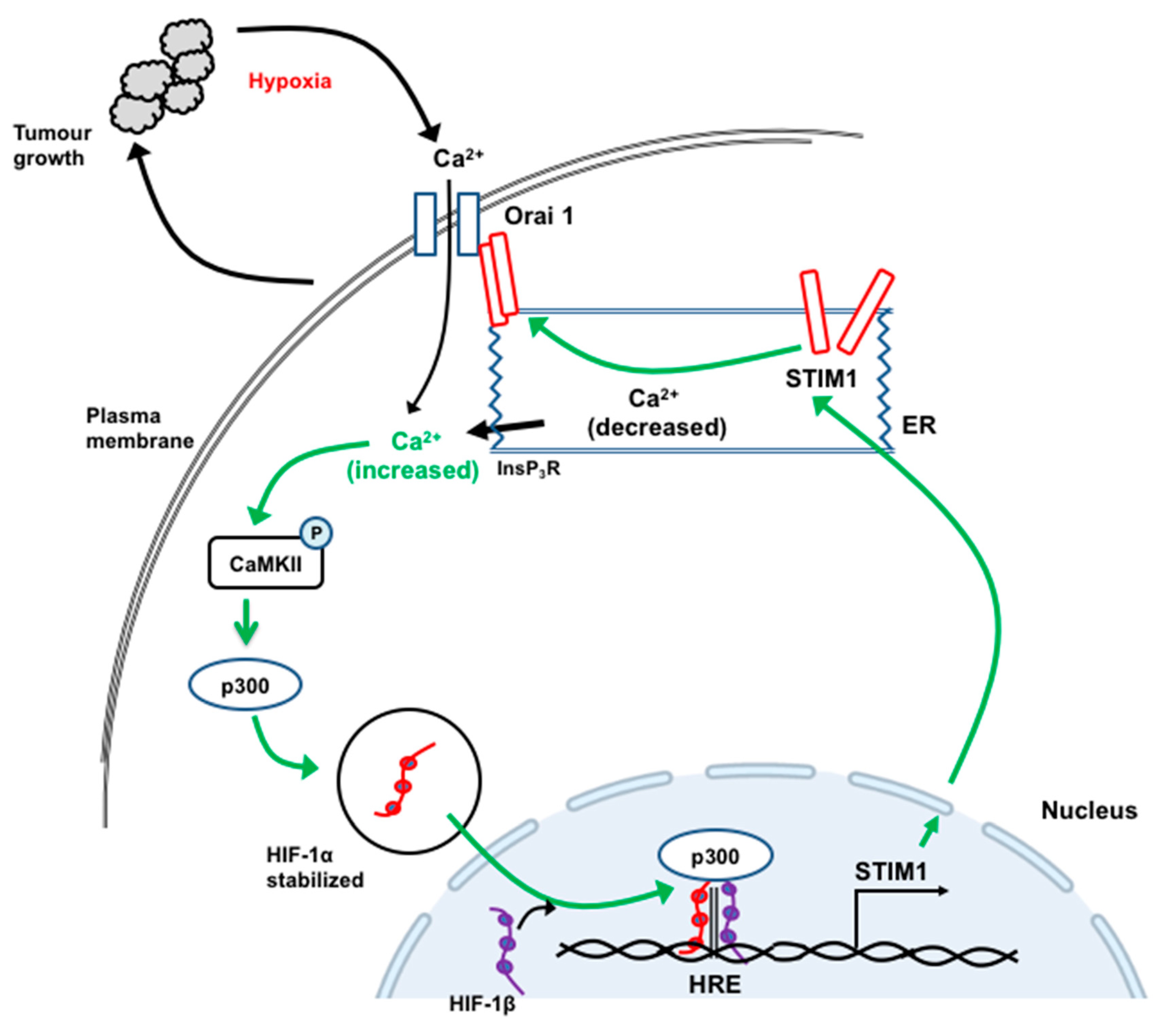
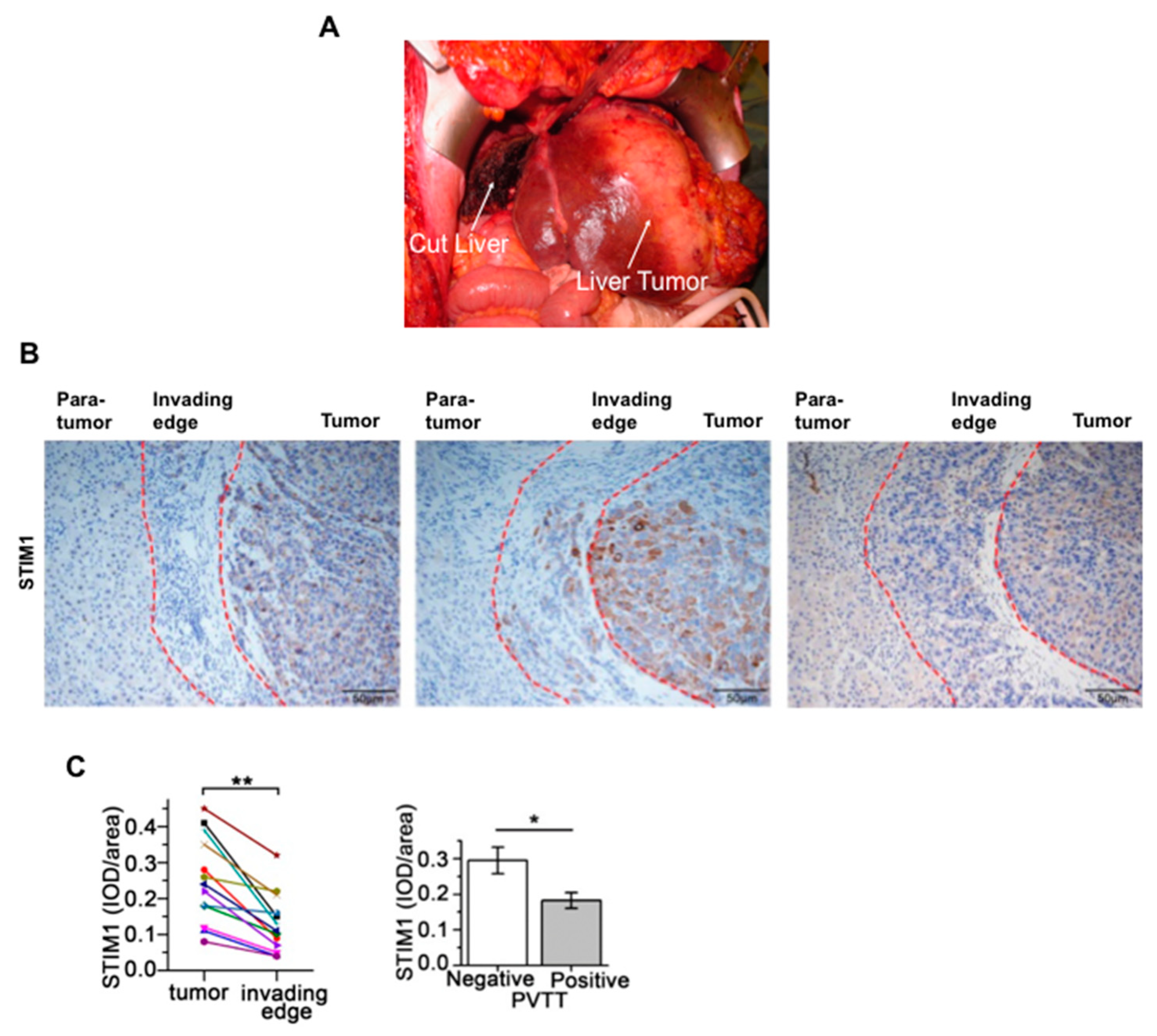
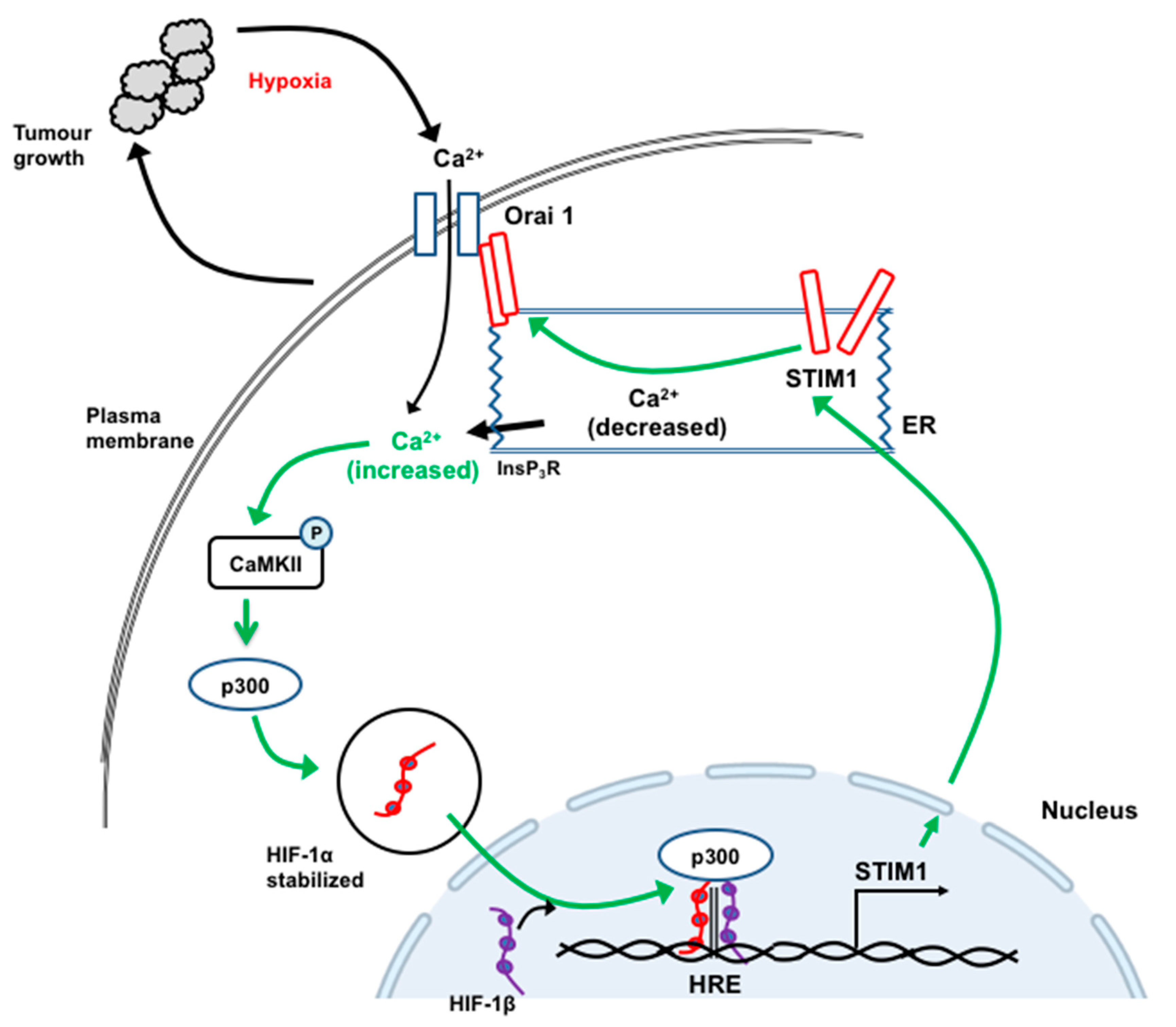
12. STIM1 and Orai1 in the Progression and Metastasis of Hepatocellular Carcinoma
13. TRPC6, TRPV4 and TRPV1 in the Progression, Metastasis and Apoptosis of Hepatocellular Carcinoma
14. Type 3 InsP3 Receptors in the Progression of Hepatocellular Carcinoma
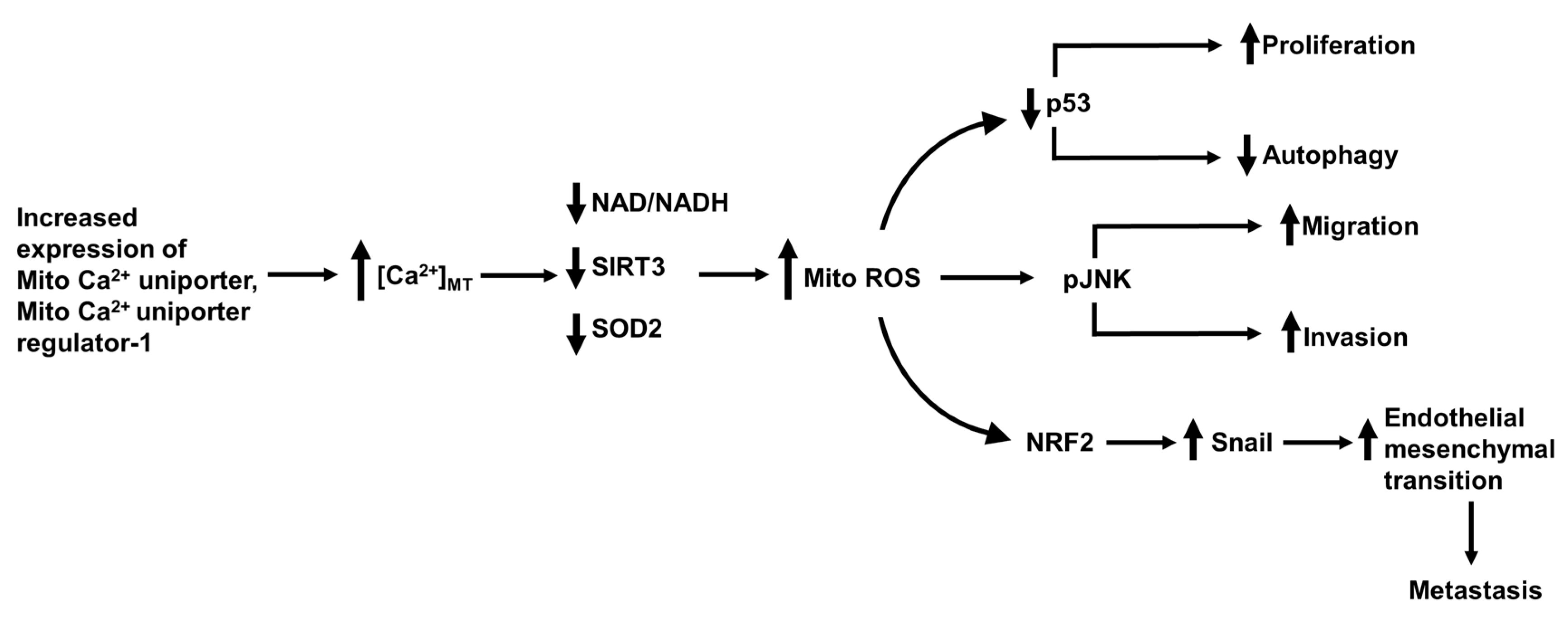
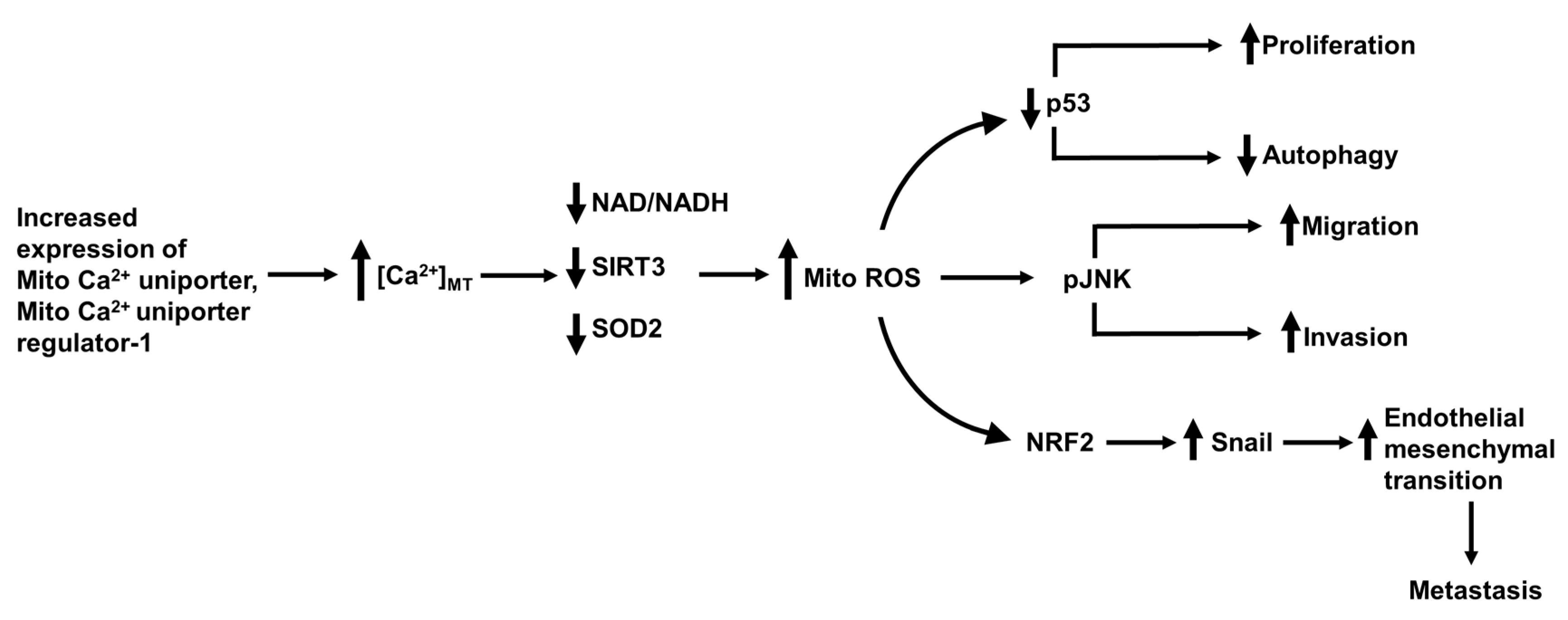
15. The Mitochondrial Ca2+ Uniporter, Permeability Transition Pore and Mitofusin-2 in Hepatocellular Carcinoma Metastasis
16. Tuftelin1, Ca2+ Calmodulin Kinases and Ca2+ Binding Protein 39 in the Promotion and Metastasis of Hepatocellular Carcinoma
17. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| HBV | hepatitis B |
| HCV | hepatitis C |
| [Ca2+]cyt | concentration of free Ca2+ in the cytoplasmic space |
| [Ca2+]ER | concentration of free Ca2+ in the lumen of the ER |
| [Ca2+]MT | concentration of free Ca2+ in the mitochondrial matrix |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CamKK2 | Ca2+/calmodulin-dependent protein kinase kinase 2 |
| CaMKIV | Ca2+/calmodulin-dependent protein kinase IV |
| ER | endoplasmic reticulum |
| SERCA | sarco/endoplasmic reticulum (Ca2++Mg2+)ATP-ase |
| STIM | stromal interaction molecule |
| TRP | transient receptor potential |
| InsP3 | inositol 1,4,5-trisphosphate |
| InsP3R | InsP3 receptor |
| ROS | reactive oxygen species |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
References
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef]
- Ali, E.; Rychkov, G.Y.; Barritt, G.J. Deranged hepatocyte intracellular Ca2+ homeostasis and the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma. Cell Calcium 2019. [Google Scholar] [CrossRef] [PubMed]
- Nio, K.; Yamashita, T.; Kaneko, S. The evolving concept of liver cancer stem cells. Mol. Cancer 2017, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Zheng, X.; Ji, J. Liver cancer stem cells as a hierarchical society: Yes or no? Acta Biochim. Biophys. Sin. 2020. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, J.; Zhang, X.; Zhou, H.; Liu, G.; Li, Q. Cancer Stem Cells: A Potential Breakthrough in HCC-Targeted Therapy. Front. Pharmacol. 2020, 11, 198. [Google Scholar] [CrossRef]
- Wang, N.; Wang, S.; Li, M.Y.; Hu, B.G.; Liu, L.P.; Yang, S.L.; Yang, S.; Gong, Z.; Lai, P.B.S.; Chen, G.G. Cancer stem cells in hepatocellular carcinoma: An overview and promising therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816287. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.Q.; Teoh, N.; Xu, L.; Pok, S.; Li, X.; Chu, E.S.H.; Chiu, J.; Dong, L.; Arfianti, E.; Haigh, W.G.; et al. Dietary cholesterol promotes steatohepatitis related hepatocellular carcinoma through dysregulated metabolism and calcium signaling. Nat. Commun. 2018, 9, 4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Qiu, Z.; Wei, L.; Tang, R.; Lian, B.; Zhao, Y.; He, X.; Xie, L. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PLoS ONE 2014, 9, e100854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Han, H.; Jin, K.; Lin, N.; Guo, T.; Chen, Y.; Cheng, H.; Lu, F.; Fang, W.; et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel alpha2delta1 subunit. Cancer Cell 2013, 23, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, H.; Wang, M.; Zimmerman, J.W.; Pennison, M.J.; Sharma, S.; Surratt, T.; Xu, Z.X.; Brezovich, I.; Absher, D.; Myers, R.M.; et al. Tumour-specific amplitude-modulated radiofrequency electromagnetic fields induce differentiation of hepatocellular carcinoma via targeting Cav3.2T-type voltage-gated calcium channels and Ca2+ influx. EBioMedicine 2019, 44, 209–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Li, T.; Ma, X.; Wang, X.; Van Ness, C.; Gan, Y.; Zhou, H.; Tang, J.; Lou, G.; Wang, Y.; et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca(2)(+)/calmodulin-dependent protein kinase II. Mol. Cancer Ther. 2013, 12, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Shui, B.; Zhao, W.; Liu, H.; Li, W.; Lee, J.C.; Doran, R.; Lee, F.K.; Sun, T.; Shen, Q.S.; et al. Central role of IP3R2-mediated Ca2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019, 10, 396. [Google Scholar] [CrossRef] [Green Version]
