Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
Fatty Acids Metabolism Importance in Cancer Progression: Epigenetic Processes as Novel Integral Approaches for Prevention and Treatment
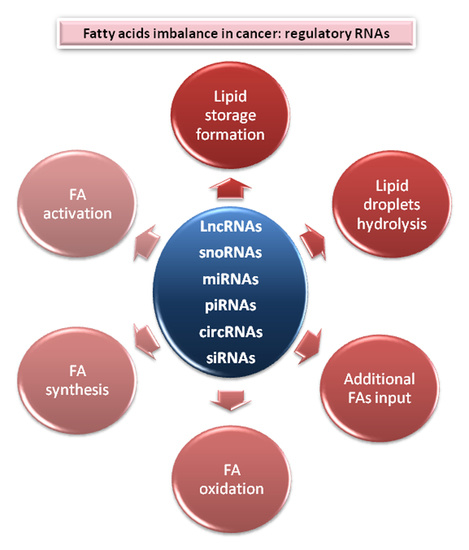
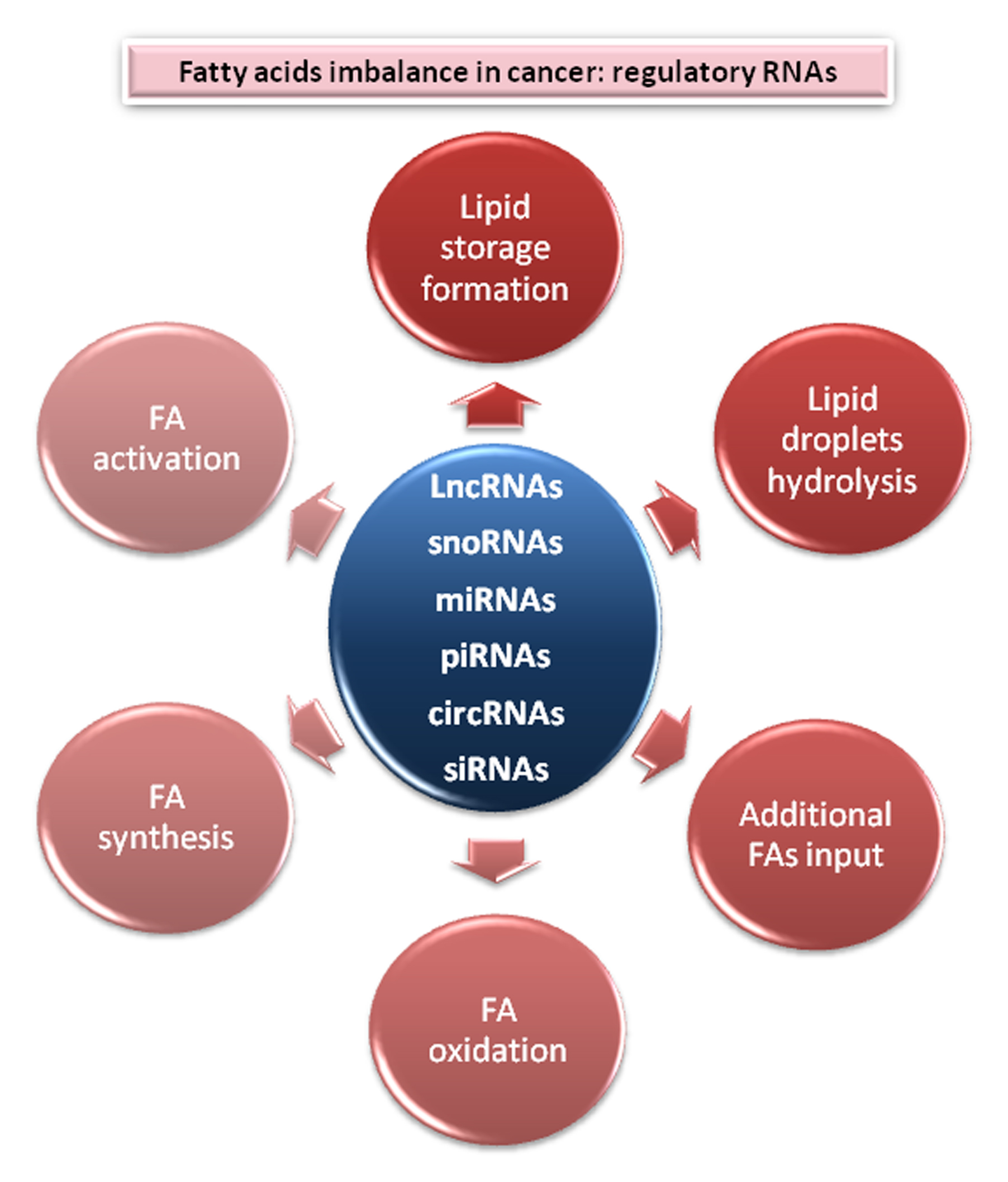
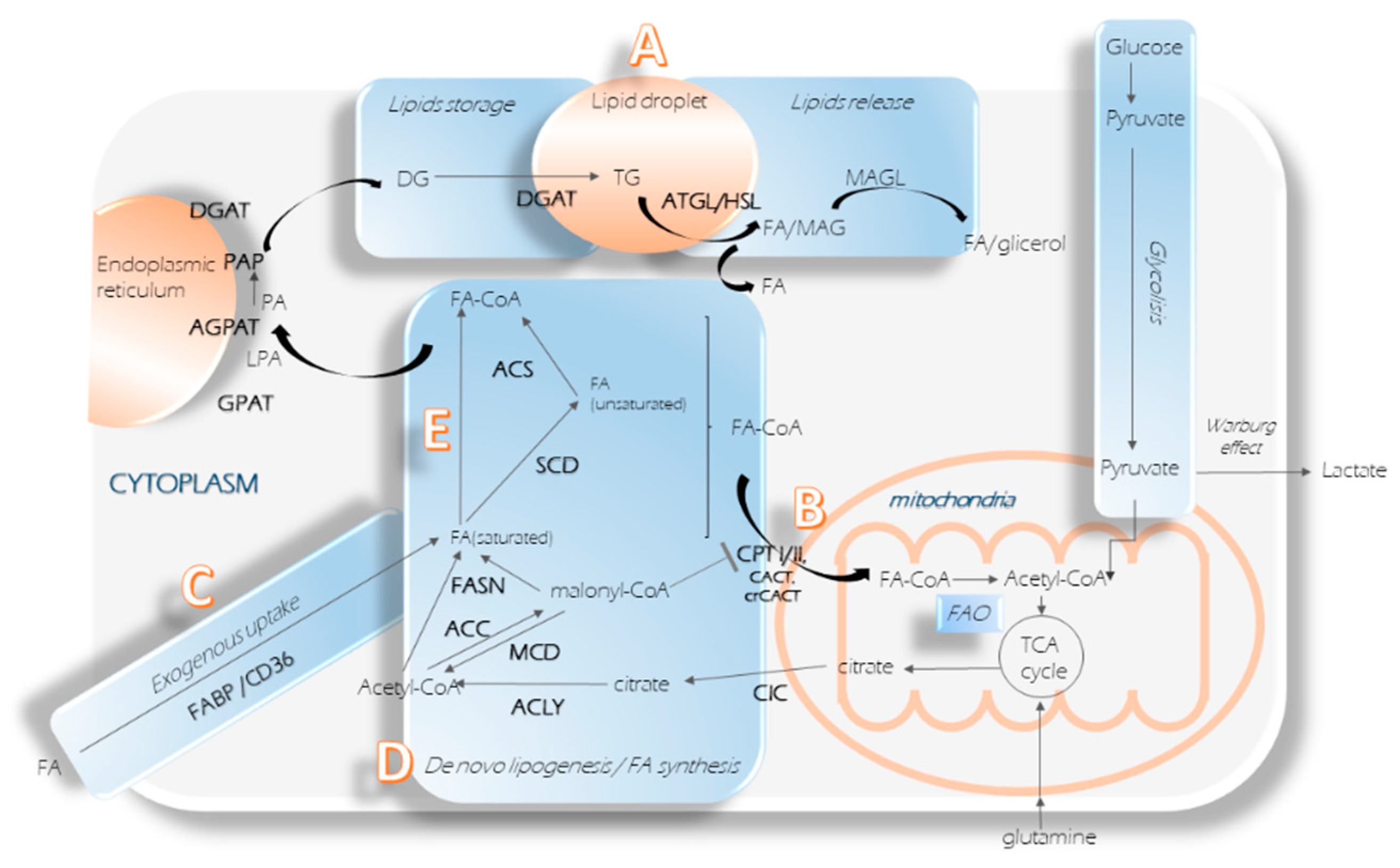
2. Central Nodes to Control Fatty Acids Imbalance in Cancer
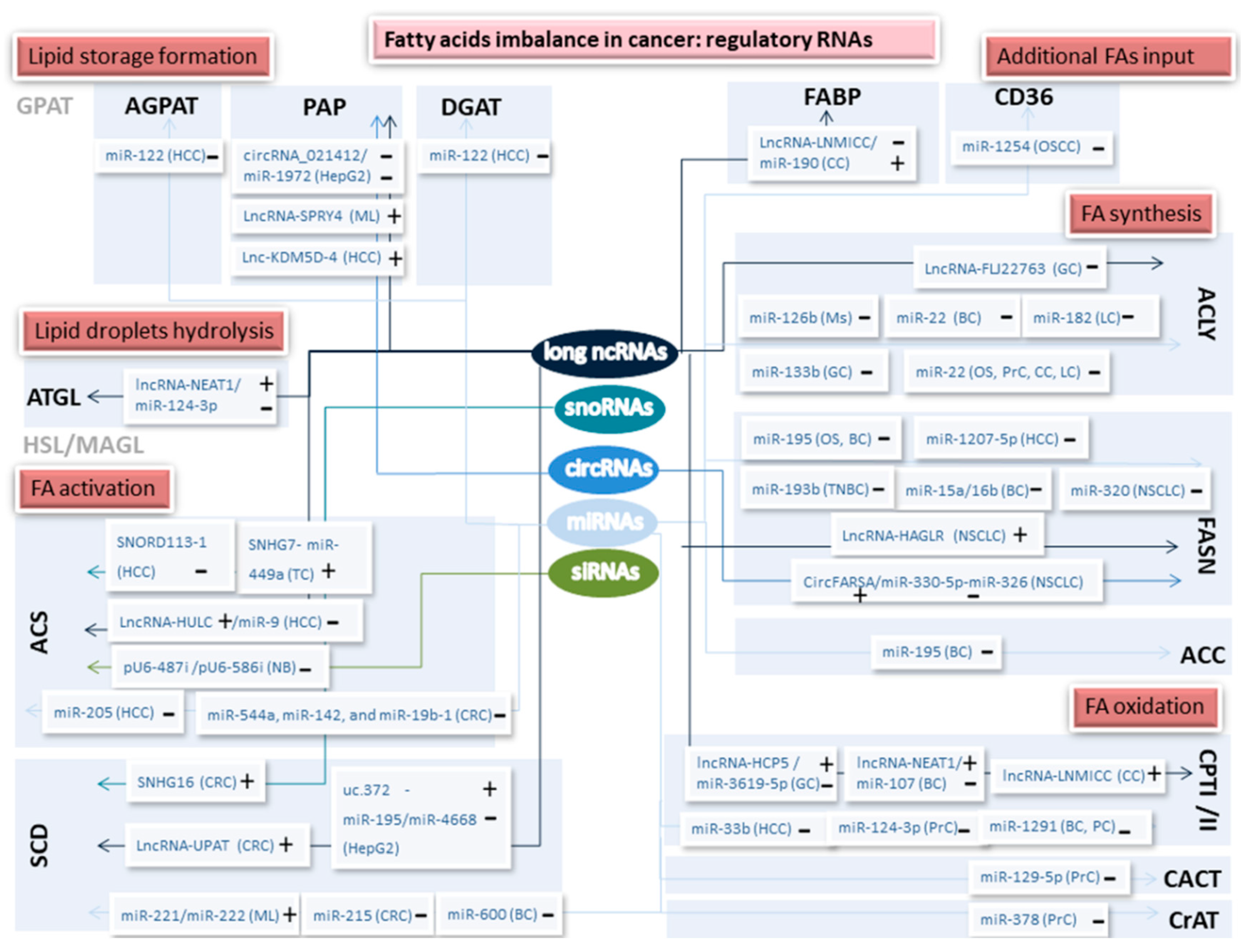
2.1. Lipids Store Formation
2.2. Lipid Droplets Hydrolysis
2.3. Fatty Acids Oxidation
2.4. Additional FAs Input
2.5. Fatty Acid Synthesis Enzymes
2.6. Fatty Acid Activation and Desaturation
2.6.1. Acyl–CoA Synthetases
2.6.2. Stearoyl-CoA Desaturase
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Cruz Gil, S. Lipid Metabolism Alterations in Colorectal Cancer: Potential Clinical Relevance in the Prognosis of the Disease. Ph.D Thesis, Universidad Autónoma de Madrid, Madrid, Spain, 2019. [Google Scholar]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Ookhtens, M.; Kannan, R.; Lyon, I.; Baker, N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am. J. Physiol. 1984, 247, R146–R153. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Zaidi, N.; Lupien, L.; Kuemmerle, N.B.; Kinlaw, W.B.; Swinnen, J.V.; Smans, K. Lipogenesis and lipolysis: The pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid Res. 2013, 52, 585–589. [Google Scholar] [CrossRef]
- Daniëls, V.W.; Smans, K.; Royaux, I.; Chypre, M.; Swinnen, J.V.; Zaidi, N. Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS ONE 2014, 9, e106913. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Fidalgo, A.; Varanda, A.S.; Oliveira, C.; Santos, M.A.S. tRNA Deregulation and Its Consequences in Cancer. Trends Mol. Med. 2019, 25, 853–865. [Google Scholar] [CrossRef]
