De Novo T790M Mutation in an L858R Epidermal Growth Factor Receptor Mutant-Associated Lung Adenocarcinoma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
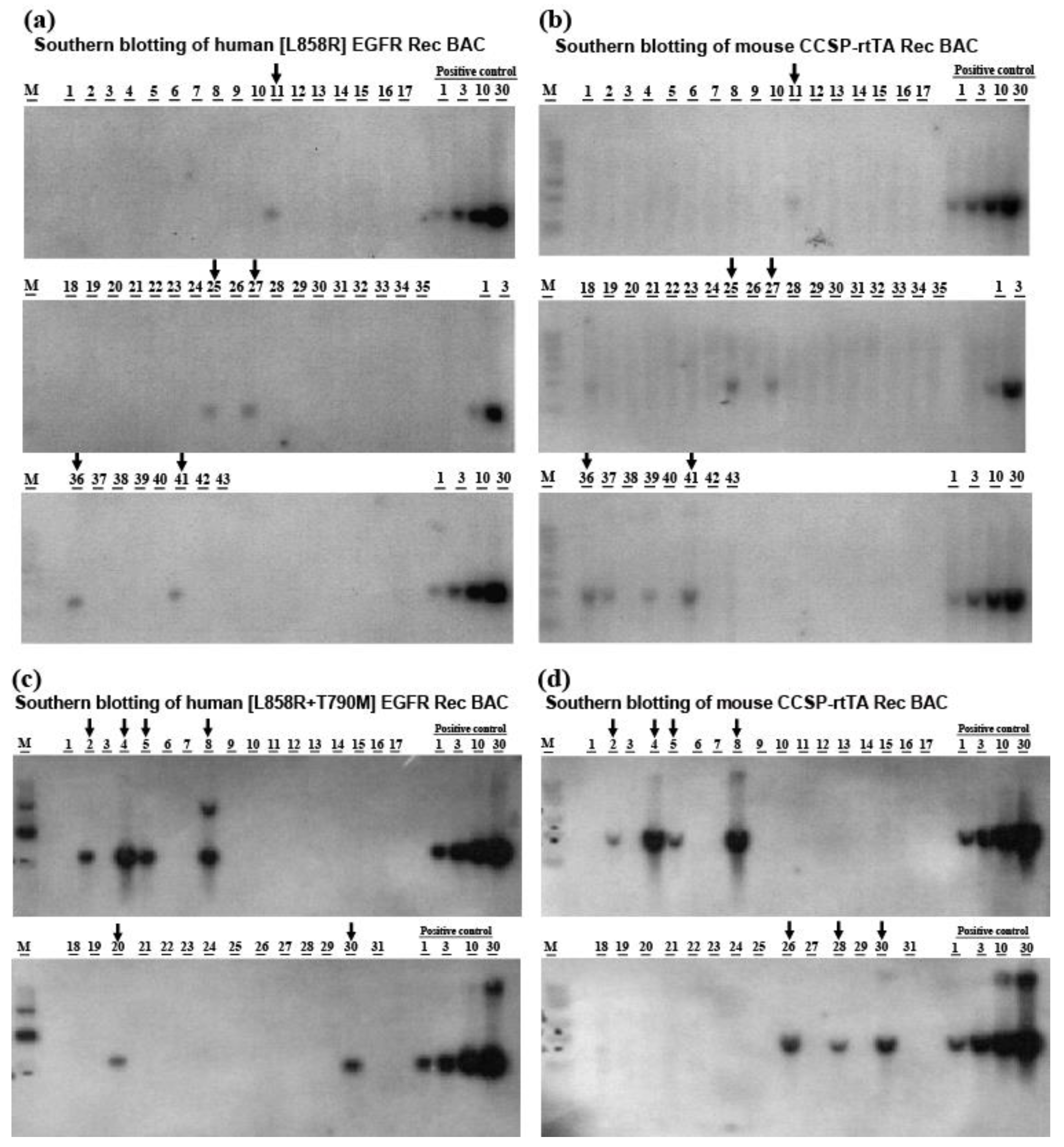
2.1. Generation of Transgenic Mice Carrying Lung-Specific TetO-[L858R]-hEGFR or TetO-[L858R+T790M]-hEGFR BAC Transgenes
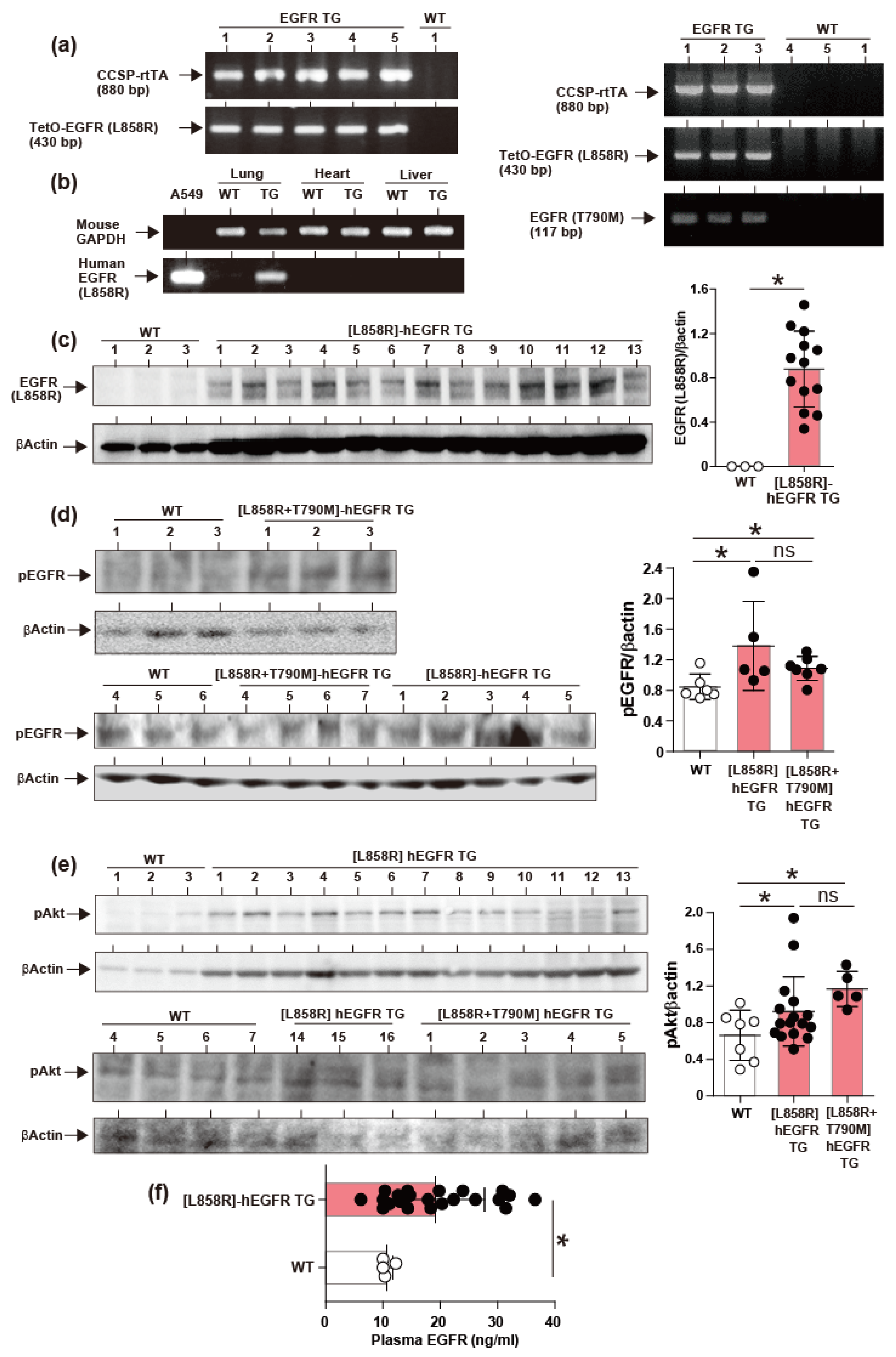
2.2. Induction of hEGFR Transgene Expression
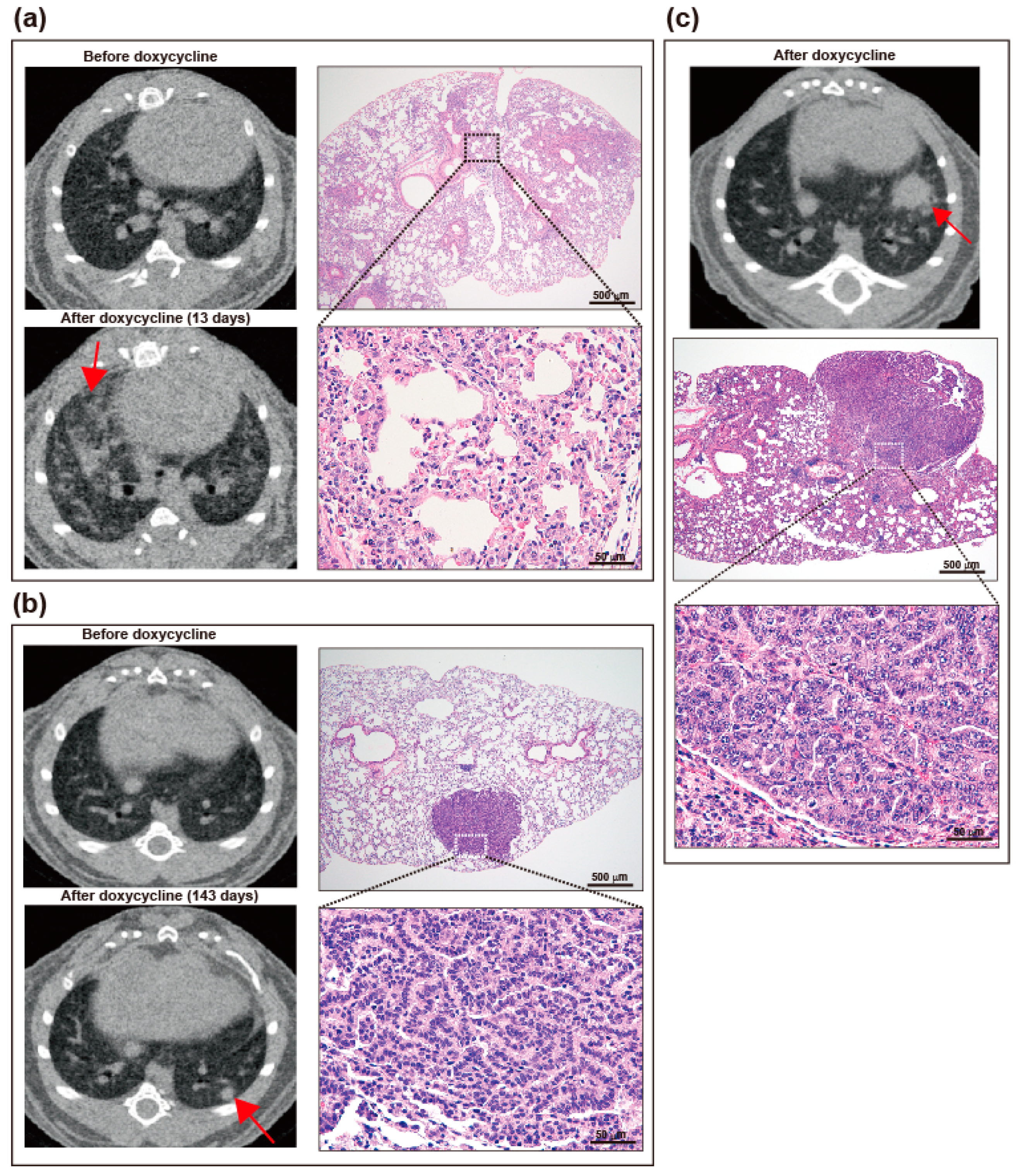
2.3. Induction of Lung Tumors in TG Mice on Doxycycline
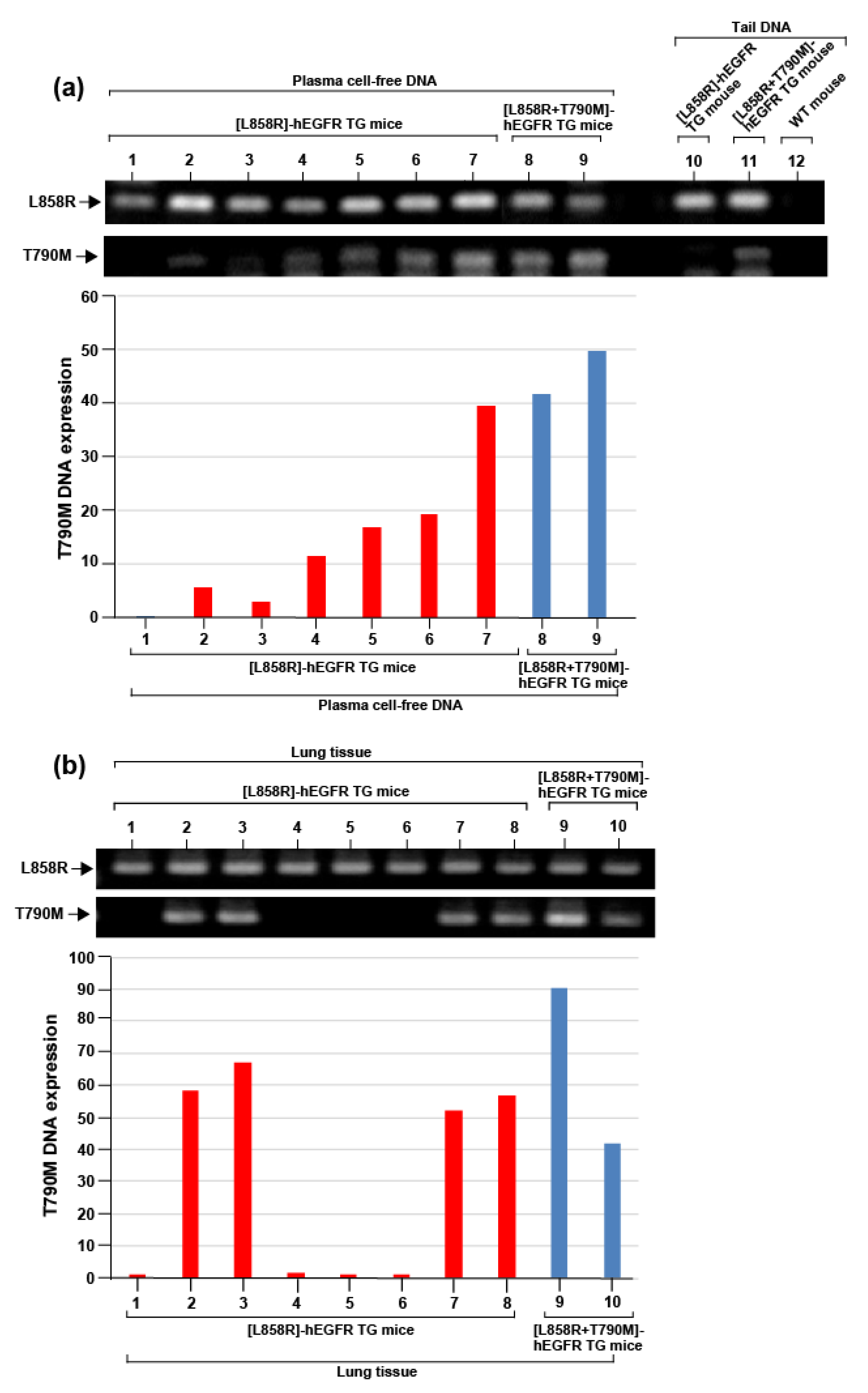
2.4. Acquisition of De Novo T790M Mutant in [L858R]-hEGFR Bitransgenic Mouse Lung Tumor
3. Discussion
4. Materials and Methods
4.1. Preparation of [L858R] Human EGFR and [L858R+T790M] Human EGFR Recombinant Bacterial Artificial Chromosome Expression Constructs
4.2. Preparation of the CCSP-rtTA Recombinant BAC Expression Construct
4.3. Ethical Statement
4.4. Screening of Lung Tumors by Micro-Computed Tomography (CT)
4.5. Plasma Sampling
4.6. Lung Tissue Examination
4.7. Biochemical Analysis
4.8. Western Blotting
4.9. Evaluation of Gene Expression by PCR