- Scrima, R.; Piccoli, C.; Moradpour, D.; Capitanio, N. Targeting Endoplasmic Reticulum and/or Mitochondrial Ca2+ Fluxes as Therapeutic Strategy for HCV Infection. Front. Chem. 2018, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [Green Version]
- Casciano, J.C.; Duchemin, N.J.; Lamontagne, R.J.; Steel, L.F.; Bouchard, M.J. Hepatitis B virus modulates store-operated calcium entry to enhance viral replication in primary hepatocytes. PLoS ONE 2017, 12, e0168328. [Google Scholar] [CrossRef]
- Ali, E.S.; Petrovsky, N. Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends Endocrinol. Metab. 2019, 30, 270–281. [Google Scholar] [CrossRef]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.K.; Kim, O.H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.Y.; et al. Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.Q.; Chen, Y.F.; Chen, J.R.; Jou, Y.S.; Wu, P.C.; Kao, C.H.; Wang, C.H.; Huang, Y.L.; Chen, C.F.; Huang, T.S.; et al. CISD2 Haploinsufficiency Disrupts Calcium Homeostasis, Causes Nonalcoholic Fatty Liver Disease, and Promotes Hepatocellular Carcinoma. Cell Rep. 2017, 21, 2198–2211. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Xu, M.; Jeong, S.; Qian, Y.; Wu, H.; Xia, Q.; Kong, X. The Role of Nrf2 in Liver Disease: Novel Molecular Mechanisms and Therapeutic Approaches. Front. Pharmacol. 2018, 9, 1428. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Marcelo, K.L.; Rajapakshe, K.; Coarfa, C.; Dean, A.; Wilganowski, N.; Robinson, H.; Sevick, E.; Bissig, K.D.; Goldie, L.C.; et al. The camKK2/camKIV relay is an essential regulator of hepatic cancer. Hepatology 2015, 62, 505–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Qing, J.; Xia, Y.; Wang, K.; Zhang, F. Suppression of stromal interaction molecule 1 inhibits SMMC7721 hepatocellular carcinoma cell proliferation by inducing cell cycle arrest. Biotechnol. Appl. Biochem. 2015, 62, 107–111. [Google Scholar] [CrossRef]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- Anand, P.; Filipenko, P.; Huaman, J.; Lyudmer, M.; Hossain, M.; Santamaria, C.; Huang, K.; Ogunwobi, O.O.; Holford, M. Selective Inhibition of Liver Cancer Cells Using Venom Peptide. Mar. Drugs 2019, 17, 587. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Liu, Q.; Liu, G.; Cao, Z.; Fu, J.; Du, H.; et al. Pharmacological inhibition of TRPV4 channel suppresses malignant biological behavior of hepatocellular carcinoma via modulation of ERK signaling pathway. Biomed. Pharmacother. 2018, 101, 910–919. [Google Scholar] [CrossRef]
- Chen, W.T.; Lin, G.B.; Lin, S.H.; Lu, C.H.; Hsieh, C.H.; Ma, B.L.; Chao, C.Y. Static magnetic field enhances the anticancer efficacy of capsaicin on HepG2 cells via capsaicin receptor TRPV1. PLoS ONE 2018, 13, e0191078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Liu, G.; Li, M.; Fang, Y.; Qian, K.; Tang, Y.; Wu, X.; Lei, X.; Li, X.; Liu, Q.; et al. Targeting TRPV1 on cellular plasticity regulated by Ovol 2 and Zeb 1 in hepatocellular carcinoma. Biomed. Pharmacother. 2019, 118, 109270. [Google Scholar] [CrossRef] [PubMed]
- Guerra, M.T.; Florentino, R.M.; Franca, A.; Lima Filho, A.C.; Dos Santos, M.L.; Fonseca, R.C.; Lemos, F.O.; Fonseca, M.C.; Kruglov, E.; Mennone, A.; et al. Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 2019, 68, 1676–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef]
- Zhou, L.J.; Mo, Y.B.; Bu, X.; Wang, J.J.; Bai, J.; Zhang, J.W.; Cheng, A.B.; Ma, J.H.; Wang, Y.W.; Xie, Y.X. Erinacine Facilitates the Opening of the Mitochondrial Permeability Transition Pore Through the Inhibition of the PI3K/ Akt/GSK-3beta Signaling Pathway in Human Hepatocellular Carcinoma. Cell Physiol. Biochem. 2018, 50, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef]
