Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer


Abstract
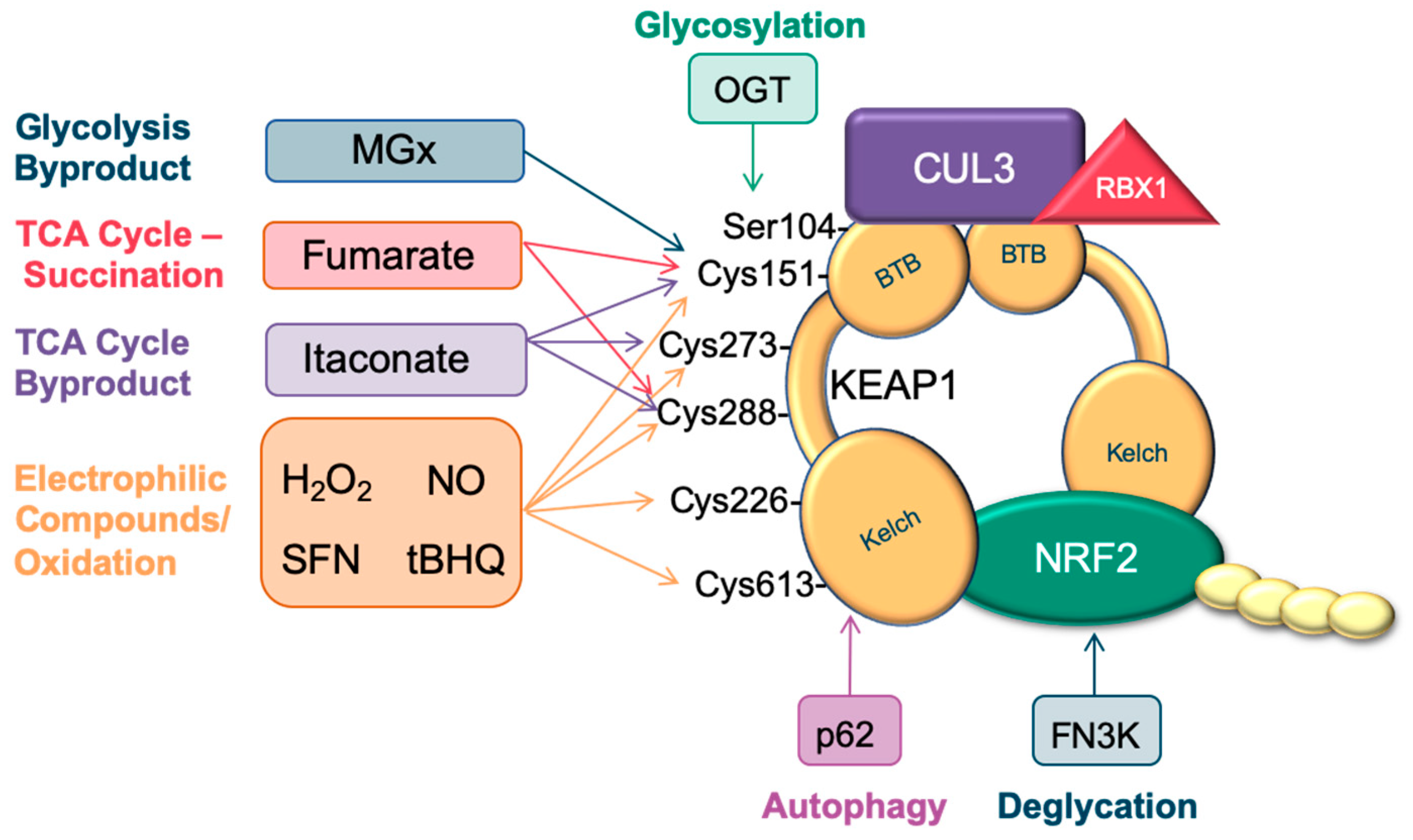
:Simple Summary
Abstract
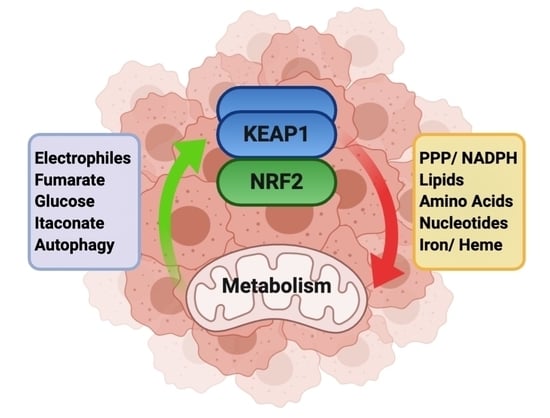
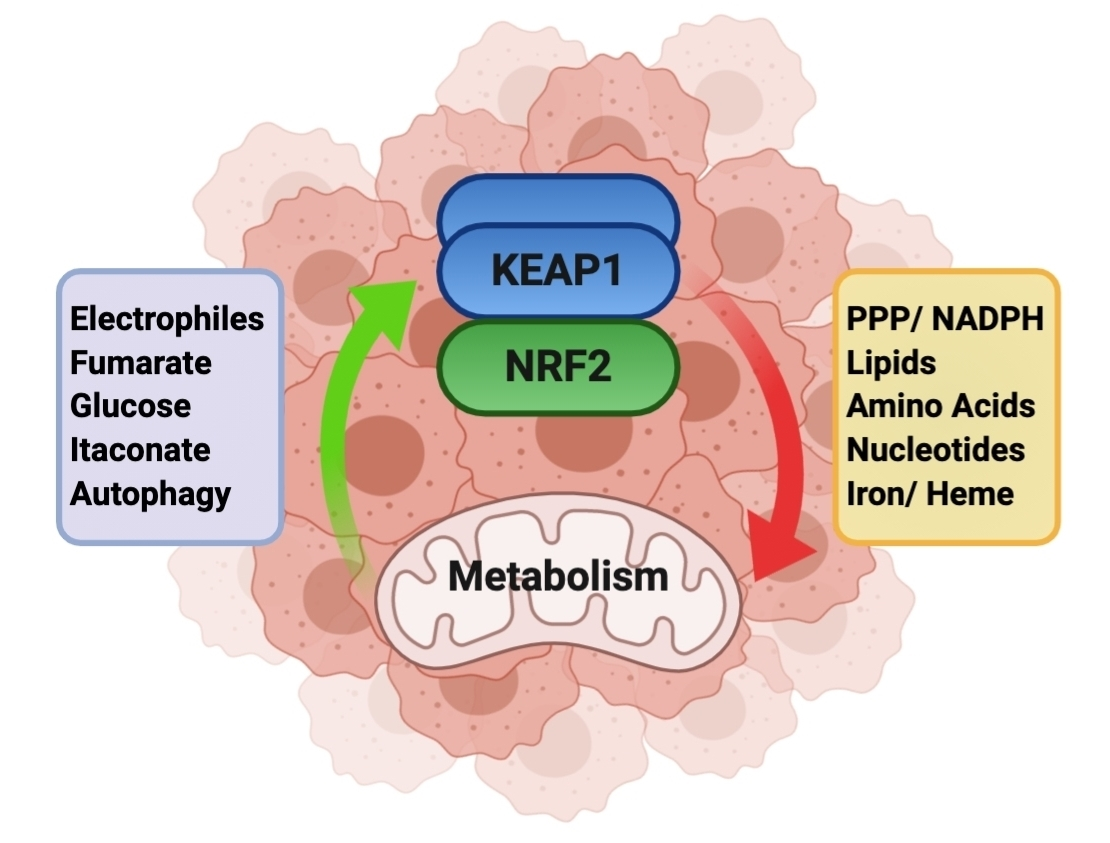{kind=link}
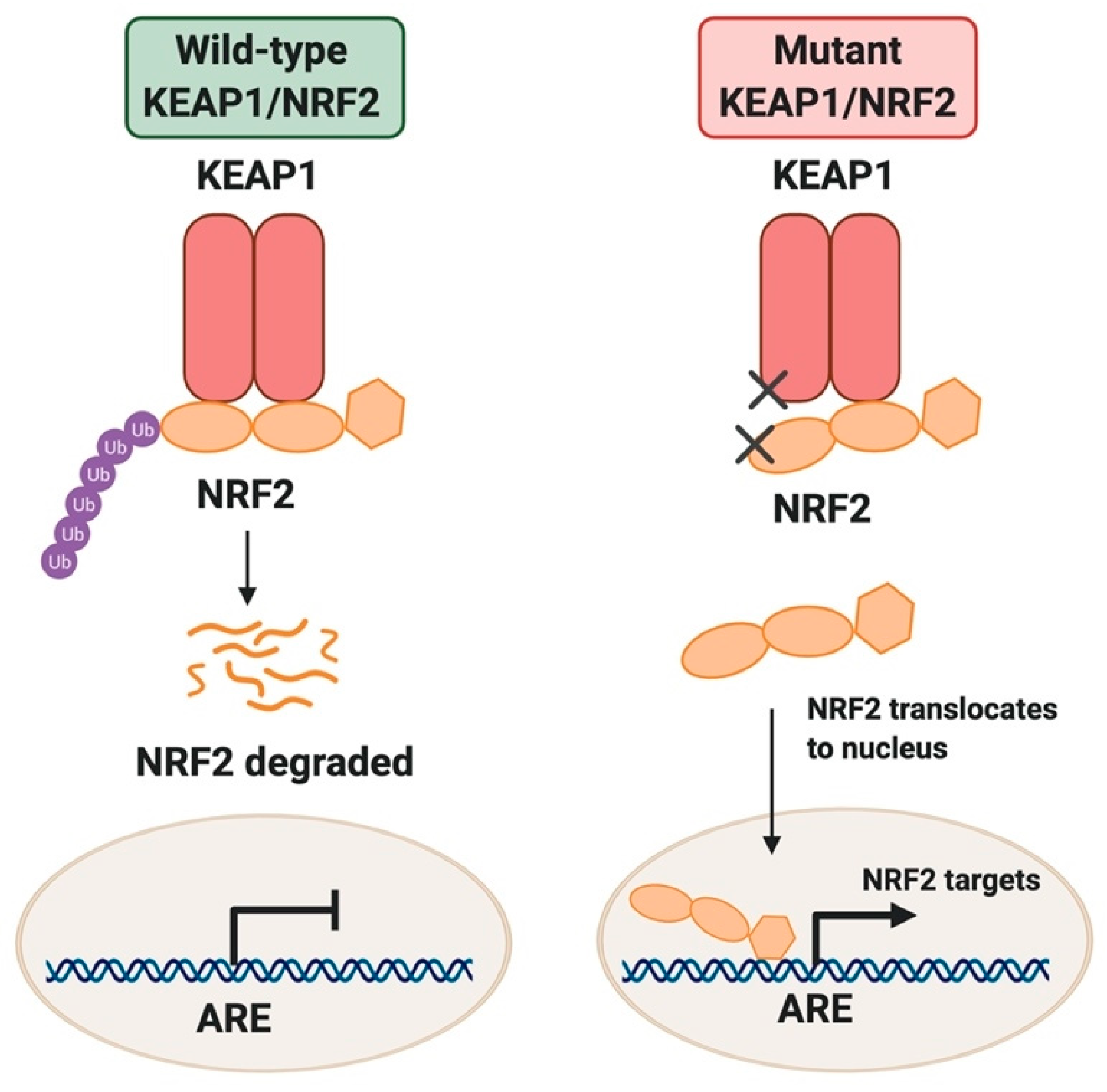{kind=link}
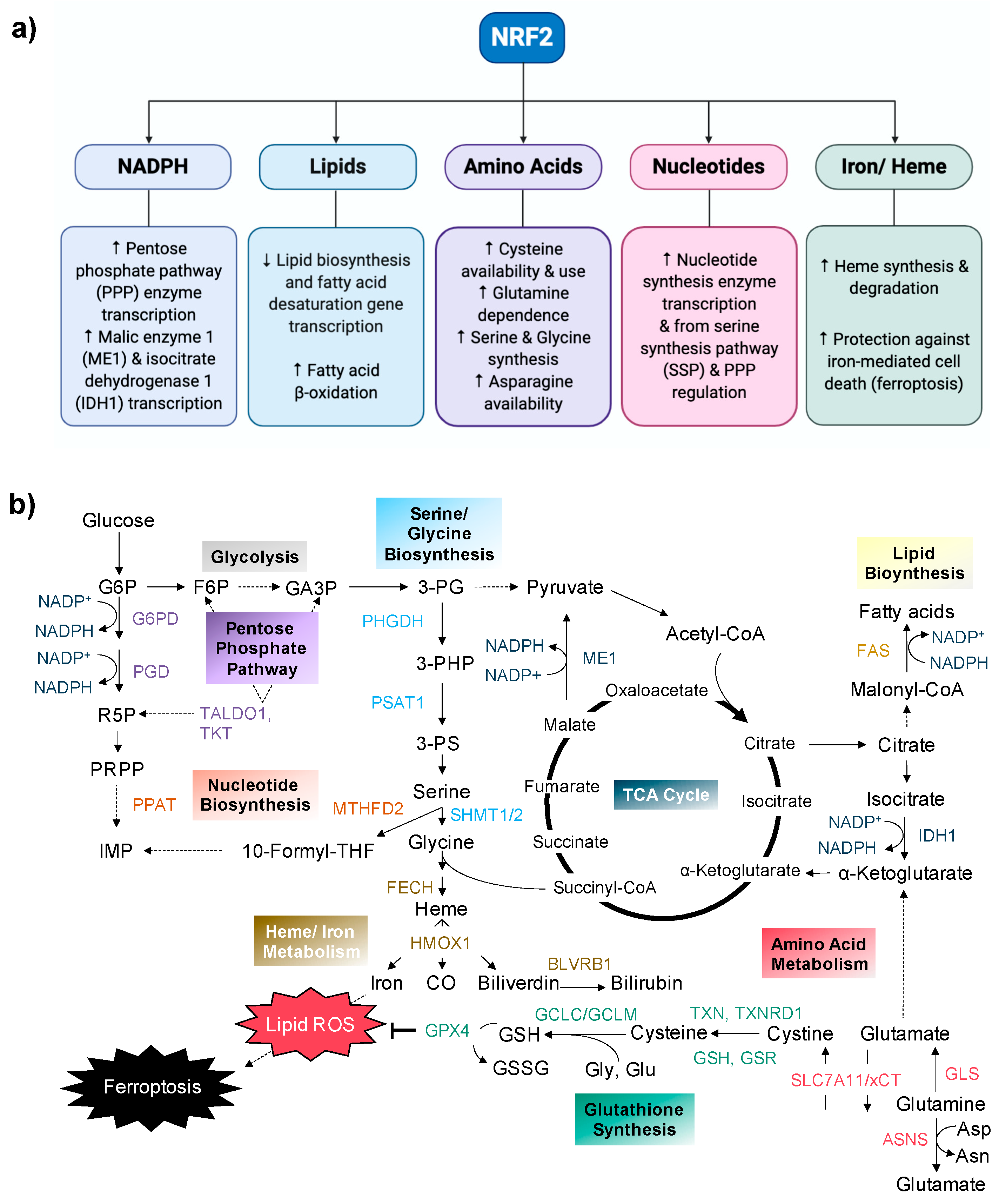{kind=link}
{kind=link}
1. Introduction
2. Modulation of Metabolic Processes by NRF2
2.1. NADPH Production
2.2. Lipid Metabolism
2.3. Amino Acid Metabolism
2.3.1. Cysteine, Cystine and Glutathione
2.3.2. Glutamine
2.3.3. Serine/Glycine
2.3.4. Asparagine
2.4. Nucleotide Metabolism
2.5. Iron/Heme
3. Metabolic Pathways that Stabilize NRF2
3.1. Electrophiles
3.2. Fumarate
3.3. Glucose
3.4. Itaconate and the Immune System
3.5. Autophagy
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Singh, A.; Bodas, M.; Wakabayashi, N.; Bunz, F.; Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal. 2010, 13, 1627–1637. [Google Scholar] [CrossRef] [Green Version]
- Torrente, L.; Prieto-Farigua, N.; Falzone, A.; Elkins, C.M.; Boothman, D.A.; Haura, E.B.; DeNicola, G.M. Inhibition of TXNRD or SOD1 overcomes NRF2-mediated resistance to β-lapachone. Redox Biol. 2020, 30, 101440. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.; Hoang, N.T.; Lovejoy, A.; Stehr, H.; Newman, A.M.; Gentles, A.J.; Kong, W.; Truong, D.; Martin, S.; Chaudhuri, A.; et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017, 7, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the KEAP1/NRF2 and PI3K Pathways Drives Non-Small-Cell Lung Cancer with an Altered Immune Microenvironment. Cell Metab. 2018, 27, 935–943.e4. [Google Scholar] [CrossRef] [Green Version]
- Rogers, Z.N.; McFarland, C.D.; Winters, I.P.; Seoane, J.A.; Brady, J.J.; Yoon, S.; Curtis, C.; Petrov, D.A.; Winslow, M.M. Mapping the in vivo fitness landscape of lung adenocarcinoma tumor suppression in mice. Nat. Genet. 