4.10. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Latimer, K.M.; Mott, T.F. Lung cancer: Diagnosis, treatment principles, and screening. Am. Fam. Physician 2015, 91, 250–256. [Google Scholar] [PubMed]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.; Yatabe, Y.; Powell, C.A.; Beer, D.; Riely, G.; Garg, K.; et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society: International multidisciplinary classification of lung adenocarcinoma: Executive summary. Proc. Am. Thorac. Soc. 2011, 8, 381–385. [Google Scholar] [CrossRef]
- Socinski, M.A.; Obasaju, C.; Gandara, D.; Hirsch, F.R.; Bonomi, P.; Bunn, P.; Kim, E.S.; Langer, C.J.; Natale, R.B.; Novello, S.; et al. Clinicopathologic Features of Advanced Squamous NSCLC. J. Thorac. Oncol. 2016, 11, 1411–1422. [Google Scholar] [CrossRef]
- Devarakonda, S.; Morgensztern, D.; Govindan, R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015, 16, e342–e351. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef]
- Regales, L.; Balak, M.N.; Gong, Y.; Politi, K.; Sawai, A.; Le, C.; Koutcher, J.A.; Solit, D.B.; Rosen, N.; Zakowski, M.F.; et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS ONE 2007, 2, e810. [Google Scholar] [CrossRef]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Li, D.; Chen, L.; Shimamura, T.; Kobayashi, S.; McNamara, K.; Mahmood, U.; Mitchell, A.; Sun, Y.; Al-Hashem, R.; et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 2006, 9, 485–495. [Google Scholar] [CrossRef]
- Xu, X.; Liu, T.; Wang, Y.; Fu, J.; Yang, Q.; Wu, J.; Zhou, H. miRNA-mRNA Associated with Survival in Endometrial Cancer. Front. Genet. 2019, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Reyes, A.; Hayer, K.E.; Bassing, C.H. Genomic Alterations of Non-Coding Regions Underlie Human Cancer: Lessons from T-ALL. Trends Mol. Med. 2016, 22, 1035–1046. [Google Scholar] [CrossRef][Green Version]
- Oldridge, D.A.; Wood, A.C.; Weichert-Leahey, N.; Crimmins, I.; Sussman, R.; Winter, C.; McDaniel, L.D.; Diamond, M.; Hart, L.S.; Zhu, S.; et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015, 528, 418–421. [Google Scholar] [CrossRef]