- Huang, T.; Xu, S.; Deo, R.; Ma, A.; Li, H.; Ma, K.; Gan, X. Targeting the Ca2+/Calmodulin-dependent protein kinase II by Tetrandrine in human liver cancer cells. Biochem. Biophys. Res. Commun. 2019, 508, 1227–1232. [Google Scholar] [CrossRef]
- Li, Z.; Lu, J.; Zeng, G.; Pang, J.; Zheng, X.; Feng, J.; Zhang, J. MiR-129-5p inhibits liver cancer growth by targeting calcium calmodulin-dependent protein kinase IV (CAMK4). Cell Death Dis. 2019, 10, 789. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Yan, Q.; Fang, S.; Liu, M.; Li, Y.; Yuan, Y.F.; Li, Y.; Zhu, Y.; Qi, J.; Yang, X.; et al. Calcium-binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating extracellular signal-regulated kinase signaling pathway. Hepatology 2017, 66, 1529–1545. [Google Scholar] [CrossRef] [Green Version]
- Brosch, M.; Kattler, K.; Herrmann, A.; von Schonfels, W.; Nordstrom, K.; Seehofer, D.; Damm, G.; Becker, T.; Zeissig, S.; Nehring, S.; et al. Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control. Nat. Commun. 2018, 9, 4150. [Google Scholar] [CrossRef]
- Bou-Nader, M.; Caruso, S.; Donne, R.; Celton-Morizur, S.; Calderaro, J.; Gentric, G.; Cadoux, M.; L’Hermitte, A.; Klein, C.; Guilbert, T.; et al. Polyploidy spectrum: A new marker in HCC classification. Gut 2020, 69, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Ainama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef]
- Exton, J.H. Mechanisms of hormonal regulation of hepatic glucose metabolism. Diabetes Metab. Rev. 1987, 3, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Woods, N.M.; Cuthbertson, K.S.; Cobbold, P.H. Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature 1986, 319, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Trampert, D.C.; Nathanson, M.H. Regulation of bile secretion by calcium signaling in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1761–1770. [Google Scholar] [CrossRef]
- Ueasilamongkol, P.; Khamphaya, T.; Guerra, M.T.; Rodrigues, M.A.; Gomes, D.A.; Kong, Y.; Wei, W.; Jain, D.; Trampert, D.C.; Ananthanarayanan, M.; et al. Type 3 Inositol 1,4,5-Trisphosphate Receptor Is Increased and Enhances Malignant Properties in Cholangiocarcinoma. Hepatology 2020, 71, 583–599. [Google Scholar] [CrossRef]
- Case, R.M.; Eisner, D.; Gurney, A.; Jones, O.; Muallem, S.; Verkhratsky, A. Evolution of calcium homeostasis: From birth of the first cell to an omnipresent signalling system. Cell Calcium 2007, 42, 345–350. [Google Scholar] [CrossRef]
- Plattner, H.; Verkhratsky, A. Inseparable tandem: Evolution chooses ATP and Ca2+ to control life, death and cellular signalling. Philos. Trans R Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [Green Version]
- Kheradpezhouh, E.; Ma, L.; Morphett, A.; Barritt, G.J.; Rychkov, G.Y. TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. USA 2014, 111, 3176–3181. [Google Scholar] [CrossRef] [Green Version]
- Echevarria, W.; Leite, M.F.; Guerra, M.T.; Zipfel, W.R.; Nathanson, M.H. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003, 5, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Rychkov, G.; Brereton, H.M.; Harland, M.L.; Barritt, G.J. Plasma membrane Ca2+ release-activated Ca2+ channels with a high selectivity for Ca2+ identified by patch-clamp recording in rat liver cells. Hepatology 2001, 33, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Kuroda, H.; Oikawa, T.; Okada, Y.; Fujiwara, Y.; Abe, T.; Sato, H.; Sawara, K.; Takikawa, Y. Efficacy of combination therapy with transcatheter arterial chemoembolization and radiofrequency ablation for intermediate-stage hepatocellular carcinoma. Scand. J. Gastroenterol. 2018, 53, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Friedman, S.L.; Goossens, N.; Hoshida, Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J. Hepatol. 