2018, 50, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Ding, S.; Kersbergen, A.; Dong, X.; Song, J.Y.; Xie, Y.; Reljic, B.; Li, K.; Vince, J.E.; Rathi, V.; et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329.e18. [Google Scholar] [CrossRef]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; Denicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. eLife 2019, 8, e45572. [Google Scholar] [CrossRef]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonrungruan, C.; Major, M.B.; Chen, K.N.; Kang, X.; et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sánchez-Rivera, F.J.; Westcott, P.M.K.; Mercer, K.L.; Bhutkar, A.; Muir, A.; González Robles, T.J.; Lamboy Rodríguez, S.; Liao, L.Z.; Ng, S.R.; et al. Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nat. Cancer 2020, 1, 589–602. [Google Scholar] [CrossRef]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 1–12. [Google Scholar] [CrossRef]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 2020, 37, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Happel, C.; Manna, S.K.; Acquaah-Mensah, G.; Carrerero, J.; Kumar, S.; Nasipuri, P.; Krausz, K.W.; Wakabayashi, N.; Dewi, R.; et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 2013, 123, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Ludtmann, M.H.R.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Slocum, S.L.; Skoko, J.J.; Wakabayashi, N.; Aja, S.; Yamamoto, M.; Kensler, T.W.; Chartoumpekis, D.V. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch. Biochem. Biophys. 2016, 591, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Bowman, B.M.; Montgomery, S.A.; Schrank, T.P.; Simon, J.M.; Ptacek, T.S.; Tamir, T.Y.; Muvlaney, K.M.; Weir, S.J.; Nguyen, T.T.; Murphy, R.M.; et al. A conditional mouse expressing an activating mutation in NRF2 displays hyperplasia of the upper gastrointestinal tract and decreased white adipose tissue. J. Pathol 2020, 252, 125–137. [Google Scholar] [CrossRef]
- Suzuki, T.; Uruno, A.; Yumoto, A.; Taguchi, K.; Suzuki, M.; Harada, N.; Ryoke, R.; Naganuma, E.; Osanai, N.; Goto, A.; et al. Nrf2 contributes to the weight gain of mice during space travel. Commun. Biol. 2020, 3, 496. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 2017, 6, e27713. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xu, J.; Zheng, Q.; He, J.; Zhou, W.; Wang, K.; Huang, X.; Fan, Q.; Ma, J.; Cheng, J.; et al. NRF2 SUMOylation promotes de novo serine synthesis and maintains HCC tumorigenesis. Cancer Lett. 2019, 466, 39–48. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.R.; Karaca, M.; Adamski, K.N.; Chorley, B.N.; Wang, X.; Bell, D.A. Novel hematopoietic target genes in the NRF2-mediated transcriptional pathway. Oxid. Med. Cell Longev. 2013, 2013, 120305. [Google Scholar] [CrossRef] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.A.; Hagel, K.R.; Hawk, M.A.; Schafer, Z.T. Metabolism during ECM Detachment: Achilles Heel of Cancer Cells? Trends Cancer 2017, 3, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Badur, M.G.; Luebeck, J.; Magana, J.H.; Birmingham, A.; Sasik, R.; Ahn, C.S.; Ideker, T.; Metallo, C.M.; Mali, P. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol. Cell 2018, 69, 699–708.e697. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svensson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J.; et al. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, R.G.; Seres, T.; Lam, G.; Johnston, R.B., Jr.; Warshaw, J.B. Characterization of multiple cysteine and cystine transporters in rat alveolar type II cells. Am. J. Physiol. 1997, 273, L1147–L1155. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef]
- Combs, J.A.; DeNicola, G.M. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers (Basel) 2019, 11, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e127. [Google Scholar] [CrossRef] [Green Version]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 2017, 6, e28083. [Google Scholar] [CrossRef]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016, 76, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Briggs, K.J.; Koivunen, P.; Cao, S.; Backus, K.M.; Olenchock, B.A.; Patel, H.; Zhang, Q.; Signoretti, S.; Gerfen, G.J.; Richardson, A.L.; et al. Paracrine Induction of HIF by Glutamate in Breast Cancer: EglN1 Senses Cysteine. Cell 2016, 166, 126–139. [Google Scholar] [CrossRef] [Green Version]