- Bratkovič, T.; Božič, J.; Rogelj, B. Functional diversity of small nucleolar RNAs. Nucleic Acids Res. 2020, 48, 1627–1651. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, M.J.; Kwon, I.C.; Roberts, T.M. Delivery Strategies and Potential Targets for siRNA in Major Cancer Types. Adv. Drug Deliv. Rev. 2016, 104, 2–15. [Google Scholar] [CrossRef]
- Gómez de Cedrón, M.; Ramírez de Molina, A. Microtargeting cancer metabolism: Opening new therapeutic windows based on lipid metabolism. J. Lipid Res. 2016, 57, 193–206. [Google Scholar] [CrossRef]
- Yu, M.; Lu, B.; Zhang, J.; Ding, J.; Liu, P.; Lu, Y. tRNA-derived RNA fragments in cancer: Current status and future perspectives. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, J.; Hann, S.S. The emerging roles of PIWI-interacting RNA in human cancers. Cancer Manag. Res. 2019, 11, 5895–5909. [Google Scholar] [CrossRef]
- Patop, I.L.; Wüst, S.; Kadener, S. Past, present, and future of circRNAs. EMBO J. 2019, 38, e100836. [Google Scholar] [CrossRef] [PubMed]
- Vo, J.N.; Cieslik, M.; Zhang, Y.; Shukla, S.; Xiao, L.; Zhang, Y.; Wu, Y.-M.; Dhanasekaran, S.M.; Engelke, C.G.; Cao, X.; et al. The Landscape of Circular RNA in Cancer. Cell 2019, 176, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Hann, S.S. Biological Roles and Mechanisms of Circular RNA in Human Cancers. Oncol. Targets Ther. 2020, 13, 2067–2092. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Raymond, V.-A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle Georget. Tex. 2018, 17, 903–916. [Google Scholar] [CrossRef]
- Hsu, S.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef]
- Guo, X.-Y.; He, C.-X.; Wang, Y.-Q.; Sun, C.; Li, G.-M.; Su, Q.; Pan, Q.; Fan, J.-G. Circular RNA Profiling and Bioinformatic Modeling Identify Its Regulatory Role in Hepatic Steatosis. Available online: https://www.hindawi.com/journals/bmri/2017/5936171/ (accessed on 13 July 2020).