2018, 68, 526–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P.; Sunkara, T.; Muralidharan, P.; Raj, J.P. Update in global trends and aetiology of hepatocellular carcinoma. Contemp. Oncol. 2018, 22, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Lafitte, M.; Sirvent, A.; Roche, S. Collagen Kinase Receptors as Potential Therapeutic Targets in Metastatic Colon Cancer. Front. Oncol. 2020, 10, 125. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837 e1822. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, E.S.M.; Rodrigues, P.D.; Alvares-da-Silva, M.R.; Scaffaro, L.A.; Farenzena, M.; Teixeira, U.F.; Waechter, F.L. Treatment strategies for locally advanced hepatocellular carcinoma. Transl. Gastroenterol. Hepatol. 2019, 4, 12. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yan, G.; Zheng, L.; Zhou, Y.; Sheng, H.; Wu, L.; Zhang, Q.; Lei, J.; Zhang, J.; Xin, R.; et al. STIM1 is a metabolic checkpoint regulating the invasion and metastasis of hepatocellular carcinoma. Theranostics 2020, 10, 6483–6499. [Google Scholar] [CrossRef]
- Bayo, J.; Fiore, E.J.; Dominguez, L.M.; Real, A.; Malvicini, M.; Rizzo, M.; Atorrasagasti, C.; Garcia, M.G.; Argemi, J.; Martinez, E.D.; et al. A comprehensive study of epigenetic alterations in hepatocellular carcinoma identifies potential therapeutic targets. J. Hepatol. 2019, 71, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M.; Fujimoto, A. Genome sequencing analysis of liver cancer for precision medicine. Semin Cancer Biol. 2019, 55, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Frades, I.; Andreasson, E.; Mato, J.M.; Alexandersson, E.; Matthiesen, R.; Martinez-Chantar, M.L. Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic Fatty liver disease. PLoS ONE 2015, 10, e0124544. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.K.; Ng, I.O. Joining the dots for better liver cancer treatment. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 74–75. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, X.; Qiu, H.; Peng, Z. An investigation of efficacy, safety, and prognostic factors of drug-eluting beads-transarterial chemoembolization operation with CalliSpheres((R)) Microspheres in treating Chinese hepatocellular carcinoma patients. J. Clin. Lab. Anal. 2019, 33, e22975. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Long, L.; Yao, Y.; Fang, Z.; Zhang, Z.; Zhang, Y. CalliSpheres Drug-Eluting Bead Transcatheter Arterial Chemoembolization Presents With Better Efficacy and Equal Safety Compared to Conventional TACE in Treating Patients With Hepatocellular Carcinoma. Technol. Cancer Res. Treat 2019, 18, 1533033819830751. [Google Scholar] [CrossRef] [Green Version]
- Lencioni, R.; Petruzzi, P.; Crocetti, L. Chemoembolization of hepatocellular carcinoma. Semin Intervent Radiol. 2013, 30, 3–11. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Caruso, S.; Calatayud, A.L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouze, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Y.; Cai, N.; Ye, L.H.; Zhang, X.D. Transformation of human liver L-O2 cells mediated by stable HBx transfection. Acta Pharmacol. Sin. 2009, 30, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, C.; Qin, J.; Liu, J.; Zheng, C. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015, 29, 4268–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef]
- Hass, H.G.; Vogel, U.; Scheurlen, M.; Jobst, J. Gene-expression Analysis Identifies Specific Patterns of Dysregulated Molecular Pathways and Genetic Subgroups of Human Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 5087–5095. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.; Matchett, K.P.; Dobie, R.; Wilson-Kanamori, J.R.; Henderson, N.C. Single-cell technologies in hepatology: New insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 457–472. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Park, Y.R.; Chun, J.N.; So, I.; Kim, H.J.; Baek, S.; Jeon, J.H.; Shin, S.Y. Data-driven Analysis of TRP Channels in Cancer: Linking Variation in Gene Expression to Clinical Significance. Cancer Genom. Proteom. 2016, 13, 83–90. [Google Scholar]