- Liu, C.; Tian, X.; Zhang, J.; Jiang, L. Long Non-coding RNA DLEU1 Promotes Proliferation and Invasion by Interacting With miR-381 and Enhancing HOXA13 Expression in Cervical Cancer. Front. Genet. 2018, 9, 629. [Google Scholar] [CrossRef]
- Abildgaard, C.; Do Canto, L.M.; Steffensen, K.D.; Rogatto, S.R. Long Non-coding RNAs Involved in Resistance to Chemotherapy in Ovarian Cancer. Front. Oncol. 2019, 9, 1549. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wei, S.; Song, Y. T790M and acquired resistance of EGFR TKI: A literature review of clinical reports. J. Thorac. Dis. 2011, 3, 10–18. [Google Scholar]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef]
- Abe, K.; Hazama, M.; Katoh, H.; Yamamura, K.; Suzuki, M. Establishment of an efficient BAC transgenesis protocol and its application to functional characterization of the mouse Brachyury locus. Exp. Anim. 2004, 53, 311–320. [Google Scholar] [CrossRef][Green Version]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Boveda-Ruiz, D.; Takagi, T.; Toda, M.; Gil-Bernabe, P.; Miyake, Y.; Yasukawa, A.; Matsuda, Y.; Suzuki, N.; et al. Development and preclinical efficacy of novel transforming growth