- Mazar, J.; Zhao, W.; Khalil, A.M.; Lee, B.; Shelley, J.; Govindarajan, S.S.; Yamamoto, F.; Ratnam, M.; Aftab, M.N.; Collins, S.; et al. The Functional Characterization of Long Noncoding RNA SPRY4-IT1 in Human Melanoma Cells. Oncotarget 2014, 5, 8959–8969. [Google Scholar] [CrossRef]
- Molina, E.; Chew, G.S.; Myers, S.A.; Clarence, E.M.; Eales, J.M.; Tomaszewski, M.; Charchar, F.J. A Novel Y-Specific Long Non-Coding RNA Associated with Cellular Lipid Accumulation in HepG2 cells and Atherosclerosis-related Genes. Sci. Rep. 2017, 7, 16710. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef]
- Zare, M.; Panahi, G.; Koushki, M.; Mostafavi-Pour, Z.; Meshkani, R. Metformin reduces lipid accumulation in HepG2 cells via downregulation of miR-33b. Arch. Physiol. Biochem. 2019, 1–8. [Google Scholar] [CrossRef]
- Wu, H.; Liu, B.; Chen, Z.; Li, G.; Zhang, Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1α/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 2020, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Z.; Li, Z.; Wang, S.; Shen, N.; Xin, Y.; Huang, T. Long non-coding RNA nuclear paraspeckle assembly transcript 1 interacts with microRNA-107 to modulate breast cancer growth and metastasis by targeting carnitine palmitoyltransferase-1. Int. J. Oncol. 2019, 55, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Wang, W.; Liao, Y.; Chen, Y.; Liu, T.; Du, Q.; Huang, J.; Liang, Y.; Liu, J.; Zhao, Y.; et al. LNMICC Promotes Nodal Metastasis of Cervical Cancer by Reprogramming Fatty Acid Metabolism. Cancer Res. 2018, 78, 877–890. [Google Scholar] [CrossRef]
- Valentino, A.; Calarco, A.; Di Salle, A.; Finicelli, M.; Crispi, S.; Calogero, R.A.; Riccardo, F.; Sciarra, A.; Gentilucci, A.; Galderisi, U.; et al. Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: Molecular basis and pathophysiological consequences. Oncogene 2017, 36, 6030–6040. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, A.; Valentino, A.; Calarco, A.; Maggi, M.; Salciccia, S.; Peluso, G.; Sciarra, A. Deregulation of micrornas mediated control of carnitine cycle in prostate cancer: Molecular basis and pathophysiological consequences. Eur. Urol. Suppl. 2018, 17, 168–169. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Y.; Han, F.; Zhao, Y.; Tu, M.; Wang, Y.; Huang, C.; Fan, S.; Chen, P.; Yao, X.; et al. A novel miR-1291-ERRα-CPT1C axis modulates tumor cell proliferation, metabolism and tumorigenesis. Theranostics 2020, 10, 7193–7210. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, Y.; Zhang, X. MiR-1254 Functions as a Tumor Suppressor in Oral Squamous Cell Carcinoma by Targeting CD36. Technol. Cancer Res. Treat. 2019, 18, 1533033819859447. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, Q.; Lu, J.; Ma, G.; Ge, Y.; Chu, H.; Du, M.; Wang, M.; Zhang, Z. Long non-coding RNA FLJ22763 is involved in the progression and prognosis of gastric cancer. Gene 2019, 693, 84–91. [Google Scholar] [CrossRef]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 Suppresses Mesothelioma Malignancy by Targeting IRS1 and Interfering with the Mitochondrial Function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: Evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef]
- Koufaris, C.; Valbuena, G.N.; Pomyen, Y.; Tredwell, G.D.; Nevedomskaya, E.; Lau, C.-H.; Yang, T.; Benito, A.; Ellis, J.K.; Keun, H.C. Systematic integration of molecular profiles identifies miR-22 as a regulator of lipid and folate metabolism in breast cancer cells. Oncogene 2016, 35, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, M.; Hu, J.; Lei, R.; Xiong, H.; Ji, H.; Yin, H.; Wei, Q.; Hu, G. The microRNA-182-PDK4 axis regulates lung tumorigenesis by modulating pyruvate dehydrogenase and lipogenesis. Oncogene 2017, 36, 989–998. [Google Scholar] [CrossRef]
- Cheng, Y.; Jia, B.; Wang, Y.; Wan, S. miR-133b acts as a tumor suppressor and negatively regulates ATP citrate lyase via PPARγ in gastric cancer. Oncol. Rep. 2017, 38, 3220–3226. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Yadav, V.; Kumar, S.; Saini, N. MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci. Rep. 2015, 5, 17454. [Google Scholar] [CrossRef]
- Song, J.; Wu, X.; Liu, F.; Li, M.; Sun, Y.; Wang, Y.; Wang, C.; Zhu, K.; Jia, X.; Wang, B.; et al. Long non-coding RNA PVT1 promotes glycolysis and tumor progression by regulating miR-497/HK2 axis in osteosarcoma. Biochem. Biophys. Res. Commun. 2017, 490, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, F.; Zhao, J.; Li, B.; Liang, Y.; Pan, W.; Zhang, S.; Wang, X.; Zheng, D. Long non-coding RNA PVT1 promotes osteosarcoma development by acting as a molecular sponge to regulate miR-195. Oncotarget 2016, 7, 82620–82633. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ma, J.; Cai, D. Increased HAGLR expression promotes non-small cell lung cancer proliferation and invasion via enhanced de novo lipogenesis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317697574. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, X.; Shi, J.; Cao, P.; Wan, M.; Zhang, Q.; Wang, Y.; Kridel, S.J.; Liu, W.; Xu, J.; et al. Fatty acid synthase is a primary target of MiR-15a and MiR-16-1 in breast cancer. Oncotarget 2016, 7, 78566–78576. [Google Scholar] [CrossRef]
- Wahdan-Alaswad, R.S.; Cochrane, D.R.; Spoelstra, N.S.; Howe, E.N.; Edgerton, S.M.; Anderson, S.M.; Thor, A.D.; Richer, J.K. Metformin-induced killing of triple-negative breast cancer cells is mediated by reduction in fatty acid synthase via miRNA-193b. Horm. Cancer 2014, 5, 374–389. [Google Scholar] [CrossRef]
- Zhao, G.; Dong, L.; Shi, H.; Li, H.; Lu, X.; Guo, X.; Wang, J. MicroRNA-1207-5p inhibits hepatocellular carcinoma cell growth and invasion through the fatty acid synthase-mediated Akt/mTOR signalling pathway. Oncol. Rep. 2016, 36, 1709–1716. [Google Scholar] [CrossRef]
- Mao, J.H.; Zhou, R.P.; Peng, A.F.; Liu, Z.L.; Huang, S.H.; Long, X.H.; Shu, Y. microRNA-195 suppresses osteosarcoma cell invasion and migration in vitro by targeting FASN. Oncol. Lett. 2012, 4, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Zhu, Y.; Jiang, C.; Wang, Y.; Fu, J.; Fan, Z.; Qin, H. MicroRNA-320 was downregulated in non-small cell lung cancer and inhibited cell proliferation, migration and invasion by targeting fatty acid synthase. Mol. Med. Rep. 2016, 14, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Hang, D.; Zhou, J.; Qin, N.; Zhou, W.; Ma, H.; Jin, G.; Hu, Z.; Dai, J.; Shen, H. A novel plasma circular RNA circFARSA is a potential biomarker for non-small cell lung cancer. Cancer Med. 2018, 7, 2783–2791. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Cheng, M.; Niu, Y.; Chi, X.; Liu, X.; Fan, J.; Fan, H.; Chang, Y.; Yang, W. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c. Int. J. Biol. Sci. 2017, 13, 349–357. [Google Scholar] [CrossRef]
