PARP1 Deficiency Reduces Tumour Growth by Decreasing E2F1 Hyperactivation: A Novel Mechanism in the Treatment of Cancer

 , , ,
, , , 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Deletion of Parp1 Rescues Embryonic Lethality in Rb-Deficient Mice
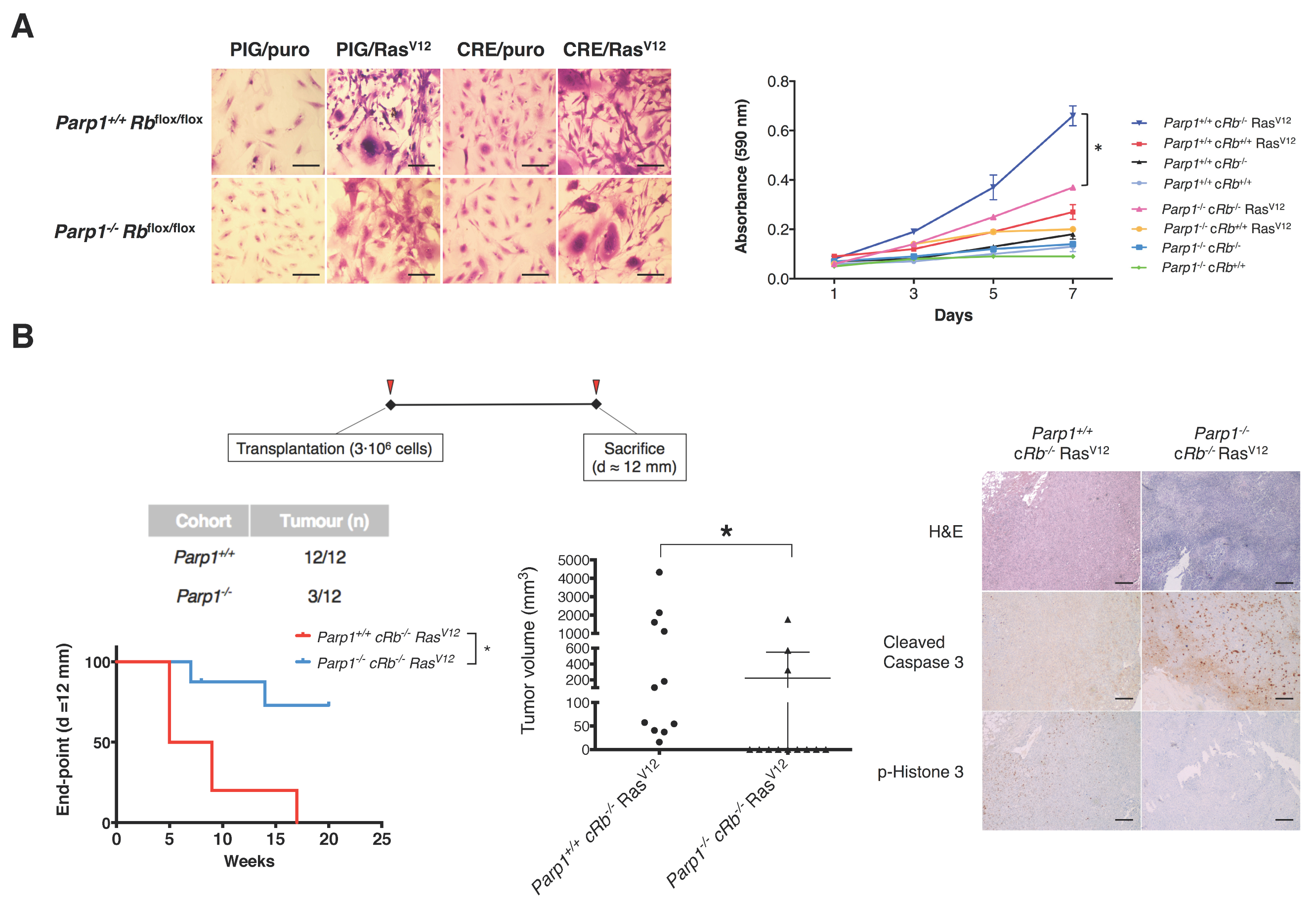
2.2. Deficiency of PARP1 Reduces Tumour Growth in Vitro and in Vivo
2.3. PARP1 Deficiency Inhibits E2F1 Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Primary Cell Cultures
4.2. Affinity Measurement
4.3. Co-Localization Studies
4.4. Chromatin Immunoprecipitation (ChIP) Assay
4.5. Incorporation of 5-Ethynyl-2’-Deoxyuridine (EdU)
4.6. Luciferase Assays
4.7. Animal Studies
4.8. Immunoblot
4.9. Immunohistochemistry
4.10. Statistics
4.11. Study Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240–2256. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Tulin, A.V. The roles of PARP1 in gene control and cell differentiation. Curr. Opin. Genet. Dev. 2010, 20, 512–518. [Google Scholar] [CrossRef] [Green Version]
- Livraghi, L.; Garber, J.E. PARP inhibitors in the management of breast cancer: Current data and future prospects. BMC Med. 2015, 13, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, G.E.; Kristeleit, R.S. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: Current practice and future directions. Br. J. Cancer 2016, 115, 1157–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Pogoriler, J. Retinoblastoma family genes. Oncogene 2006, 25, 5190–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999, 342, 249–268. [Google Scholar]
- Kraus, W.L.; Lis, J.T. PARP goes transcription. Cell 2003, l113, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Smulson, M.E. Poly(ADP- ribose) polymerase upregulates E2F-1 promoter activity and DNA pol alpha expression during early S phase. Oncogene 1999, 18, 5015–5023. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Luo, R.; Samara, R.L.; Espinoza, A.; Hassa, P.O.; Hottiger, M.O.; Smulson, M.E. PARP-1 binds E2F-1 independently of its DNA binding and catalytic domains, and acts as a novel coactivator of E2F-1-mediated transcription during re-entry of quiescent cells into S phase. Oncogene 2003, 22, 8460–8471. [Google Scholar] [CrossRef] [Green Version]
- Clarke, A.R.; Maandag, E.R.; Van Roon, M.; Van der Lugt, N.M.; Van der Valk, M.; Hooper, M.L.; Berns, A.; te Riele, H. Requirement for a functional Rb-1 gene in murine development. Nature 1992, 359, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.A.; Weinberg, R.A. Effects of an Rb mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Chang, C.Y.; Hu, N.; Wang, Y.C.; Lai, C.C.; Herrup, K.; Lee, W.H.; Bradley, A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992, 359, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; De Bruin, A.; Saavedra, H.I.; Starovic, M.; Trimboli, A.; Yang, Y.; Opavska, J.; Wilson, P.; Thompson, J.C.; Ostrowski, M.C.; et al. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 2003, 421, 942–947. [Google Scholar] [CrossRef]
- Tsai, K.Y.; Hu, Y.; Macleod, K.F.; Crowley, D.; Yamasaki, L.; Jacks, T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell 1998, 2, 293–304. [Google Scholar] [CrossRef]