- Sleire, L.; Skeie, B.S.; Netland, I.A.; Forde, H.E.; Dodoo, E.; Selheim, F.; Leiss, L.; Heggdal, J.I.; Pedersen, P.H.; Wang, J.; et al. Drug repurposing: Sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene 2015, 34, 5951–5959. [Google Scholar] [CrossRef]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased Expression of System xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, H.J.; Karlenius, T.C.; Tonissen, K.F. Regulation of the human thioredoxin gene promoter and its key substrates: A study of functional and putative regulatory elements. Biochim. Biophys. Acta 2014, 1840, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef]
- Moinova, H.R.; Mulcahy, R.T. Up-regulation of the human gamma-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem. Biophys. Res. Commun. 1999, 261, 661–668. [Google Scholar] [CrossRef]
- Solis, W.A.; Dalton, T.P.; Dieter, M.Z.; Freshwater, S.; Harrer, J.M.; He, L.; Shertzer, H.G.; Nebert, D.W. Glutamate-cysteine ligase modifier subunit: Mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem. Pharmacol. 2002, 63, 1739–1754. [Google Scholar] [CrossRef]
- Bea, F.; Hudson, F.N.; Chait, A.; Kavanagh, T.J.; Rosenfeld, M.E. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ. Res. 2003, 92, 386–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekhar, K.R.; Crooks, P.A.; Sonar, V.N.; Friedman, D.B.; Chan, J.Y.; Meredith, M.J.; Starnes, J.H.; Kelton, K.R.; Summar, S.R.; Sasi, S.; et al. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003, 63, 5636–5645. [Google Scholar]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBoeuf, S.E.; Wu, W.L.; Karakousi, T.R.; Karadal, B.; Jackson, S.R.; Davidson, S.M.; Wong, K.K.; Koralov, S.B.; Sayin, V.I.; Papagiannakopoulos, T. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020, 31, 339–350.e334. [Google Scholar] [CrossRef]
- Agyeman, A.S.; Chaerkady, R.; Shaw, P.G.; Davidson, N.E.; Visvanathan, K.; Pandey, A.; Kensler, T.W. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res. Treat. 2012, 132, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.M.; Ye, J. Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165841. [Google Scholar] [CrossRef]
