Dendrogenin A Enhances Anti-Leukemic Effect of Anthracycline in Acute Myeloid Leukemia


 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
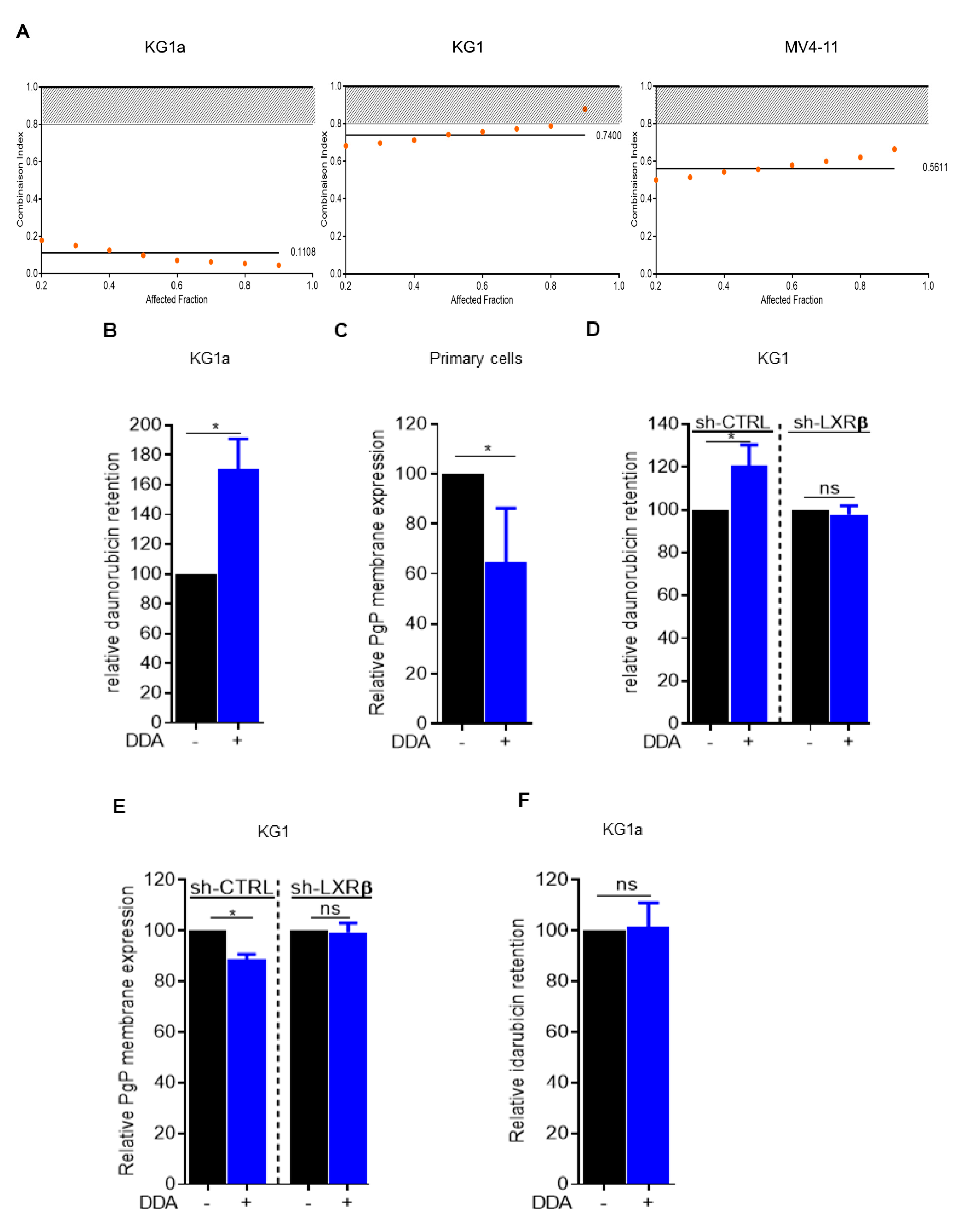
2.1. Daunorubicin Combined with DDA Synergistically Induce AML Cell Death
2.2. Idarubicin Combined with DDA Synergistically Induce AML Cell Death
2.3. Dendrogenin A Increases DNA Damage Induced by Idarubicin Treatment
2.4. DDA–Idarubicin-Induced Cell Death is Mediated by Autophagy
3. Discussion
4. Materials and Methods
4.1. Combinaison Index DDA-Daunorubicin
4.2. Antibodies and Reagents
4.3. Cell Lines and AML Primary Samples
4.4. Trypan Bleu Viability Test
4.5. Chemotherapy Efflux Test
4.6. PgP Membrane Expression
4.7. Apoptosis Quantitation by Flow Cytometry
4.8. Western Blot
4.9. Lentiviral Infection of KG-1
4.10. May-Grünwald Giemsa Staining (MGG)
4.11. Immunofluorescence
4.12. Alkalin Comet Assay
4.13. Xenograft Model and In Vivo Antitumor Activity Assay
4.14. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Medina, P.; Paillasse, M.R.; Segala, G.; Voisin, M.; Mhamdi, L.; Dalenc, F.; Lacroix-Triki, M.; Filleron, T.; Pont, F.; Saati, T.A.; et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat. Commun. 2013, 4, 1840. [Google Scholar] [CrossRef] [Green Version]
- Silvente-Poirot, S.; Poirot, M. Cholesterol and cancer, in the balance. Science 2014, 343, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903. [Google Scholar] [CrossRef] [PubMed]
- Serhan, N.; Mouchel, P.-L.; de Medina, P.; Segala, G.; Mougel, A.; Saland, E.; Rives, A.; Lamaziere, A.; Despres, G.; Sarry, J.-E.; et al. Dendrogenin A synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo. Cancers (Basel) 2020, 12, 1725. [Google Scholar] [CrossRef] [PubMed]
- de Medina, P.; Paillasse, M.R.; Payré, B.; Silvente-Poirot, S.; Poirot, M. Synthesis of new alkylaminooxysterols with potent cell differentiating activities: identification of leads for the treatment of cancer and neurodegenerative diseases. J. Med. Chem. 2009, 52, 7765–7777. [Google Scholar] [CrossRef] [PubMed]
- de Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Bailly, J.D.; Muller, C.; Jaffrézou, J.P.; Demur, C.; Gassar, G.; Bordier, C.; Laurent, G. Lack of correlation between expression and function of P-glycoprotein in acute myeloid leukemia cell lines. Leukemia 1995, 9, 799–807. [Google Scholar]