- Wu, H.; Ng, R.; Chen, X.; Steer, C.J.; Song, G. MicroRNA-21 is a potential link between non-alcoholic fatty liver disease and hepatocellular carcinoma via modulation of the HBP1-p53-Srebp1c pathway. Gut 2016, 65, 1850–1860. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.-T.; Josson, S.; Mukhopadhyay, N.K.; Kim, J.; Freeman, M.R.; Huang, W.-C. MicroRNA-185 and 342 inhibit tumorigenicity and induce apoptosis through blockade of the SREBP metabolic pathway in prostate cancer cells. PLoS ONE 2013, 8, e70987. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; He, J.; Zhou, W.; Xiang, G.; Xu, R. MicroRNA-132 cause apoptosis of glioma cells through blockade of the SREBP-1c metabolic pathway related to SIRT1. Biomed. Pharmacother. 2016, 78, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, Z.; Huang, R.; Xia, Z.; Xiang, G.; Zhang, J. MicroRNA-449 suppresses proliferation of hepatoma cell lines through blockade lipid metabolic pathway related to SIRT1. Int. J. Oncol. 2014, 45, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Wang, Y.; Sun, B.; Xiao, Z.; Ye, L.; Zhang, X. MiR-205 modulates abnormal lipid metabolism of hepatoma cells via targeting acyl-CoA synthetase long-chain family member 1 (ACSL1) mRNA. Biochem. Biophys. Res. Commun. 2014, 444, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lu, J.; Gao, J.; Li, M.; Wang, H.; Zhan, X. The function of SNHG7/miR-449a/ACSL1 axis in thyroid cancer. J. Cell. Biochem. 2020. [Google Scholar] [CrossRef]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Xiao, Z.; Sun, B.; Wang, Y.; Zheng, M.; Ye, L.; Zhang, X. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochem. Biophys. Res. Commun. 2014, 445, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Yang, F.; Ding, C.-L.; Zhao, L.-J.; Ren, H.; Zhao, P.; Wang, W.; Qi, Z.-T. Small nucleolar RNA 113–1 suppresses tumorigenesis in hepatocellular carcinoma. Mol. Cancer 2014, 13. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gil, S.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Martin-Hernandez, R.; Vargas, T.; Molina, S.; Herranz, J.; Davalos, A.; Reglero, G.; Ramirez de Molina, A. Targeting the lipid metabolic axis ACSL/SCD in colorectal cancer progression by therapeutic miRNAs: miR-19b-1 role. J. Lipid Res. 2018, 59, 14–24. [Google Scholar] [CrossRef] [PubMed]
- MicroRNA-Dependent Regulation of Transcription in Non-Small Cell Lung Cancer. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0090524 (accessed on 15 July 2020).
- Angius, A.; Uva, P.; Pira, G.; Muroni, M.R.; Sotgiu, G.; Saderi, L.; Uleri, E.; Caocci, M.; Ibba, G.; Cesaraccio, M.R.; et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int. J. Mol. Sci. 2019, 20, 4067. [Google Scholar] [CrossRef]
- Kim, H.C.; Lee, S.-W.; Cho, Y.-Y.; Lim, J.M.; Ryoo, Z.Y.; Lee, E.J. RNA interference of long-chain acyl-CoA synthetase 6 suppresses the neurite outgrowth of mouse neuroblastoma NB41A3 cells. Mol. Med. Rep. 2009, 2, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ding, Y.; Yao, J.; Wei, Z.; Jin, H.; Chen, C.; Feng, J.; Ying, R. miR-215 Inhibits Colorectal Cancer Cell Migration and Invasion via Targeting Stearoyl-CoA Desaturase. Comput. Math. Methods Med. 2020, 2020. [Google Scholar] [CrossRef]
- El Helou, R.; Pinna, G.; Cabaud, O.; Wicinski, J.; Bhajun, R.; Guyon, L.; Rioualen, C.; Finetti, P.; Gros, A.; Mari, B.; et al. miR-600 Acts as a Bimodal Switch that Regulates Breast Cancer Stem Cell Fate through WNT Signaling. Cell Rep. 2017, 18, 2256–2268. [Google Scholar] [CrossRef]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef]
- Guo, J.; Fang, W.; Sun, L.; Lu, Y.; Dou, L.; Huang, X.; Tang, W.; Yu, L.; Li, J. Ultraconserved element uc.372 drives hepatic lipid accumulation by suppressing miR-195/miR4668 maturation. Nat. Commun. 2018, 9, 612. [Google Scholar] [CrossRef]
- Christensen, L.L.; True, K.; Hamilton, M.P.; Nielsen, M.M.; Damas, N.D.; Damgaard, C.K.; Ongen, H.; Dermitzakis, E.; Bramsen, J.B.; Pedersen, J.S.; et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol. Oncol. 2016, 10, 1266–1282. [Google Scholar] [CrossRef]
- Puglisi, R.; Bellenghi, M.; Pontecorvi, G.; Gulino, A.; Petrini, M.; Felicetti, F.; Bottero, L.; Mattia, G.; Carè, A. SCD5 restored expression favors differentiation and epithelial-mesenchymal reversion in advanced melanoma. Oncotarget 2018, 9, 7567–7581. [Google Scholar] [CrossRef]
- Vanni, S. Intracellular Lipid Droplets: From Structure to Function. Lipid Insights 2017, 10. [Google Scholar] [CrossRef]
- Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo-Calvo, D.; López-Vilaró, L.; Nasarre, L.; Perez-Olabarria, M.; Vázquez, T.; Escuin, D.; Badimon, L.; Barnadas, A.; Lerma, E.; Llorente-Cortés, V. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: A molecular and clinicopathological study. BMC Cancer 2015, 15, 460. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.-Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Tirinato, L.; Pagliari, F.; Limongi, T.; Marini, M.; Falqui, A.; Seco, J.; Candeloro, P.; Liberale, C.; Di Fabrizio, E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Bhujwalla, Z.M.; Glunde, K. Targeting Phospholipid Metabolism in Cancer. Front. Oncol. 2016, 6. [Google Scholar] [CrossRef]
- Yamashita, A.; Hayashi, Y.; Matsumoto, N.; Nemoto-Sasaki, Y.; Oka, S.; Tanikawa, T.; Sugiura, T. Glycerophosphate/Acylglycerophosphate acyltransferases. Biology 2014, 3, 801–830. [Google Scholar] [CrossRef] [PubMed]
- Marchan, R.; Büttner, B.; Lambert, J.; Edlund, K.; Glaeser, I.; Blaszkewicz, M.; Leonhardt, G.; Marienhoff, L.; Kaszta, D.; Anft, M.; et al. Glycerol-3-phosphate Acyltransferase 1 Promotes Tumor Cell Migration and Poor Survival in Ovarian Carcinoma. Cancer Res. 2017, 77, 4589–4601. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.M.; Paul, D.S.; Depetrillo, M.A.; Singh, B.P.; Malarkey, D.E.; Coleman, R.A. Mice deficient in glycerol-3-phosphate acyltransferase-1 have a reduced susceptibility to liver cancer. Toxicol. Pathol. 2012, 40, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Pellon-Maison, M.; Montanaro, M.A.; Lacunza, E.; Garcia-Fabiani, M.B.; Soler-Gerino, M.C.; Cattaneo, E.R.; Quiroga, I.Y.; Abba, M.C.; Coleman, R.A.; Gonzalez-Baro, M.R. Glycerol-3-phosphate acyltranferase-2 behaves as a cancer testis gene and promotes growth and tumorigenicity of the breast cancer MDA-MB-231 cell line. PLoS ONE 2014, 9, e100896. [Google Scholar] [CrossRef]
