Dexamethasone Inhibits TRAIL-Induced Apoptosis through c-FLIP(L) Upregulation and DR5 Downregulation by GSK3β Activation in Cancer Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. DEX Inhibits TRAIL-Induced Apoptosis
2.2. Effect of DEX on the Expression Level of Apoptosis-Related Proteins
2.3. DEX-Induced c-FLIP(L) Upregulation Reduces TRAIL Sensitivity
2.4. GSK-3β Plays a Critical Role on Upregulation of c-FLIP(L) Expression by DEX Treatment
2.5. DEX Treatment Decreases the Expression of DR5
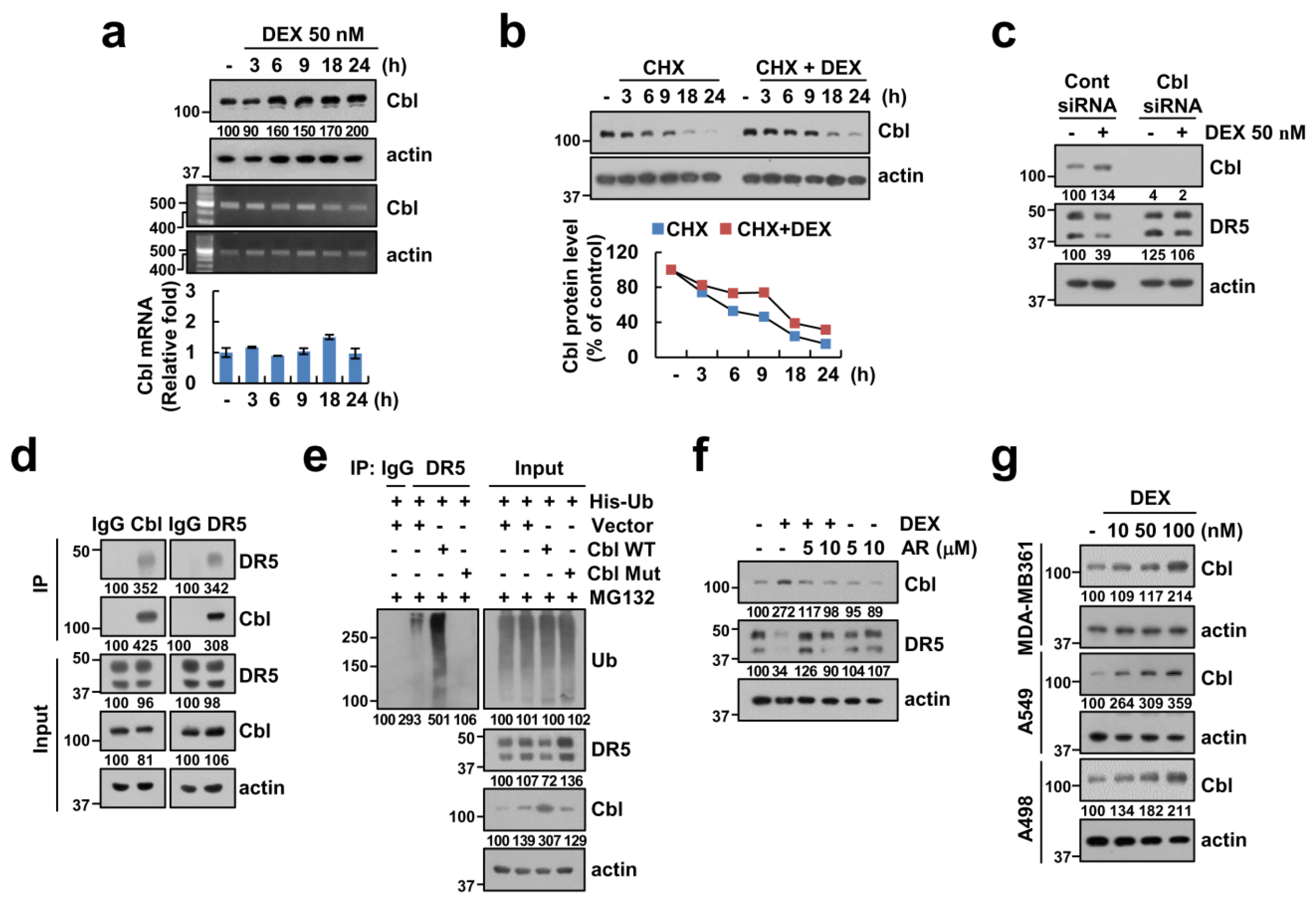
2.6. Upregulation of Cbl Plays a Critical Role in DEX-Induced DR5 Downregulation
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Materials
4.2. Flow Cytometry Analysis
4.3. Western Blotting
4.4. IETDse (Caspase-8) Activity
4.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Quantitative PCR (qPCR)
4.6. Transfection
4.7. Detection of DR5 on Cell Surface
4.8. Immunoprecipitation
4.9. Ubiquitination Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hao, L.; Zhao, Y.; Li, Z.G.; He, H.G.; Liang, Q.; Zhang, Z.G.; Shi, Z.D.; Zhang, P.Y.; Han, C.H. Tumor necrosis factor-related apoptosis-inducing ligand inhibits proliferation and induces apoptosis of prostate and bladder cancer cells. Oncol. Lett. 2017, 13, 3638–3640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene 2008, 27, 6207–6215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, S.A.; Cole, T.J.; Schmid, W.; Schütz, G. Molecular genetic analysis of glucocorticoid and mineralocorticoid signaling in development and physiological processes. Steroids 1996, 61, 236–239. [Google Scholar] [CrossRef]
- Wintermantel, T.M.; Bock, D.; Fleig, V.; Greiner, E.F.; Schütz, G. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol. Endocrinol. 2005, 19, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J.R.; Rogatsky, I. Minireview: Glucocorticoids in autoimmunity: Unexpected targets and mechanisms. Mol. Endocrinol. (Baltim. Md.) 2011, 25, 1075–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Nonomura, N.; Satoh, E.; Harada, Y.; Nakayama, M.; Tokizane, T.; Fukui, T.; Ono, Y.; Inoue, H.; Shin, M.; et al. Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J. Natl. Cancer Inst. 2001, 93, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Zhang, N.; Zhao, R. Dexamethasone downregulates SLC7A5 expression and promotes cell cycle arrest, autophagy and apoptosis in BeWo cells. J. Cell. Physiol. 2016, 231, 233–242. [Google Scholar] [CrossRef]
- Nieuwenhuis, B.; Lüth, A.; Kleuser, B. Dexamethasone protects human fibroblasts from apoptosis via an S1P3-receptor subtype dependent activation of PKB/Akt and Bcl XL. Pharmacol. Res. 2010, 61, 449–459. [Google Scholar] [CrossRef]
- Haake, S.M.; Dinh, C.T.; Chen, S.; Eshraghi, A.A.; Van De Water, T.R. Dexamethasone protects auditory hair cells against TNFalpha-initiated apoptosis via activation of PI3K/Akt and NFkappaB signaling. Hear. Res. 2009, 255, 22–32. [Google Scholar] [CrossRef]