factor-beta1 short interfering RNAs for pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 397–406. [Google Scholar] [CrossRef]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Yasuma, T.; Toda, M.; Kim, H.; Fujimoto, H.; Hataji, O.; Takeshita, A.; Nishihama, K.; Okano, T.; et al. A Staphylococcus pro-apoptotic peptide induces acute exacerbation of pulmonary fibrosis. Nat. Commun. 2020, 11, 1539. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Kobayashi, T.; Fujimoto, H.; Nakahara, H.; D’Alessandro-Gabazza, C.N.; Hinneh, J.A.; Takahashi, Y.; Yasuma, T.; Nishihama, K.; Toda, M.; et al. Inhibition of Cell Apoptosis and Amelioration of Pulmonary Fibrosis by Thrombomodulin. Am. J. Pathol. 2017, 187, 2312–2322. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiwara, T.; Kobayashi, T.; Yasuma, T.; D’Alessandro-Gabazza, C.N.; Toda, M.; Fujimoto, H.; Fujiwara, K.; Takeshita, A.; Nishihama, K.; Okano, T.; et al. De Novo T790M Mutation in an L858R Epidermal Growth Factor Receptor Mutant-Associated Lung Adenocarcinoma. Cancers 2020, 12, 3074. https://doi.org/10.3390/cancers12103074