- Agarwal, A.K. Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 2012, 23, 290–302. [Google Scholar] [CrossRef]
- Takeuchi, K.; Reue, K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1195–E1209. [Google Scholar] [CrossRef]
- Vargas, T.; Moreno-Rubio, J.; Herranz, J.; Cejas, P.; Molina, S.; González-Vallinas, M.; Mendiola, M.; Burgos, E.; Aguayo, C.; Custodio, A.B.; et al. ColoLipidGene: Signature of lipid metabolism-related genes to predict prognosis in stage-II colon cancer patients. Oncotarget 2015, 6, 7348–7363. [Google Scholar] [CrossRef]
- Song, L.; Duan, P.; Gan, Y.; Li, P.; Zhao, C.; Xu, J.; Zhang, Z.; Zhou, Q. Silencing LPAATβ inhibits tumor growth of cisplatin-resistant human osteosarcoma in vivo and in vitro. Int. J. Oncol. 2017, 50, 535–544. [Google Scholar] [CrossRef]
- Springett, G.M.; Bonham, L.; Hummer, A.; Linkov, I.; Misra, D.; Ma, C.; Pezzoni, G.; Di Giovine, S.; Singer, J.; Kawasaki, H.; et al. Lysophosphatidic acid acyltransferase-beta is a prognostic marker and therapeutic target in gynecologic malignancies. Cancer Res. 2005, 65, 9415–9425. [Google Scholar] [CrossRef]
- Diefenbach, C.S.M.; Soslow, R.A.; Iasonos, A.; Linkov, I.; Hedvat, C.; Bonham, L.; Singer, J.; Barakat, R.R.; Aghajanian, C.; Dupont, J. Lysophosphatidic acid acyltransferase-beta (LPAAT-beta) is highly expressed in advanced ovarian cancer and is associated with aggressive histology and poor survival. Cancer 2006, 107, 1511–1519. [Google Scholar] [CrossRef]
- Sumantran, V.N.; Mishra, P.; Sudhakar, N. Microarray analysis of differentially expressed genes regulating lipid metabolism during melanoma progression. Indian J. Biochem. Biophys. 2015, 52, 125–131. [Google Scholar]
- Gatto, F.; Miess, H.; Schulze, A.; Nielsen, J. Flux balance analysis predicts essential genes in clear cell renal cell carcinoma metabolism. Sci. Rep. 2015, 5, 10738. [Google Scholar] [CrossRef]
- Zhou, X.; Lawrence, T.J.; He, Z.; Pound, C.R.; Mao, J.; Bigler, S.A. The expression level of lysophosphatidylcholine acyltransferase 1 (LPCAT1) correlates to the progression of prostate cancer. Exp. Mol. Pathol. 2012, 92, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, F.; da Costa, K.-A.; Wang, S.; Kruhøffer, M.; Lewin, T.M.; Orntoft, T.F.; Coleman, R.A.; Birkenkamp-Demtröder, K. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) overexpression in human colorectal cancer. J. Mol. Med. Berl. Ger. 2009, 87, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Garg, A. Enzymatic activity of the human 1-acylglycerol-3-phosphate-O-acyltransferase isoform 11: Upregulated in breast and cervical cancers. J. Lipid Res. 2010, 51, 2143–2152. [Google Scholar] [CrossRef]
- Fan, X.; Weng, Y.; Bai, Y.; Wang, Z.; Wang, S.; Zhu, J.; Zhang, F. Lipin-1 determines lung cancer cell survival and chemotherapy sensitivity by regulation of endoplasmic reticulum homeostasis and autophagy. Cancer Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhang, F.; Tay, L.W.R.; Boroda, S.; Nian, W.; Levental, K.R.; Levental, I.; Harris, T.E.; Chang, J.T.; Du, G. Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 2893–2904. [Google Scholar] [CrossRef] [PubMed]
- Brohée, L.; Demine, S.; Willems, J.; Arnould, T.; Colige, A.C.; Deroanne, C.F. Lipin-1 regulates cancer cell phenotype and is a potential target to potentiate rapamycin treatment. Oncotarget 2015, 6, 11264–11280. [Google Scholar] [CrossRef]
- Bagnato, C.; Igal, R.A. Overexpression of diacylglycerol acyltransferase-1 reduces phospholipid synthesis, proliferation, and invasiveness in simian virus 40-transformed human lung fibroblasts. J. Biol. Chem. 2003, 278, 52203–52211. [Google Scholar] [CrossRef]
- Benjamin, D.I.; Li, D.S.; Lowe, W.; Heuer, T.; Kemble, G.; Nomura, D.K. Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells. ACS Chem. Biol. 2015, 10, 1616–1623. [Google Scholar] [CrossRef]
- Luo, X.; Zhao, X.; Cheng, C.; Li, N.; Liu, Y.; Cao, Y. The implications of signaling lipids in cancer metastasis. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef] [PubMed]
- Purow, B. Molecular Pathways: Targeting Diacylglycerol Kinase Alpha in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5008–5012. [Google Scholar] [CrossRef]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 631. [Google Scholar] [CrossRef]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Z.; Sheng, W.; Xu, M. Emerging roles of long non-coding RNAs in tumor metabolism. J. Hematol. Oncol. 2018, 11. [Google Scholar] [CrossRef]
- Zagani, R.; El-Assaad, W.; Gamache, I.; Teodoro, J.G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget 2015, 6, 28282–28295. [Google Scholar] [CrossRef]
- Al-Zoughbi, W.; Pichler, M.; Gorkiewicz, G.; Guertl-Lackner, B.; Haybaeck, J.; Jahn, S.W.; Lackner, C.; Liegl-Atzwanger, B.; Popper, H.; Schauer, S.; et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget 2016, 7, 33832–33840. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, R.; Leo, L.D.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Tomin, T.; Fritz, K.; Gindlhuber, J.; Waldherr, L.; Pucher, B.; Thallinger, G.G.; Nomura, D.K.; Schittmayer, M.; Birner-Gruenberger, R. Deletion of Adipose Triglyceride Lipase Links Triacylglycerol Accumulation to a More-Aggressive Phenotype in A549 Lung Carcinoma Cells. J. Proteome Res. 2018, 17, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Eder, S.; Schauer, S.; Diwoky, C.; Temmel, H.; Guertl, B.; Gorkiewicz, G.; Tamilarasan, K.P.; Kumari, P.; Trauner, M.; et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011, 333, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Funakoshi, T.; Mori, M.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2017, 31, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef]
- Kulyté, A.; Lorente-Cebrián, S.; Gao, H.; Mejhert, N.; Agustsson, T.; Arner, P.; Rydén, M.; Dahlman, I. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E267–E274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.; Li, H.; Song, Y.; Zhao, Y.; Zhai, L.; Wang, H.; Zhong, R.; Tang, H.; Zhu, D. miR-378 Activates the Pyruvate-PEP Futile Cycle and Enhances Lipolysis to Ameliorate Obesity in Mice. EBioMedicine 2016, 5, 93–104. [Google Scholar] [CrossRef]