- Zhao, B.; Xie, G.J.; Li, R.F.; Chen, Q.; Zhang, X.Q. Dexamethasone protects normal human liver cells from apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand by upregulating the expression of P-glycoproteins. Mol. Med. Rep. 2015, 12, 8093–8100. [Google Scholar] [CrossRef] [Green Version]
- Dinh, C.T.; Chen, S.; Bas, E.; Dinh, J.; Goncalves, S.; Telischi, F.; Angeli, S.; Eshraghi, A.A.; Van De Water, T. Dexamethasone protects against apoptotic cell death of cisplatin-exposed auditory hair cells in vitro. Otol. Neurotol. 2015, 36, 1566–1571. [Google Scholar] [CrossRef] [PubMed]
- Crozier, M.; Porter, L.A. Paclitaxel-induced transcriptional regulation of Fas signaling pathway is antagonized by dexamethasone. Breast Cancer Res. Treat. 2015, 154, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Drewniok, C.; Hupe, M.; Haas, T.L.; Diessenbacher, P.; Sprick, M.R.; Schon, M.P.; Henkler, F.; Gollnick, H.; Walczak, H.; et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL-and CD95L-mediated apoptosis. Oncogene 2008, 27, 3211–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panka, D.J.; Mano, T.; Suhara, T.; Walsh, K.; Mier, J.W. Phosphatidylinositol 3-kinase/Akt activity regulates c-FLIP(L) expression in tumor cells. J. Biol. Chem. 2001, 276, 6893–6896. [Google Scholar] [CrossRef] [Green Version]
- Micheau, O.; Lens, S.; Gaide, O.; Alevizopoulos, K.; Tschopp, J. NF-kappaB signals induce the expression of c-FLIP(L). Mol. Cell. Biol. 2001, 21, 5299–5305. [Google Scholar] [CrossRef] [Green Version]
- Seidelin, J.B.; Coskun, M.; Vainer, B.; Riis, L.; Soendergaard, C.; Nielsen, O.H. ERK controls epithelial cell death receptor signalling and cellular FLICE-like inhibitory protein (c-FLIP(L)) in ulcerative colitis. J. Mol. Med. (Berl.) 2013, 91, 839–849. [Google Scholar] [CrossRef]
- Spokoini, R.; Kfir-Erenfeld, S.; Yefenof, E.; Sionov, R.V. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol. Endocrinol. 2010, 24, 1136–1150. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhong, Z.; Zheng, Z.; Shi, X.M.; Zhang, W. Inhibition of glycogen synthase kinase-3beta attenuates glucocorticoid-induced suppression of myogenic differentiation in vitro. PLoS ONE 2014, 9, e105528. [Google Scholar]
- Oh, Y.T.; Deng, L.; Deng, J.; Sun, S.Y. The proteasome deubiquitinase inhibitor b-AP15 enhances DR5 activation-induced apoptosis through stabilizing DR5. Sci. Rep. 2017, 7, 8027. [Google Scholar] [CrossRef] [Green Version]