- Smeets, M.E.; Raymakers, R.A.; Vierwinden, G.; Pennings, A.H.; Boezeman, J.; Minderman, H.; de Witte, T.M. Idarubicin DNA intercalation is reduced by MRP1 and not Pgp. Leukemia 1999, 13, 1390–1398. [Google Scholar] [CrossRef] [Green Version]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Lee, J.-H.; Cheong, H.-M.; Kang, M.-Y.; Kim, S.-Y.; Kang, Y. Ser1778 of 53BP1 Plays a Role in DNA Double-strand Break Repairs. Korean J. Physiol. Pharmacol. 2009, 13, 343–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Fernandez, H.F.; Sun, Z.; Yao, X.; Litzow, M.R.; Luger, S.M.; Paietta, E.M.; Racevskis, J.; Dewald, G.W.; Ketterling, R.P.; Bennett, J.M.; et al. Anthracycline dose intensification in acute myeloid leukemia. N. Engl. J. Med. 2009, 361, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Jaffrézou, J.P.; Levade, T.; Bettaïeb, A.; Andrieu, N.; Bezombes, C.; Maestre, N.; Vermeersch, S.; Rousse, A.; Laurent, G. Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis. EMBO J. 1996, 15, 2417–2424. [Google Scholar] [CrossRef]
- Laurent, G.; Jaffrézou, J.P. Signaling pathways activated by daunorubicin. Blood 2001, 98, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M.W. Understanding the causes of multidrug resistance in cancer: a comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-E.; Hayashi, Y.K.; Bonne, G.; Arimura, T.; Noguchi, S.; Nonaka, I.; Nishino, I. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009, 5, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rello-Varona, S.; Lissa, D.; Shen, S.; Niso-Santano, M.; Senovilla, L.; Mariño, G.; Vitale, I.; Jemaá, M.; Harper, F.; Pierron, G.; et al. Autophagic removal of micronuclei. Cell Cycle 2012, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Voisin, M.; de Medina, P.; Mallinger, A.; Dalenc, F.; Huc-Claustre, E.; Leignadier, J.; Serhan, N.; Soules, R.; Ségala, G.; Mougel, A.; et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2017, 114, E9346–E9355. [Google Scholar] [CrossRef] [Green Version]
- Bauriaud-Mallet, M.; Vija-Racaru, L.; Brillouet, S.; Mallinger, A.; de Medina, P.; Rives, A.; Payre, B.; Poirot, M.; Courbon, F.; Silvente-Poirot, S. The cholesterol-derived metabolite dendrogenin A functionally reprograms breast adenocarcinoma and undifferentiated thyroid cancer cells. J. Steroid Biochem. Mol. Biol. 2019, 192, 105390. [Google Scholar] [CrossRef]
- Poirot, M.; Silvente-Poirot, S. The tumor-suppressor cholesterol metabolite, dendrogenin A, is a new class of LXR modulator activating lethal autophagy in cancers. Biochem. Pharmacol. 2018, 153, 75–81. [Google Scholar] [CrossRef]
- Toulany, M.; Lee, K.-J.; Fattah, K.R.; Lin, Y.-F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saland, E.; Boutzen, H.; Castellano, R.; Pouyet, L.; Griessinger, E.; Larrue, C.; de Toni, F.; Scotland, S.; David, M.; Danet-Desnoyers, G.; et al. A robust and rapid xenograft model to assess efficacy of chemotherapeutic agents for human acute myeloid leukemia. Blood Cancer J. 2015, 5, e297. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Karyotype | FLT3-ITD | FLT3-TKD | NPM1 | CEBPA | IDH1 | IDH2 | DNMT3A |