- Shaw, T.A.; Singaravelu, R.; Powdrill, M.H.; Nhan, J.; Ahmed, N.; Özcelik, D.; Pezacki, J.P. MicroRNA-124 Regulates Fatty Acid and Triglyceride Homeostasis. iScience 2018, 10, 149–157. [Google Scholar] [CrossRef]
- Das, S.K.; Stadelmeyer, E.; Schauer, S.; Schwarz, A.; Strohmaier, H.; Claudel, T.; Zechner, R.; Hoefler, G.; Vesely, P.W. Micro RNA-124a regulates lipolysis via adipose triglyceride lipase and comparative gene identification 58. Int. J. Mol. Sci. 2015, 16, 8555–8568. [Google Scholar] [CrossRef]
- Qi, R.; Wang, J.; Wang, Q.; Qiu, X.; Yang, F.; Liu, Z.; Huang, J. MicroRNA-425 controls lipogenesis and lipolysis in adipocytes. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 744–755. [Google Scholar] [CrossRef]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R.; et al. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 2004, 279, 52361–52365. [Google Scholar] [CrossRef]
- You, L.; Wang, Y.; Gao, Y.; Wang, X.; Cui, X.; Zhang, Y.; Pang, L.; Ji, C.; Guo, X.; Chi, X. The role of microRNA-23b-5p in regulating brown adipogenesis and thermogenic program. Endocr. Connect. 2020, 9, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Rockstroh, D.; Löffler, D.; Kiess, W.; Landgraf, K.; Körner, A. Regulation of human adipogenesis by miR125b-5p. Adipocyte 2016, 5, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Xiao, L.; Tang, M.; Bai, F.; Li, J.; Li, L.; Shi, F.; Li, N.; Li, Y.; Du, Q.; et al. Targeting CPT1A-mediated fatty acid oxidation sensitizes nasopharyngeal carcinoma to radiation therapy. Theranostics 2018, 8, 2329–2347. [Google Scholar] [CrossRef]
- Sung, G.-J.; Choi, H.-K.; Kwak, S.; Song, J.-H.; Ko, H.; Yoon, H.-G.; Kang, H.-B.; Choi, K.-C. Targeting CPT1A enhances metabolic therapy in human melanoma cells with the BRAF V600E mutation. Int. J. Biochem. Cell Biol. 2016, 81, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Yun, S.J.; Yan, C.; Jeong, P.; Kim, Y.H.; Lee, I.S.; Kang, H.W.; Park, S.; Moon, S.K.; Choi, Y.H.; et al. Metabolic Pathway Signatures Associated with Urinary Metabolite Biomarkers Differentiate Bladder Cancer Patients from Healthy Controls. Yonsei Med. J. 2016, 57, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zeng, W.; Feng, L.; Yu, X.; Li, P.; Zhang, K.; Zhou, Z.; Cheng, S. Integrated transcriptomic analysis of distance-related field cancerization in rectal cancer patients. Oncotarget 2017, 8, 61107–61117. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.-P.; Liu, J.; Jiang, W.-Q.; Carew, J.S.; Ogasawara, M.A.; Pelicano, H.; Croce, C.M.; Estrov, Z.; Xu, R.-H.; Keating, M.J.; et al. Elimination of chronic lymphocytic leukemia cells in stromal microenvironment by targeting CPT with an antiangina drug perhexiline. Oncogene 2016, 35, 5663–5673. [Google Scholar] [CrossRef]
- Gu, J.-J.; Yao, M.; Yang, J.; Cai, Y.; Zheng, W.-J.; Wang, L.; Yao, D.-B.; Yao, D.-F. Mitochondrial carnitine palmitoyl transferase-II inactivity aggravates lipid accumulation in rat hepatocarcinogenesis. World J. Gastroenterol. 2017, 23, 256–264. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.-O.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Chmurzyńska, A. The multigene family of fatty acid-binding proteins (FABPs): Function, structure and polymorphism. J. Appl. Genet. 2006, 47, 39–48. [Google Scholar] [CrossRef]
- Thompson, K.J.; Austin, R.G.; Nazari, S.S.; Gersin, K.S.; Iannitti, D.A.; McKillop, I.H. Altered fatty acid-binding protein 4 (FABP4) expression and function in human and animal models of hepatocellular carcinoma. Liver Int. Off. J. Int. Assoc. Study Liver 2017. [Google Scholar] [CrossRef]
- Guaita-Esteruelas, S.; Gumà, J.; Masana, L.; Borràs, J. The peritumoural adipose tissue microenvironment and cancer. The roles of fatty acid binding protein 4 and fatty acid binding protein 5. Mol. Cell. Endocrinol. 2018, 462, 107–118. [Google Scholar] [CrossRef]
- Kim, S.; Lee, Y.; Koo, J.S. Differential expression of lipid metabolism-related proteins in different breast cancer subtypes. PLoS ONE 2015, 10, e0119473. [Google Scholar] [CrossRef]
- Elsherbiny, M.E.; Emara, M.; Godbout, R. Interaction of brain fatty acid-binding protein with the polyunsaturated fatty acid environment as a potential determinant of poor prognosis in malignant glioma. Prog. Lipid Res. 2013, 52, 562–570. [Google Scholar] [CrossRef]
- Hui, X.; Li, H.; Zhou, Z.; Lam, K.S.L.; Xiao, Y.; Wu, D.; Ding, K.; Wang, Y.; Vanhoutte, P.M.; Xu, A. Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH2-terminal kinases and activator protein-1. J. Biol. Chem. 2010, 285, 10273–10280. [Google Scholar] [CrossRef]
- Terra, X.; Quintero, Y.; Auguet, T.; Porras, J.A.; Hernández, M.; Sabench, F.; Aguilar, C.; Luna, A.M.; Del Castillo, D.; Richart, C. FABP 4 is associated with inflammatory markers and metabolic syndrome in morbidly obese women. Eur. J. Endocrinol. 2011, 164, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Hoo, R.L.C.; Wu, X.; Pan, Y.; Lee, I.P.C.; Cheong, L.Y.; Bornstein, S.R.; Rong, X.; Guo, J.; Xu, A. A-FABP mediates adaptive thermogenesis by promoting intracellular activation of thyroid hormones in brown adipocytes. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.H.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Kolukula, V.K.; Sahu, G.; Wellstein, A.; Rodriguez, O.C.; Preet, A.; Iacobazzi, V.; D’Orazi, G.; Albanese, C.; Palmieri, F.; Avantaggiati, M.L. SLC25A1, or CIC, is a novel transcriptional target of mutant p53 and a negative tumor prognostic marker. Oncotarget 2014, 5, 1212–1225. [Google Scholar] [CrossRef]
- Ozkaya, A.B.; Ak, H.; Atay, S.; Aydin, H.H. Targeting mitochondrial citrate transport in breast cancer cell lines. Anticancer Agents Med. Chem. 2015, 15, 374–381. [Google Scholar] [CrossRef]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef]
- Cheng, L.; Lu, W.; Kulkarni, B.; Pejovic, T.; Yan, X.; Chiang, J.-H.; Hood, L.; Odunsi, K.; Lin, B. Analysis of chemotherapy response programs in ovarian cancers by the next-generation sequencing technologies. Gynecol. Oncol. 2010, 117, 159–169. [Google Scholar] [CrossRef]
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP Citrate Lyase (ACLY): A Promising Target for Cancer Prevention and Treatment. Available online: http://www.eurekaselect.com/127128/article (accessed on 14 March 2018).