- Song, J.J.; Szczepanski, M.J.; Kim, S.Y.; Kim, J.H.; An, J.Y.; Kwon, Y.T.; Alcala, M.A., Jr.; Bartlett, D.L.; Lee, Y.J. c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell. Signal. 2010, 22, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Harmon, J.M.; Norman, M.R.; Fowlkes, B.J.; Thompson, E.B. Dexamethasone induces irreversible G1 arrest and death of a human lymphoid cell line. J. Cell. Physiol. 1979, 98, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Liang, Q. Low-dose dexamethasone affects osteoblast viability by inducing autophagy via intracellular ROS. Mol. Med. Rep. 2018, 17, 4307–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly-Maitre, B.; de Sousa, G.; Boulukos, K.; Gugenheim, J.; Rahmani, R. Dexamethasone inhibits spontaneous apoptosis in primary cultures of human and rat hepatocytes via Bcl-2 and Bcl-xL induction. Cell Death Differ. 2001, 8, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roosmalen, I.A.; Quax, W.J.; Kruyt, F.A. Two death-inducing human TRAIL receptors to target in cancer: Similar or distinct regulation and function? Biochem. Pharmacol. 2014, 91, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Min, K.J.; Woo, S.M.; Shahriyar, S.A.; Kwon, T.K. Elucidation for modulation of death receptor (DR) 5 to strengthen apoptotic signals in cancer cells. Arch. Pharm. Res. 2019, 42, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Min, K.J.; Choi, K.S.; Kubatka, P.; Kruzliak, P.; Kim, D.E.; Kwon, T.K. Chloroquine enhances TRAIL-mediated apoptosis through up-regulation of DR5 by stabilization of mRNA and protein in cancer cells. Sci. Rep. 2016, 6, 22921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, K.; Kolpakov, M.A.; Seqqat, R.; Guo, J.; Guo, X.; Qi, Z.; Yu, D.; Mohapatra, B.; Zutshi, N.; An, W.; et al. c-Cbl inhibition improves cardiac function and survival in response to myocardial ischemia. Circulation 2014, 129, 2031–2043. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.H.; Sun, X.Y.; Liu, J.X.; Zhang, Q.Y.; Liu, P.; Huang, Q.H.; Li, K.K.; Chen, Q.; Chen, Z.; Chen, S.J. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc. Natl Acad Sci USA 2010, 107, 21683–21688. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L)(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, J.; Zhang, Y.; Qu, J.; Xu, L.; Zheng, H.; Liu, Y.; Qu, X. Cbl-b-dependent degradation of FLIP(L) is involved in ATO-induced autophagy in leukemic K562 and gastric cancer cells. FEBS Lett. 2012, 586, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Kleinesudeik, L.; Rohde, K.; Fulda, S. Regulation of the antiapoptotic protein cFLIP by the glucocorticoid Dexamethasone in ALL cells. Oncotarget 2018, 9, 16521–16532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.Y.; Namkoong, S.; Lee, S.J.; Por, E.; Kim, C.K.; Billiar, T.R.; Han, J.A.; Ha, K.S.; Chung, H.T.; Kwon, Y.G.; et al. Dexamethasone protects primary cultured hepatocytes from death receptor-mediated apoptosis by upregulation of cFLIP. Cell Death Differ. 