- Clarke, A.R. Murine models of neoplasia: Functional analysis of the tumour suppressor genes Rb-1 and p53. Cancer Metastasis Rev. 1995, 14, 125–148. [Google Scholar] [CrossRef]
- Seoane, M.; Iglesias, P.; Gonzalez, T.; Dominguez, F.; Fraga, M.; Aliste, C.; Forteza, J.; Costoya, J.A. Retinoblastoma loss modulates DNA damage response favoring tumor progression. PLoS ONE 2008, 3, e3632. [Google Scholar] [CrossRef]
- Magnaghi-Jaulin, L.; Groisman, R.; Naguibneva, I.; Robin, P.; Lorain, S.; Le Villain, J.P.; Troalen, F.; Trouche, D.; Harel-Bellan, A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 1998, 391, 601–605. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [Green Version]
- Henglein, B.; Chenivesse, X.; Wang, J.; Eick, D.; Bréchot, C. Structure and cell cycle-regulated transcription of the human cyclin A gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5490–5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, G.; Ohtani, K.; Nevins, J.R. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 1994, 8, 1514–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhrbom, L.; Nerio, E.; Holland, E.C. Dissecting tumor maintenance requirements using bioluminescence imaging of cell proliferation in a mouse glioma model. Nat. Med. 2004, 10, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, S.J.; Prater, C.A.; Dean, D.C. Retinoblastoma protein switches the E2F site from positive to negative element. Nature 1992, 358, 259–261. [Google Scholar] [CrossRef]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef] [Green Version]
- Swift, S.; Lorens, J.; Achacoso, P.; Nolan, G.P. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr. Protoc. Immunol. 2001. Chapter 10, Unit 10.17C. [Google Scholar]
- Dick, F.; Dyson, N. Three Regions of the pRB Pocket Domain Affect Its Inactivation by Human Papillomavirus E7 Proteins. J. Virol. 2002, 76, 6224–6234. [Google Scholar] [CrossRef] [Green Version]
- Rayman, J.B.; Takahashi, Y.; Indjeian, V.B.; Dannenberg, J.H.; Catchpole, S.; Watson, R.J.; te Riele, H.; Dynlacht, B.D. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002, 16, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Euhus, D.M.; Hudd, C.; LaRegina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef]
- Marino, S.; Vooijs, M.; Van Der Gulden, H.; Jonkers, J.; Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000, 14, 994–1004. [Google Scholar]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iglesias, P.; Seoane, M.; Golán, I.; Castro-Piedras, I.; Fraga, M.; Arce, V.M.; Costoya, J.A. PARP1 Deficiency Reduces Tumour Growth by Decreasing E2F1 Hyperactivation: A Novel Mechanism in the Treatment of Cancer. Cancers 2020, 12, 2907. https://doi.org/10.3390/cancers12102907
Iglesias P, Seoane M, Golán I, Castro-Piedras I, Fraga M, Arce VM, Costoya JA. PARP1 Deficiency Reduces Tumour Growth by Decreasing E2F1 Hyperactivation: A Novel Mechanism in the Treatment of Cancer. Cancers. 2020; 12(10):2907. https://doi.org/10.3390/cancers12102907
Chicago/Turabian StyleIglesias, Pablo, Marcos Seoane, Irene Golán, Isabel Castro-Piedras, Máximo Fraga, Víctor M. Arce, and Jose A. Costoya. 2020. "PARP1 Deficiency Reduces Tumour Growth by Decreasing E2F1 Hyperactivation: A Novel Mechanism in the Treatment of Cancer" Cancers 12, no. 10: 2907. https://doi.org/10.3390/cancers12102907