- Teng, L.; Chen, Y.; Cao, Y.; Wang, W.; Xu, Y.; Wang, Y.; Lv, J.; Li, C.; Su, Y. Overexpression of ATP citrate lyase in renal cell carcinoma tissues and its effect on the human renal carcinoma cells in vitro. Oncol. Lett. 2018, 15, 6967–6974. [Google Scholar] [CrossRef]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The acetyl-CoA carboxylase enzyme: A target for cancer therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasegawa, K.; Ohashi, K.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur. J. Cancer Oxf. Engl. 1990 2005, 41, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Sinilnikova, O.M.; Ginolhac, S.M.; Magnard, C.; Léoné, M.; Anczukow, O.; Hughes, D.; Moreau, K.; Thompson, D.; Coutanson, C.; Hall, J.; et al. Acetyl-CoA carboxylase alpha gene and breast cancer susceptibility. Carcinogenesis 2004, 25, 2417–2424. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef]
- Chajès, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef]
- Raimondo, S.; Saieva, L.; Cristaldi, M.; Monteleone, F.; Fontana, S.; Alessandro, R. Label-free quantitative proteomic profiling of colon cancer cells identifies acetyl-CoA carboxylase alpha as antitumor target of Citrus limon-derived nanovesicles. J. Proteomics 2018, 173, 1–11. [Google Scholar] [CrossRef]
- Jones, J.E.C.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Luo, J.; Hong, Y.; Lu, Y.; Qiu, S.; Chaganty, B.K.R.; Zhang, L.; Wang, X.; Li, Q.; Fan, Z. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett. 2017, 384, 39–49. [Google Scholar] [CrossRef]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2115–2120. [Google Scholar]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. Lond. Engl. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Kuhajda, F.P. Fatty acid synthase and cancer: New application of an old pathway. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Oncogenic properties of the endogenous fatty acid metabolism: Molecular pathology of fatty acid synthase in cancer cells. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R.; Colomer, R. Targeting fatty acid synthase: Potential for therapeutic intervention in her-2/neu-overexpressing breast cancer. Drug News Perspect. 2005, 18, 375–385. [Google Scholar] [CrossRef]
- Rossi, S.; Ou, W.; Tang, D.; Bhattacharya, N.; Dei Tos, A.P.; Fletcher, J.A.; Loda, M. Gastrointestinal stromal tumours overexpress fatty acid synthase. J. Pathol. 2006, 209, 369–375. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Loftus, T.M.; Jaworsky, D.E.; Frehywot, G.L.; Townsend, C.A.; Ronnett, G.V.; Lane, M.D.; Kuhajda, F.P. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 2000, 288, 2379–2381. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W.K. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. MCR 2012, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Zhu, L.; Zhu, Q.; Su, J.; Liu, M.; Huang, W. SREBP-1 is an independent prognostic marker and promotes invasion and migration in breast cancer. Oncol. Lett. 2016, 12, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, L.; Yang, H.; Li, J.; Min, X.; Yang, F.; Liu, J.; Huang, G. Pyruvate kinase M2 interacts with nuclear sterol regulatory element–binding protein 1a and thereby activates lipogenesis and cell proliferation in hepatocellular carcinoma. J. Biol. Chem. 2018, 293, 6623–6634. [Google Scholar] [CrossRef]
- Wen, Y.-A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef]
- Yan, S.; Yang, X.-F.; Liu, H.-L.; Fu, N.; Ouyang, Y.; Qing, K. Long-chain acyl-CoA synthetase in fatty acid metabolism involved in liver and other diseases: An update. World J. Gastroenterol. 2015, 21, 3492–3498. [Google Scholar] [CrossRef]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA Metabolism and Partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef]
- Soupene, E.; Kuypers, F.A. Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. Maywood NJ 2008, 233, 507–521. [Google Scholar] [CrossRef]
- Heimerl, S.; Moehle, C.; Zahn, A.; Boettcher, A.; Stremmel, W.; Langmann, T.; Schmitz, G. Alterations in intestinal fatty acid metabolism in inflammatory bowel disease. Biochim. Biophys. Acta 2006, 1762, 341–350. [Google Scholar] [CrossRef]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. WJG 2014, 20, 9872–9881. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-C.; Wang, C.-Y.; Hung, Y.-H.; Weng, T.-Y.; Yen, M.-C.; Lai, M.-D. Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Fraisl, P.; Shi, X.; Gabrielson, E.; Forss-Petter, S.; Berger, J.; Watkins, P.A. Very long-chain acyl-CoA synthetase 3: Overexpression and growth dependence in lung cancer. PLoS ONE 2013, 8, e69392. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Dits, N.F.; Erkens-Schulze, S.; van Ijcken, W.F.J.; van Weerden, W.M.; Jenster, G. Modulation of androgen receptor signaling in hormonal therapy-resistant prostate cancer cell lines. PLoS ONE 2011, 6, e23144. [Google Scholar] [CrossRef]
- Cao, Y.; Pearman, A.T.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 11280–11285. [Google Scholar] [CrossRef]
- Cao, Y.; Dave, K.B.; Doan, T.P.; Prescott, S.M. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 2001, 61, 8429–8434. [Google Scholar]
- Sung, Y.K.; Park, M.K.; Hong, S.H.; Hwang, S.Y.; Kwack, M.H.; Kim, J.C.; Kim, M.K. Regulation of cell growth by fatty acid-CoA ligase 4 in human hepatocellular carcinoma cells. Exp. Mol. Med. 2007, 39, 477–482. [Google Scholar] [CrossRef]
- Sun, X.-J.; Xu, G.-L. Overexpression of Acyl-CoA Ligase 4 (ACSL4) in Patients with Hepatocellular Carcinoma and its Prognosis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 4343–4350. [Google Scholar] [CrossRef]
- Wu, X.; Deng, F.; Li, Y.; Daniels, G.; Du, X.; Ren, Q.; Wang, J.; Wang, L.H.; Yang, Y.; Zhang, V.; et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget 2015, 6, 44849–44863. [Google Scholar] [CrossRef]
- Ye, X.; Zhang, Y.; Wang, X.; Li, Y.; Gao, Y. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 2016, 68, 320–327. [Google Scholar] [CrossRef]
- Pitule, P.; Vycital, O.; Bruha, J.; Novak, P.; Hosek, P.; Treska, V.; Hlavata, I.; Soucek, P.; Kralickova, M.; Liska, V. Differential expression and prognostic role of selected genes in colorectal cancer patients. Anticancer Res. 2013, 33, 4855–4865. [Google Scholar] [PubMed]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal. Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Bebber, C.M.; Müller, F.; Prieto Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Li, H.; Zhang, L.; Zheng, X.; Cheng, W. Crosstalk between noncoding RNAs and ferroptosis: New dawn for overcoming cancer progression. Cell Death Dis. 2020, 11, 1–22. [Google Scholar] [CrossRef]
- Enoch, H.G.; Catalá, A.; Strittmatter, P. Mechanism of rat liver microsomal stearyl-CoA desaturase. Studies of the substrate specificity, enzyme-substrate interactions, and the function of lipid. J. Biol. Chem. 1976, 251, 5095–5103. [Google Scholar]