- McMillan, E.A.; Ryu, M.J.; Diep, C.H.; Mendiratta, S.; Clemenceau, J.R.; Vaden, R.M.; Kim, J.H.; Motoyaji, T.; Covington, K.R.; Peyton, M.; et al. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 2018, 173, 864–878.e829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.P.; Falzone, A.; Liu, M.; Gonzalez-Sanchez, P.; Choi, B.H.; Coloff, J.L.; Saller, J.J.; Karreth, F.A.; DeNicola, G.M. PHGDH supports liver ceramide synthesis and sustains lipid homeostasis. Cancer Metab. 2020, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Gantner, M.L.; Eade, K.; Wallace, M.; Handzlik, M.K.; Fallon, R.; Trombley, J.; Bonelli, R.; Giles, S.; Harkins-Perry, S.; Heeren, T.F.C.; et al. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N. Engl. J. Med. 2019, 381, 1422–1433. [Google Scholar] [CrossRef]
- Muthusamy, T.; Cordes, T.; Handzlik, M.K.; You, L.; Lim, E.W.; Gengatharan, J.; Pinto, A.F.M.; Badur, M.G.; Kolar, M.J.; Wallace, M.; et al. Serine restriction alters sphingolipid diversity to constrain tumour growth. Nature 2020, 10, 1440. [Google Scholar] [CrossRef]
- Gao, X.; Lee, K.; Reid, M.A.; Sanderson, S.M.; Qiu, C.; Li, S.; Liu, J.; Locasale, J.W. Serine Availability Influences Mitochondrial Dynamics and Function through Lipid Metabolism. Cell Rep. 2018, 22, 3507–3520. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Tao, Y.; Duran, A.; Llado, V.; Galvez, A.; Barger, J.F.; Castilla, E.A.; Chen, J.; Yajima, T.; Porollo, A.; et al. Control of nutrient stress-induced metabolic reprogramming by PKCζ in tumorigenesis. Cell 2013, 152, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Ou, Y.; Wang, S.J.; Jiang, L.; Zheng, B.; Gu, W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J. Biol. Chem. 2015, 290, 457–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef]
- Ngo, B.; Kim, E.; Osorio-Vasquez, V.; Doll, S.; Bustraan, S.; Liang, R.J.; Luengo, A.; Davidson, S.M.; Ali, A.; Ferraro, G.B.; et al. Limited Environmental Serine and Glycine Confer Brain Metastasis Sensitivity to PHGDH Inhibition. Cancer Discov. 2020, 10, 1352–1373. [Google Scholar] [CrossRef]