- Vriens, J.; Janssens, A.; Prenen, J.; Nilius, B.; Wondergem, R. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 2004, 36, 19–28. [Google Scholar] [CrossRef]
- Yang, N.; Tang, Y.; Wang, F.; Zhang, H.; Xu, D.; Shen, Y.; Sun, S.; Yang, G. Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013, 330, 163–169. [Google Scholar] [CrossRef]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Han, P.; Li, M.; Yan, W.; Liu, J.; Liu, J.; He, J.; Tu, W.; Xia, Y.; Zhou, Z.; et al. The histidine-rich calcium binding protein (HRC) promotes tumor metastasis in hepatocellular carcinoma and is upregulated by SATB1. Oncotarget 2015, 6, 6811–6824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuette, D.; Moore, L.M.; Robert, M.E.; Taddei, T.H.; Ehrlich, B.E. Hepatocellular Carcinoma Outcome Is Predicted by Expression of Neuronal Calcium Sensor 1. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Bautista, W.; Lipschitz, J.; McKay, A.; Minuk, G.Y. Cancer Stem Cells are Depolarized Relative to Normal Stem Cells Derived from Human Livers. Ann. Hepatol. 2017, 16, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Badr, S.A.; Fahmi, M.W.; Nomir, M.M.; El-Shishtawy, M.M. Calcium channel alpha2 delta1 subunit as a novel biomarker for diagnosis of hepatocellular carcinoma. Cancer Biol. Med. 2018, 15, 52–60. [Google Scholar]
- Hu, Z.; Cao, X.; Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Cao, Z.; Du, H.; Cheng, X.; et al. Transient receptor potential vanilloid-type 2 targeting on stemness in liver cancer. Biomed. Pharmacother. 2018, 105, 697–706. [Google Scholar] [CrossRef]
- Casciano, J.C.; Bouchard, M.J. Hepatitis B virus X protein modulates cytosolic Ca2+ signaling in primary human hepatocytes. Virus Res. 2018, 246, 23–27. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Ersoy, B.A.; Maner-Smith, K.M.; Li, Y.; Alpertunga, I.; Cohen, D.E. Thioesterase-mediated control of cellular calcium homeostasis enables hepatic ER stress. J. Clin Investig. 2018, 128, 141–156. [Google Scholar] [CrossRef]
- Jung, T.W.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J. Biol. Chem. 2018, 293, 3981–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maus, M.; Cuk, M.; Patel, B.; Lian, J.; Ouimet, M.; Kaufmann, U.; Yang, J.; Horvath, R.; Hornig-Do, H.T.; Chrzanowska-Lightowlers, Z.M.; et al. Store-Operated Ca2+ Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metab. 2017, 25, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Han, D.; Kang, H.G.; Jeong, S.J.; Jo, J.E.; Shin, J.; Kim, D.K.; Park, H.W. Intravenous sustained-release nifedipine ameliorates nonalcoholic fatty liver disease by restoring autophagic clearance. Biomaterials 2019, 197, 1–11. [Google Scholar] [CrossRef]
- Wires, E.S.; Trychta, K.A.; Back, S.; Sulima, A.; Rice, K.C.; Harvey, B.K. High fat diet disrupts endoplasmic reticulum calcium homeostasis in the rat liver. J. Hepatol. 2017, 67, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selli, C.; Erac, Y.; Kosova, B.; Erdal, E.S.; Tosun, M. Silencing of TRPC1 regulates store-operated calcium entry and proliferation in Huh7 hepatocellular carcinoma cells. Biomed. Pharmacother. 2015, 71, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Selli, C.; Pearce, D.A.; Sims, A.H.; Tosun, M. Differential expression of store-operated calcium- and proliferation-related genes in hepatocellular carcinoma cells following TRPC1 ion channel silencing. Mol. Cell Biochem. 2016, 420, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yang, Y.; Xie, R.; Liu, J.; Nie, X.; An, J.; Wen, G.; Liu, X.; Jin, H.; Tuo, B. The NCX1/TRPC6 Complex Mediates TGFbeta-Driven Migration and Invasion of Human Hepatocellular Carcinoma Cells. Cancer Res. 2018, 78, 2564–2576. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Du, H.; Zhang, X.; Xu, X. Pharmacological Inhibition of Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Alleviates Carbon Tetrachloride-Induced Liver Fibrosis in Mice. J. Nippon Med. Sch. 2019, 86, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Bort, A.; Spinola, E.; Rodriguez-Henche, N.; Diaz-Laviada, I. Capsaicin exerts synergistic antitumor effect with sorafenib in hepatocellular carcinoma cells through AMPK activation. Oncotarget 2017, 8, 87684–87698. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kang, Y.S.; Lee, J.S.; Nicolova, S.; Kim, J.A. Involvement of NADPH oxidase-mediated generation of reactive oxygen species in the apototic cell death by capsaicin in HepG2 human hepatoma cells. Free Radic. Res. 2004, 38, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.P.; Chen, J.C.; Wu, C.C.; Chen, C.T.; Tang, N.Y.; Ho, Y.T.; Lo, C.; Lin, J.P.; Chung, J.G.; Lin, J.G. Capsaicin-induced apoptosis in human hepatoma HepG2 cells. Anticancer Res. 2009, 29, 165–174. [Google Scholar] [PubMed]
- Miao, X.; Liu, G.; Xu, X.; Xie, C.; Sun, F.; Yang, Y.; Zhang, T.; Hua, S.; Fan, W.; Li, Q.; et al. High expression of vanilloid receptor-1 is associated with better prognosis of patients with hepatocellular carcinoma. Cancer Genet. Cytogenet. 2008, 186, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef] [Green Version]
- Dou, C.; Zhou, Z.; Xu, Q.; Liu, Z.; Zeng, Y.; Wang, Y.; Li, Q.; Wang, L.; Yang, W.; Liu, Q.; et al. Hypoxia-induced TUFT1 promotes the growth and metastasis of hepatocellular carcinoma by activating the Ca2+/PI3K/AKT pathway. Oncogene 2019, 38, 1239–1255. [Google Scholar] [CrossRef]
- Liu, C.; Gong, K.; Mao, X.; Li, W. Tetrandrine induces apoptosis by activating reactive oxygen species and repressing Akt activity in human hepatocellular carcinoma. Int. J. Cancer 2011, 129, 1519–1531. [Google Scholar] [CrossRef]
- Lan, J.; Wang, N.; Huang, L.; Liu, Y.; Ma, X.; Lou, H.; Chen, C.; Feng, Y.; Pan, W. Design and synthesis of novel tetrandrine derivatives as potential anti-tumor agents against human hepatocellular carcinoma. Eur. J. Med. Chem. 2017, 127, 554–566. [Google Scholar] [CrossRef]
- Meng, Z.; Ma, X.; Du, J.; Wang, X.; He, M.; Gu, Y.; Zhang, J.; Han, W.; Fang, Z.; Gan, X.; et al. CAMK2gamma antagonizes mTORC1 activation during hepatocarcinogenesis. Oncogene 2017, 36, 2446–2456. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
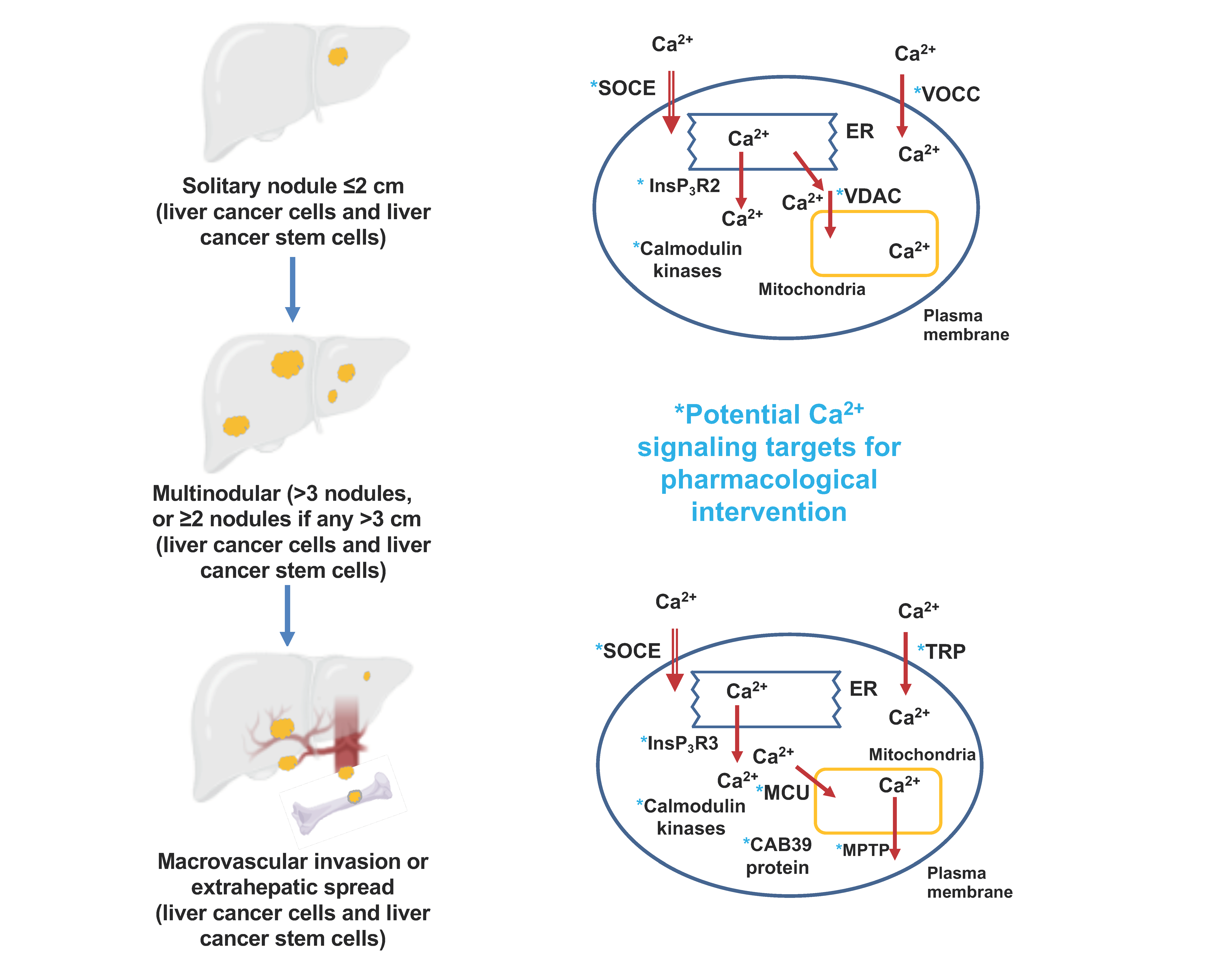
| Stage of Hepatocellular Carcinoma | Targeted Ca2+ Signaling Process | Specific Protein Targeted | Proposed Pharmacological Intervention | References |
|---|---|---|---|---|
| Liver cancer stem cells: initiation, promotion and resistance to systemic drugs | Ca2+ entry | L- and N-type voltage operated Ca2+ channels | Inhibition | [13] |
| T-type voltage operated Ca2+ channels | Activation | [14] | ||
| TRPV2 channels | Activation | [15] | ||
| Ca2+ release from the endoplasmic reticulum | InsP3R2 | Inhibition | [16] | |
| Ca2+-binding proteins | CaMKIIγ | Activation | [15] | |
| Hepatitis B and C infection: initiation and promotion | Ca2+ uptake by mitochondria | Ca2+ transfer from endoplasmic reticulum to mitochondria (voltage-dependent anion channel, VDAC) | Inhibition | [17,18] |
| Ca2+ entry | Store-operated Ca2+ entry | Inhibition | [19] | |
| Non-alcoholic fatty liver disease: initiation and promotion | Ca2+ entry | Orai1 | Activation | [5,20] |
| STIM1 | ||||
| Ca2+ entry | Voltage-operated Ca2+ channels | Inhibition (verapamil) | [21] | |
| Ca2+ uptake by the endoplasmic reticulum | SERCA2b | Activation | [22,23] | |
| Ca2+-binding proteins | CaMKII | Inhibition | [24] | |
| CaMKK2 | Inhibition | [25] | ||
| Progression, migration and metastasis | Ca2+ entry | Orai1 | Inhibition | [26,27] |
| STIM1 | ||||
| TRPC6 | Inhibition | [28,29] | ||
| TRPV4 | Inhibition | [30] | ||
| TRPV1 | Activation | [31,32] | ||
| Ca2+ release from the endoplasmic reticulum | InsP3R3 | Inhibition | [33] | |