2006, 13, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.J.; Um, H.J.; Min do, S.; Park, J.W.; Choi, K.S.; Kwon, T.K. Withaferin A sensitizes TRAIL-induced apoptosis through reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP(L). Free Radic. Biol. Med. 2009, 46, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Min, K.J.; Seo, B.R.; Kwon, T.K. YM155 sensitizes TRAIL-induced apoptosis through cathepsin S-dependent down-regulation of Mcl-1 and NF-κB-mediated down-regulation of c-FLIP(L) expression in human renal carcinoma Caki cells. Oncotarget 2016, 7, 61520–61532. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Kim, E.A.; Park, H.J.; Sung, E.G.; Song, I.H.; Kim, J.Y.; Woo, C.H.; Doh, K.O.; Kim, K.H.; Lee, T.J. Methylglyoxal-induced apoptosis is dependent on the suppression of c-FLIP(L)(L) expression via down-regulation of p65 in endothelial cells. J. Cell. Mol. Med. 2017, 21, 2720–2731. [Google Scholar] [CrossRef]
- Lang, U.E.; Kocabayoglu, P.; Cheng, G.Z.; Ghiassi-Nejad, Z.; Munoz, U.; Vetter, D.; Eckstein, D.A.; Hannivoort, R.A.; Walsh, M.J.; Friedman, S.L. GSK3beta phosphorylation of the KLF6 tumor suppressor promotes its transactivation of p21. Oncogene 2013, 32, 4557–4564. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Woo, S.M.; Min, K.J.; Seo, S.U.; Lee, T.J.; Kubatka, P.; Kim, D.E.; Kwon, T.K. WP1130 Enhances TRAIL-induced apoptosis through USP9X-dependent miR-708-mediated downregulation of c-FLIP(L). Cancers (Basel) 2019, 11, 334. [Google Scholar] [CrossRef] [Green Version]
- Min, K.J.; Shahriyar, S.A.; Kwon, T.K. Arylquin 1, a potent Par-4 secretagogue, induces lysosomal membrane permeabilization-mediated non-apoptotic cell death in cancer cells. Toxicol. Res. 2020, 36, 167–173. [Google Scholar] [CrossRef]
- Woo, S.M.; Seo, S.U.; Kubatka, P.; Min, K.J.; Kwon, T.K. Honokiol Enhances TRAIL-Mediated Apoptosis through STAMBPL1-Induced Survivin and c-FLIP(L) Degradation. Biomolecules 2019, 9, 838. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, M.-Y.; Woo, S.M.; Seo, S.U.; Kim, S.H.; Nam, J.-O.; Kim, S.; Park, J.-W.; Kubatka, P.; Min, K.-j.; Kwon, T.K. Dexamethasone Inhibits TRAIL-Induced Apoptosis through c-FLIP(L) Upregulation and DR5 Downregulation by GSK3β Activation in Cancer Cells. Cancers 2020, 12, 2901. https://doi.org/10.3390/cancers12102901
Jeon M-Y, Woo SM, Seo SU, Kim SH, Nam J-O, Kim S, Park J-W, Kubatka P, Min K-j, Kwon TK. Dexamethasone Inhibits TRAIL-Induced Apoptosis through c-FLIP(L) Upregulation and DR5 Downregulation by GSK3β Activation in Cancer Cells. Cancers. 2020; 12(10):2901. https://doi.org/10.3390/cancers12102901
Chicago/Turabian StyleJeon, Mi-Yeon, Seon Min Woo, Seung Un Seo, Sang Hyun Kim, Ju-Ock Nam, Shin Kim, Jong-Wook Park, Peter Kubatka, Kyoung-jin Min, and Taeg Kyu Kwon. 2020. "Dexamethasone Inhibits TRAIL-Induced Apoptosis through c-FLIP(L) Upregulation and DR5 Downregulation by GSK3β Activation in Cancer Cells" Cancers 12, no. 10: 2901. https://doi.org/10.3390/cancers12102901