- Shin, D.Y.; Na, I.I.; Kim, C.H.; Park, S.; Baek, H.; Yang, S.H. EGFR mutation and brain metastasis in pulmonary adenocarcinomas. J. Thorac. Oncol. 2014, 9, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, L.E.; Smit, E.F.; Vosse, B.A.; Mellema, W.W.; Heideman, D.A.; Bootsma, G.P.; Westenend, M.; Pitz, C.; de Vries, G.J.; Houben, R.; et al. EGFR mutated non-small cell lung cancer patients: More prone to development of bone and brain metastases? Lung Cancer 2014, 84, 86–91. [Google Scholar] [CrossRef]
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific organ metastases and survival in metastatic non-small-cell lung cancer. Mol. Clin. Oncol. 2015, 3, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, M.N.; Butterworth, E.A.; Kilberg, M.S. Asparagine synthetase: Regulation by cell stress and involvement in tumor biology. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E789–E799. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef] [Green Version]
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Kammerscheit, X.; Hecker, A.; Rouhier, N.; Chauvat, F.; Cassier-Chauvat, C. Methylglyoxal Detoxification Revisited: Role of Glutathione Transferase in Model Cyanobacterium Synechocystis sp. Strain PCC 6803. mBio 2020, 11, e00882-20. [Google Scholar] [CrossRef]
- Nishimoto, S.; Koike, S.; Inoue, N.; Suzuki, T.; Ogasawara, Y. Activation of Nrf2 attenuates carbonyl stress induced by methylglyoxal in human neuroblastoma cells: Increase in GSH levels is a critical event for the detoxification mechanism. Biochem. Biophys. Res. Commun. 2017, 483, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Smith, T.J.; Wu, J.; Siesser, P.F.; Bisnett, B.J.; Khan, F.; Hogue, M.; Soderblom, E.; Tang, F.; Marks, J.R.; et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017, 36, 2233–2250. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; de Stanchina, E.; et al. The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819.e821. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef]