| Uptake and release of Ca2+ from mitochondria | Mitochondrial uniporter (MCU) and MCU regulator protein 1 (Ca2+ uptake to mitochondria) | Inhibition | [34] | |
| Mitochondrial permeability transition pore (Ca2+ release from mitochondria) | Activation | [35] | ||
| Mitofusin-2 | Activation | [36] | ||
| Progression, migration and metastasis | Ca2+-binding proteins | CaMKIIγ | Inhibition (tetrandrine, berbamine) | [15,37] |
| CaMKIV | Activation | [38] | ||
| Ca2+-binding protein 39 | Inhibition | [39] | ||
| Increased [Ca2+]cyt (tuftelin1) | Inhibition | [39] |
| Model | Attributes of the Model a |
|---|---|
| Isolated hepatocytes either freshly isolated or in culture | Hepatocytes isolated from human non-diseased liver tissue, subsequently grown in culture for periods of about 1 h to 5 days. |
| Immortalized human liver cell lines derived from non-diseased human liver | L01 and L02 (HL-7702) liver cells. Immortalized cells originally obtained from normal fetal or adult human liver. |
| Human HCC cells lines derived from human HCC tissue | Commonly used: HepG2 cells and Huh-7 cells |
| Examples of other HCC cell lines include: MHCC97H, SK-Hep-1, SNU398, PLC/PRF/5, SMMC-7721. | |
| Liver cancer stem cells | Hep-12 cells which exhibit liver cancer stem cell marker proteins and Hep-11 control cells which do not exhibit liver cancer stem cell marker proteins; |
| Subsets of HCC cells (often HepG2 cells and Huh-7 cells) which exhibit liver cancer stem cell marker proteins. | |
| Mouse liver HCC model in which HCC tumors are induced by a chemical mutagen | Diethylnitrosamine (DEN)-induced liver tumors |
| Subcutaneous mouse xenograft models | HCC cells (immortalized cell line or cells isolated from human liver HCC tissue) implanted subcutaneously into immunodeficient mice: Nude mice, severe combined immunodeficient (SCID) mice, and non-obese diabetic-severe combined immunodeficiency disease (NOD/SCID) mice |
| Ca2+-Signaling Pathway | Gene | Protein | References |
|---|---|---|---|
| Ca2+ entry channels in plasma membrane | CACNA1B | Voltage-dependent N-type Ca2+ channel subunit α-1B | [12] |
| CACNA1E | Voltage-dependent R-type Ca2+ channel subunit α-1E | ||
| CACNA1H | Voltage-dependent T-type Ca2+ channel subunit α-1 H | [11] | |
| CACNA1I | Voltage-dependent T-type Ca2+ channel subunit α-1I | [12] | |
| CACNA1A | Voltage-dependent P/Q-type Ca2+ channel subunit α-1A | ||
| CACNA1C | Voltage-dependent L-type Ca2+ channel subunit α-1C | ||
| CACNA1D | Voltage-dependent L-type Ca2+ channel subunit α-1D | [11] | |
| CACNA1G | Voltage-dependent T-type Ca2+ channel subunit α-1G | [11,12,74] | |
| CACNA1S | Voltage-dependent L-type Ca2+ channel subunit α-1S | ||
| ORAI1 | Calcium release-activated Ca2+ channel protein 1 | ||
| Ca2+ transporters and exchange proteins in plasma membrane | SLC8A1 | Na+ -Ca2+ exchanger 1 | [11,12,74] |
| SLC8A2 | Na+ -Ca2+ exchanger 2 | ||
| ATP2B1 | Plasma membrane Ca2+-transporting ATPase 1 (PMCA1) | ||
| ATP2B2 | Plasma membrane Ca2+-transporting ATPase 2 (PMCA2) | ||
| ATP2B3 | Plasma membrane Ca2+-transporting ATPase 3 (PMCA3) | ||
| ATP2B4 | Plasma membrane Ca2+-transporting ATPase 4 (PMCA4) | ||
| Ca2+ channels and transporters in endoplasmic reticulum | ITPR1 | Inositol 1,4,5-trisphosphate receptor type 1 (IP3R 1) | [12] |
| ITPR2 | Inositol 1,4,5-trisphosphate receptor type 2 (IP3R 2) | [11,12] | |
| ITPR3 | Inositol 1,4,5-trisphosphate receptor type 3 (IP3R 3) | [33] | |
| STIM1 | Stromal interaction molecule 1 | [11,12,33] | |
| STIM2 | Stromal interaction molecule 2 | ||
| RYR1 | Ryanodine receptor 1 | ||
| RYR2 | Ryanodine receptor 2 | [11] | |
| RYR3 | Ryanodine receptor 3 | [12] | |
| ATP2A1 | Sarcoplasmic/endoplasmic reticulum Ca2+ATPase 1 (SERCA1) | ||
| ATP2A2 | Sarcoplasmic/endoplasmic reticulum Ca2+ATPase 2 (SERCA2) | ||
| ATP2A3 | Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 3 (SERCA3) | ||
| Ca2+-binding proteins | CALML3 | Calmodulin-like protein 3 (CaM-like protein) | [12] |