|---|---|---|---|---|---|---|---|---|
| AML1 | 46, XY<20> | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| AML2 | 46, XX<20> | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| AML3 | 46, XX<20> | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| AML4 | 46, XX, del(20)(q11q13)<20> | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| AML5 | 46, XX<20> | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| AML6 | 46, XX, t(1;16)(010;q10),inv(3)(014q24)<20> | ND | ND | ND | ND | ND | ND | ND |
| AML7 | 46, XX<20> | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| AML8 | 46, XY, r(7)<16> | ND | ND | ND | ND | ND | ND | ND |
| AML9 | 46, XX<20> | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| AML10 | 46, XX<20> | 1 | 0 | 0 | 0 | 1 | 1 | 0 |
| AML11 | 46, XY<22> | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| AML12 | 46, XX<20> | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| AML13 | 46, XY<20> | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| AML14 | 46,XX,+8<1>/47,der(11)t(1;11)(q24;q24)<1>/48,sd1,+8<2>/46,XX<7> | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| AML15 | 46, XY<20> | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| AML16 | 46, XY<20> | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| AML17 | 46, XY, inv(16)(p13q22)<10>/46,XY<10> | ND | ND | ND | ND | ND | ND | ND |
| AML18 | 46, XX<22> | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| AML19 | 47, XY, inv(16)(p13q22),+22<20> | ND | ND | ND | ND | ND | ND | ND |
| AML20 | 47, XY, +8, inv(16)(p13q22)<20> | ND | ND | ND | ND | ND | ND | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mouchel, P.-L.; Serhan, N.; Betous, R.; Farge, T.; Saland, E.; De Medina, P.; Hoffmann, J.-S.; Sarry, J.-E.; Poirot, M.; Silvente-Poirot, S.; et al. Dendrogenin A Enhances Anti-Leukemic Effect of Anthracycline in Acute Myeloid Leukemia. Cancers 2020, 12, 2933. https://doi.org/10.3390/cancers12102933
Mouchel P-L, Serhan N, Betous R, Farge T, Saland E, De Medina P, Hoffmann J-S, Sarry J-E, Poirot M, Silvente-Poirot S, et al. Dendrogenin A Enhances Anti-Leukemic Effect of Anthracycline in Acute Myeloid Leukemia. Cancers. 2020; 12(10):2933. https://doi.org/10.3390/cancers12102933
Chicago/Turabian StyleMouchel, Pierre-Luc, Nizar Serhan, Rémy Betous, Thomas Farge, Estelle Saland, Philippe De Medina, Jean-Sébastien Hoffmann, Jean-Emmanuel Sarry, Marc Poirot, Sandrine Silvente-Poirot, and et al. 2020. "Dendrogenin A Enhances Anti-Leukemic Effect of Anthracycline in Acute Myeloid Leukemia" Cancers 12, no. 10: 2933. https://doi.org/10.3390/cancers12102933