- Hiramoto, K.; Satoh, H.; Suzuki, T.; Moriguchi, T.; Pi, J.; Shimosegawa, T.; Yamamoto, M. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. (Phila) 2014, 7, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental Activation of Nrf2 Restricts the Progression of Nrf2-Activated Malignant Tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. IRG1/Itaconate Activates Nrf2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology 2020. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.A.; Li, Y.; Meliton, A.Y.; Woods, P.S.; Kimmig, L.M.; Cetin-Atalay, R.; Hamanaka, R.B.; Mutlu, G.M. Endogenous itaconate is not required for particulate matter-induced NRF2 expression or inflammatory response. Elife 2020, 9, e54877. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient kras-driven lung tumorigenesis. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839.e828. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Karsli-Uzunbas, G.; Poillet-Perez, L.; Sawant, A.; Hu, Z.S.; Zhao, Y.; Moore, D.; Hu, W.; White, E. Autophagy promotes mammalian survival by suppressing oxidative stress and p53. Genes Dev. 2020, 34, 688–700. [Google Scholar] [CrossRef]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Kageyama, S.; Sou, Y.S.; Uemura, T.; Kametaka, S.; Saito, T.; Ishimura, R.; Kouno, T.; Bedford, L.; Mayer, R.J.; Lee, M.S.; et al. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J. Biol. Chem. 2014, 289, 24944–24955. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Jiang, C.; Gao, J. Transcriptional factor Nrf2 is essential for aggresome formation during proteasome inhibition. Biomed. Rep. 2019, 11, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.N.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell Biol. 2012, 32, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Mulvaney, K.M.; Matson, J.P.; Siesser, P.F.; Tamir, T.Y.; Goldfarb, D.; Jacobs, T.M.; Cloer, E.W.; Harrison, J.S.; Vaziri, C.; Cook, J.G.; et al. Identification and characterization of MCM3 as a kelch-like ECH-associated protein 1 (KEAP1) substrate. J. Biol. Chem. 2016, 291, 23719–23733. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.C.; Hannink, M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res. 2008, 314, 1789–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.; Srivastava, S.; Li, Z.; Vaeth, M.; Fuhs, S.R.; Hunter, T.; Skolnik, E.Y. Identification of PGAM5 as a Mammalian Protein Histidine Phosphatase that Plays a Central Role to Negatively Regulate CD4(+) T Cells. Mol. Cell 2016, 63, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, A.; Ishikawa, H.; Furuoka, M.; Yokozeki, M.; Matsuda, N.; Tanimura, S.; Takeda, K. Cleaved PGAM5 is released from mitochondria depending on proteasome-mediated rupture of the outer mitochondrial membrane during mitophagy. J. Biochem. 2019, 165, 19–25. [Google Scholar] [CrossRef]
- O’Mealey, G.B.; Plafker, K.S.; Berry, W.L.; Janknecht, R.; Chan, J.Y.; Plafker, S.M. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J. Cell Sci. 2017, 130, 3467–3480. [Google Scholar] [CrossRef] [Green Version]
- Holze, C.; Michaudel, C.; MacKowiak, C.; Haas, D.A.; Benda, C.; Hubel, P.; Pennemann, F.L.; Schnepf, D.; Wettmarshausen, J.; Braun, M.; et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat. Immunol. 2018, 19, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gu, L.; Lin, X.; Liu, C.; Lu, B.; Cui, K.; Zhou, F.; Zhao, Q.; Prochownik, E.V.; Fan, C.; et al. Dynamic Regulation of ME1 Phosphorylation and Acetylation Affects Lipid Metabolism and Colorectal Tumorigenesis. Mol. Cell 2020, 77, 138–149.e135. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeBlasi, J.M.; DeNicola, G.M. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers 2020, 12, 3023. https://doi.org/10.3390/cancers12103023
DeBlasi JM, DeNicola GM. Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers. 2020; 12(10):3023. https://doi.org/10.3390/cancers12103023
Chicago/Turabian StyleDeBlasi, Janine M., and Gina M. DeNicola. 2020. "Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer" Cancers 12, no. 10: 3023. https://doi.org/10.3390/cancers12103023
APA StyleDeBlasi, J. M., & DeNicola, G. M. (2020). Dissecting the Crosstalk between NRF2 Signaling and Metabolic Processes in Cancer. Cancers, 12(10), 3023. https://doi.org/10.3390/cancers12103023