| PTK2B | Ca2+-dependent tyrosine kinase 2β | [12] | |
| S100A8, S100A9, S100A11, S100P | S100 Ca2+-binding protein A8, A9, A11 and P | [75] | |
| ITPKB | Inositol-trisphosphate 3-kinase B | [12] | |
| PDF1A | Ca2+/calmodulin-dependent 3’,5’-cyclic nucleotide phosphodiesterase 1A | ||
| PDE1C | Ca2+/calmodulin-dependent 3’,5’-cyclic nucleotide phosphodiesterase 1C | ||
| PDE1B | Ca2+/calmodulin-dependent 3’,5’-cyclic nucleotide phosphodiesterase 1B | ||
| PPP3CB | Calmodulin-dependent calcineurin A subunitβ isoform | ||
| PPP3CC | Calmodulin-dependent calcineurin A subunit γ isoform |
| Ca2+ Signaling Pathway | Gene | Protein | Change in Protein Expression | References |
|---|---|---|---|---|
| Ca2+ channels and transporters in plasma membrane | ORAI1 | Orai 1 | Increased | [27] |
| CACNA1H | Voltage-operated Ca2+ channel subunit α-1 H | Increased | [77] | |
| TRPC6 | Transient receptor potential cation channel subfamily C member 6 | Increased | [28] | |
| TRPM2 | Transient receptor potential cation channel subfamily M member 2 | Increased | [78] | |
| TRPV2 | Transient receptor potential cation channel subfamily V member 2 | Increased | [79] | |
| TRPV4 | Transient receptor potential cation channel subfamily V member 4 | Decreased | [78] | |
| Ca2+ channels and transporters in endoplasmic reticulum | STIM1 | Stromal interaction molecule 1 | Increased | [80] |
| SERCA2 | Sarco/endoplasmic reticulum (Ca2+, Mg2+) ATP-ase | Decreased (in non-alcoholic steatohepatitis-induced HCC) | [23] | |
| Ca2+ channels and transporters in mitochondria | MCUR1 | Mitochondrial Ca2+ uniporter regulator 1 | Increased | [81] |
| Ca2+-binding proteins | HRC | Histidine-rich Ca2+-binding protein | Increased | [82] |
| NCS1 | Neuronal Ca2+ sensor 1 | Increased | [83] | |
| CAB39 | Ca2+-binding protein 39 | Increased | [39] |
| Proposed Ca2+ Transporter, Channel or Ca2+-Binding Protein | Proposed Intervention Strategy | References |
|---|---|---|
| SERCA2b | Activation using small molecule activator such as the allosteric activator CDN1163 | [22] |
| Activation by modification of ER membrane fluidity affected by altering thioesterase superfamily member 2/phosphatidyl transfer protein | [90] | |
| Increased expression induced by maresin 1 leading to increased AMPK activity | [91] | |
| Activation by modulation of the SERCA2b regulator protein Cisd2 | [23] | |
| Ca2+ entry | Activation of store-operated Ca2+ entry by small molecule activator of Orai1 or STIM1. | [20] |
| Inhibition of PKC leading to de-phosphorylation and activation of Orai1 | [92] | |
| Inhibition of Ca2+ entry using Ca2+-channel blockers verapamil and nifedipine | [21,93] | |
| Ryanodine receptors (RYR1 and RYR2) | Activation using small molecules such as caffeine and caffeine analogs | [11] |
| InsP3R | Inhibition using small molecule inhibitors such as heparin and caffeine | [11,94] |
| CaMKII | Inhibition by natural product tetrandrine of phosphorylation of CaMKII | [37] |
| CaMKK2 | Inhibition using small molecule inhibitor such as STO-609 | [25] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, E.S.; Rychkov, G.Y.; Barritt, G.J. Targeting Ca2+ Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma. Cancers 2020, 12, 2755. https://doi.org/10.3390/cancers12102755
Ali ES, Rychkov GY, Barritt GJ. Targeting Ca2+ Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma. Cancers. 2020; 12(10):2755. https://doi.org/10.3390/cancers12102755
Chicago/Turabian StyleAli, Eunus S., Grigori Y. Rychkov, and Greg J. Barritt. 2020. "Targeting Ca2+ Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma" Cancers 12, no. 10: 2755. https://doi.org/10.3390/cancers12102755
APA StyleAli, E. S., Rychkov, G. Y., & Barritt, G. J. (2020). Targeting Ca2+ Signaling in the Initiation, Promotion and Progression of Hepatocellular Carcinoma. Cancers, 12(10), 2755. https://doi.org/10.3390/cancers12102755