- Igal, R.A. Stearoyl-CoA desaturase-1: A novel key player in the mechanisms of cell proliferation, programmed cell death and transformation to cancer. Carcinogenesis 2010, 31, 1509–1515. [Google Scholar] [CrossRef]
- Roongta, U.V.; Pabalan, J.G.; Wang, X.; Ryseck, R.-P.; Fargnoli, J.; Henley, B.J.; Yang, W.-P.; Zhu, J.; Madireddi, M.T.; Lawrence, R.M.; et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. MCR 2011, 9, 1551–1561. [Google Scholar] [CrossRef]
- Holder, A.M.; Gonzalez-Angulo, A.M.; Chen, H.; Akcakanat, A.; Do, K.-A.; Fraser Symmans, W.; Pusztai, L.; Hortobagyi, G.N.; Mills, G.B.; Meric-Bernstam, F. High stearoyl-CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 137, 319–327. [Google Scholar] [CrossRef]
- Wood, C.B.; Habib, N.A.; Thompson, A.; Bradpiece, H.; Smadja, C.; Hershman, M.; Barker, W.; Apostolov, K. Increase of oleic acid in erythrocytes associated with malignancies. Br. Med. J. Clin. Res. Ed. 1985, 291, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Igal, R.A. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim. Biophys. Acta 2016, 1861, 1865–1880. [Google Scholar] [CrossRef] [PubMed]
- Arous, C.; Naïmi, M.; Van Obberghen, E. Oleate-mediated activation of phospholipase D and mammalian target of rapamycin (mTOR) regulates proliferation and rapamycin sensitivity of hepatocarcinoma cells. Diabetologia 2011, 54, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.C.; Pouwer, R.H.; Gunter, J.H.; Lubik, A.A.; Quinn, R.J.; Nelson, C.C. The fatty acid synthase inhibitor triclosan: Repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget 2014, 5, 9362–9381. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Carrozzino, F.; Peyrou, M.; Carlone, S.; Montesano, R.; Benelli, R.; Foti, M. Unsaturated fatty acids promote hepatoma proliferation and progression through downregulation of the tumor suppressor PTEN. J. Hepatol. 2009, 50, 1132–1141. [Google Scholar] [CrossRef]
- Peck, B.; Schug, Z.T.; Zhang, Q.; Dankworth, B.; Jones, D.T.; Smethurst, E.; Patel, R.; Mason, S.; Jiang, M.; Saunders, R.; et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 2016, 4, 6. [Google Scholar] [CrossRef]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef]
- Ran, H.; Zhu, Y.; Deng, R.; Zhang, Q.; Liu, X.; Feng, M.; Zhong, J.; Lin, S.; Tong, X.; Su, Q. Stearoyl-CoA desaturase-1 promotes colorectal cancer metastasis in response to glucose by suppressing PTEN. J. Exp. Clin. Cancer Res. CR 2018, 37, 54. [Google Scholar] [CrossRef]
- Qiu, Y.; Cai, G.; Zhou, B.; Li, D.; Zhao, A.; Xie, G.; Li, H.; Cai, S.; Xie, D.; Huang, C.; et al. A distinct metabolic signature of human colorectal cancer with prognostic potential. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2136–2146. [Google Scholar] [CrossRef]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar] [CrossRef]
- Dong, W.; Dai, Z.; Liu, F.; Guo, X.; Ge, C.; Ding, J.; Liu, H.; Yang, F. The RNA-binding protein RBM3 promotes cell proliferation in hepatocellular carcinoma by regulating circular RNA SCD-circRNA 2 production. EBioMedicine 2019, 45, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-X.; Chen, X.-M.; Zhang, Y.-Q.; Peng, L.; Xue, X.-Y.; Li, G.-X. Comprehensive analysis of long noncoding RNA and mRNA in five colorectal cancer tissues and five normal tissues. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Gene | Regulatory RNA | Tumor Type/ Cell Type | Sense | Reference |
|---|---|---|---|---|
| Lipid droplets formation | ||||
| AGPAT (-1,-3,-9) | miR-122 | HCC | - | [27] |
| DGAT-1 | miR-122 | HCC | - | [27] |
| Lipin1 | circRNA_021412/ miR-1972 | HepG2-based hepatic steatosis | - - | [28] |
| Lipin2/DGAT-2 | LncRNA-SPRY4 | ML | + | [29] |
| Lipin2 | Lnc-KDM5D-4 | HCC | + | [30] |
| Lipid droplets hydrolysis | ||||
| ATGL | lncRNA-NEAT1/ miR-124-3p | HCC | + - | [31] |
| Fatty acids oxidation | ||||
| CPT1 | miR-33b | HCC | - | [32] |
| CPT1 | lncRNA-HCP5/ miR-3619-5p | GC | + - | [33] |
| CPT1 | lncRNA-NEAT1/ miR-107 | BC | + - | [34] |
| CPT1 | lncRNA-LNMICC | CC | + | [35] |
| CPT1A | miR-124-3p | PrC | - | [36,37] |
| CPT1C | miR-1291 | BC, PC | - | [38] |
| CACT | miR-129-5p | PrC | - | [36,37] |
| CrAT | miR-378 | PrC | - | [36,37] |
| Extra FAs input | ||||
| CD36 | miR-1254 | OSCC | - | [39] |
| FABP5 | LncRNA-LNMICC/ miR-190 | CC | - + | [35] |
| Fatty Acid Synthesis | ||||
| ACLY | LncRNA-FLJ22763 | GC | - | [40] |
| ACLY | miR-126b | Ms | - | [41] |
| ACLY | miR-22 | OS, PrC, CC, LC | - | [42] |
| ACLY | miR-22 | BC | - | [43] |
| ACLY | miR-182 | LC | - | [44] |
| ACLY | miR-133b | GC | - | [45] |
| ACC1/2 | miR-195 | BC | - | [46] |
| FASN | LncRNA-PVT1/ miR-195 | OS | + - | [47,48] |
| FASN | LncRNA-HAGLR | NSCLC | + | [49] |
| FASN | miR-15a/miR-16b | BC | - | [50] |
| FASN | miR-193b | TNBC | - | [51] |
| FASN | miR-1207-5p | HCC | - | [52] |
| FASN | miR-195 | OS, BC | - | [46,53] |
| FASN | miR-320 | NSCLC | - | [54] |
| FASN | CircFARSA/ miR-330-5p-miR-326 | NSCLC | + - | [55] |
| SREBP1 | LncRNA-HR1 | HCC | - | [56] |
| SREBP1 | miR-21 | HCC | + | [57] |
| SREBP1 | miR-185, miR-342 | PrC | - | [58] |
| SREBP1c | miR-132 | Gl | - | [59] |
| SREBP1 | miR-449 | HCC | - | [60] |
| Fatty acid activation | ||||
| ACSL1 | miR-205 | HCC | - | [61] |
| ACSL1 | SNHG7- miR-449a | TC | + | [62] |
| ACSL1 | LncRNA-HULC/ miR-9 | HCC | + - | [63] |
| ACSL4 | miR-205 | HCC | - | [64] |
| ACSL4 | SNORD113-1 | HCC | - | [65] |
| ACSL (-1; -4)/SCD | miR-544a, miR-142, and miR-19b-1 | CRC | - | [66] |
| ACSL5 | miR-149, miR-205, miR-375, miR-378, miR-422a and miR-708 (signature) | NSCLC | - | [67] |
| ACSL6 | let-7c, let-7e, miR-133a, miR-133b, miR-191-5p and miR-222-3p (part of a 20 miRs signature) | CRC | - | [68] |
| ACSL6 | pU6-487i /pU6-586i | NB | - | [69] |
| Fatty acid desaturation | ||||
| SCD-1 | miR-215 | CRC | - | [70] |
| SCD-1 | miR-600 | BC | - | [71] |
| SCD-1 | LncRNA-UPAT | CRC | + | [72] |
| SCD-1 | uc.372 miR-195/miR-4668 | HepG2 | + - | [73] |
| SCD-1 | SNHG16 | CRC | + | [74] |
| SCD-5 | miR-221/miR-222 | ML | + | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz-Gil, S.; Fernández, L.P.; Sánchez-Martínez, R.; Gómez de Cedrón, M.; Ramírez de Molina, A. Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer. Cancers 2020, 12, 2890. https://doi.org/10.3390/cancers12102890
Cruz-Gil S, Fernández LP, Sánchez-Martínez R, Gómez de Cedrón M, Ramírez de Molina A. Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer. Cancers. 2020; 12(10):2890. https://doi.org/10.3390/cancers12102890
Chicago/Turabian StyleCruz-Gil, Silvia, Lara P. Fernández, Ruth Sánchez-Martínez, Marta Gómez de Cedrón, and Ana Ramírez de Molina. 2020. "Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer" Cancers 12, no. 10: 2890. https://doi.org/10.3390/cancers12102890
APA StyleCruz-Gil, S., Fernández, L. P., Sánchez-Martínez, R., Gómez de Cedrón, M., & Ramírez de Molina, A. (2020). Non-Coding and Regulatory RNAs as Epigenetic Remodelers of Fatty Acid Homeostasis in Cancer. Cancers, 12(10), 2890. https://doi.org/10.3390/cancers12102890