Fujiwara T, Kobayashi T, Yasuma T, D’Alessandro-Gabazza CN, Toda M, Fujimoto H, Fujiwara K, Takeshita A, Nishihama K, Okano T, et al. De Novo T790M Mutation in an L858R Epidermal Growth Factor Receptor Mutant-Associated Lung Adenocarcinoma. Cancers. 2020; 12(10):3074. https://doi.org/10.3390/cancers12103074
Chicago/Turabian StyleFujiwara, Takumi, Tetsu Kobayashi, Taro Yasuma, Corina N. D’Alessandro-Gabazza, Masaaki Toda, Hajime Fujimoto, Kentaro Fujiwara, Atsuro Takeshita, Kota Nishihama, Tomohito Okano, and et al. 2020. "De Novo T790M Mutation in an L858R Epidermal Growth Factor Receptor Mutant-Associated Lung Adenocarcinoma" Cancers 12, no. 10: 3074. https://doi.org/10.3390/cancers12103074
APA StyleFujiwara, T., Kobayashi, T., Yasuma, T., D’Alessandro-Gabazza, C. N., Toda, M., Fujimoto, H., Fujiwara, K., Takeshita, A., Nishihama, K., Okano, T., D’Alessandro, V. F., Takei, Y., Hataji, O., & Gabazza, E. C. (2020). De Novo T790M Mutation in an L858R Epidermal Growth Factor Receptor Mutant-Associated Lung Adenocarcinoma. Cancers, 12(10), 3074. https://doi.org/10.3390/cancers12103074