TSPAN1: A Novel Protein Involved in Head and Neck Squamous Cell Carcinoma Chemoresistance

 ,
,  , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Drug Assay
2.3. Proteomic Study
2.4. Sample Preparation and LC-MS/MS Analyses
2.5. Peptide 10-Plex TMT Labeling
2.6. Peptide Fractionation
2.7. NanoLC-(Orbitrap) MS Analysis
2.8. Protein Analysis
2.9. Transfections
2.10. Colony Formation Assay
2.11. Quantitative Real-Time PCR (qRT-PCR)
2.12. Animal Models
2.13. Image Platform
2.14. Patients
2.15. IHC
2.16. Experimental Design and Statistical Rationale
3. Results
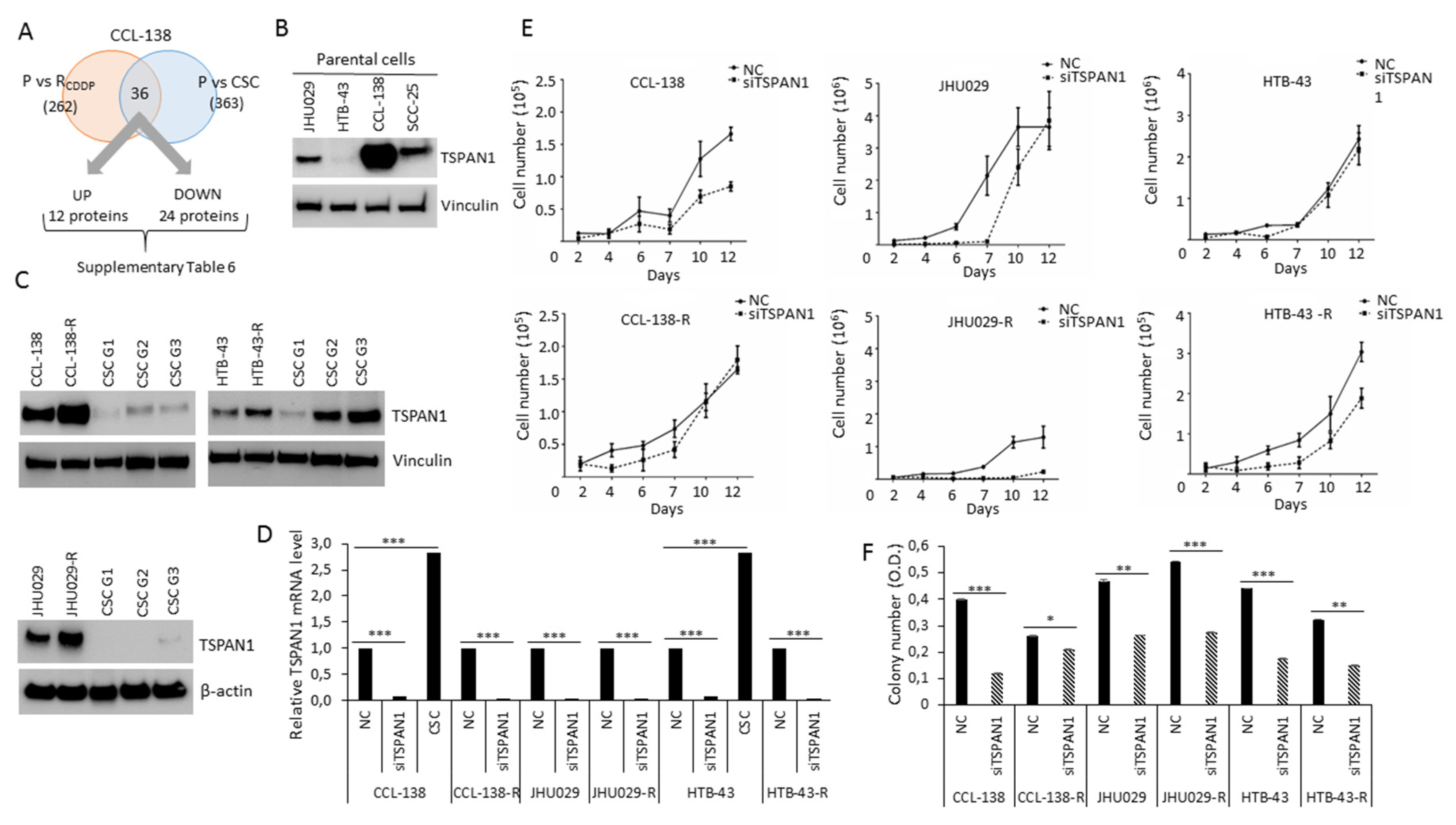
3.1. TSPAN1 Protein Is Upregulated in CDDP-Resistant HNSCC Cells
3.2. TSPAN1 Inhibition Induces Sensitivity of HNSCC Cells to Chemotherapeutic Agents
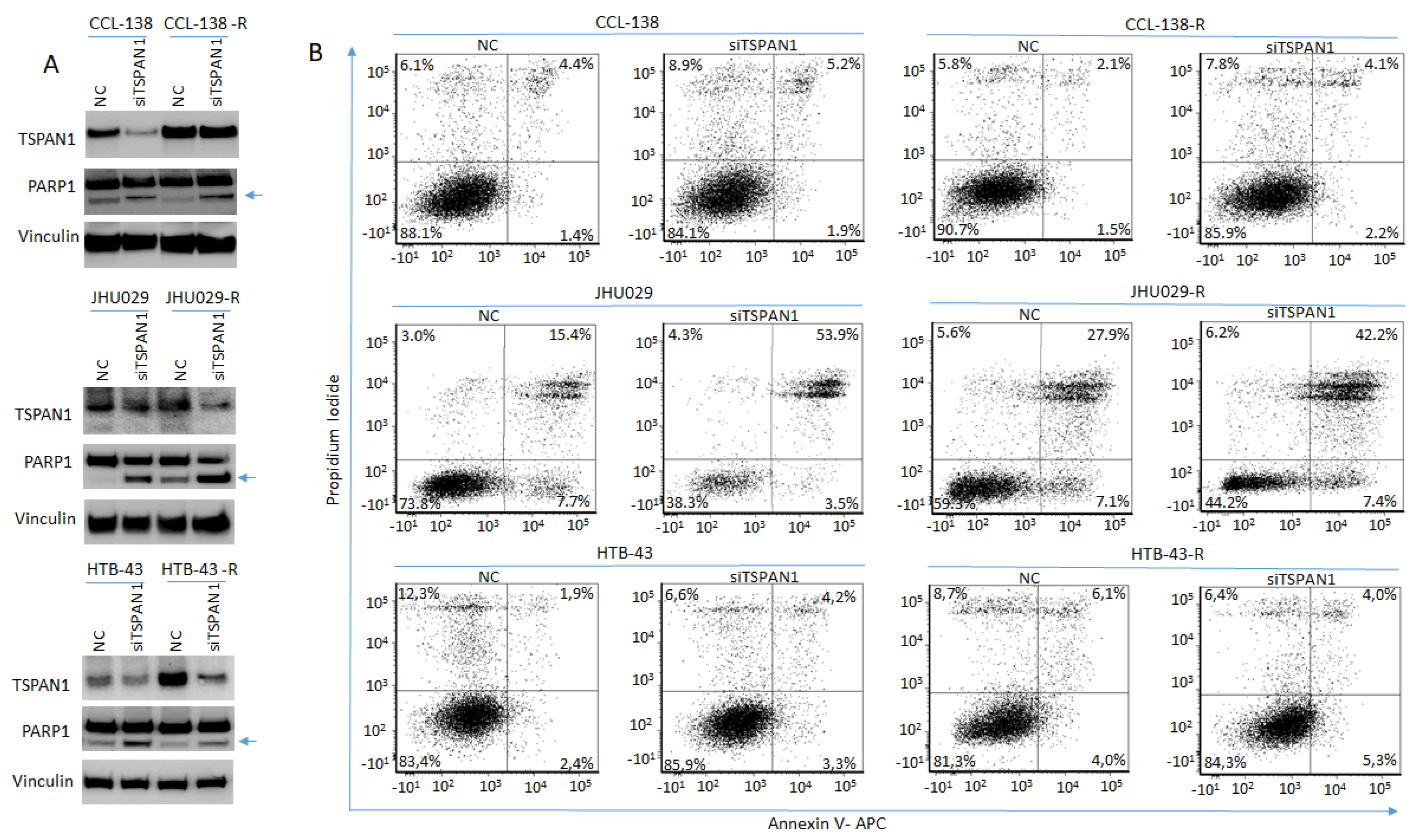
3.3. TSPAN1 Depletion Induces Apoptosis in HNSCC Cells
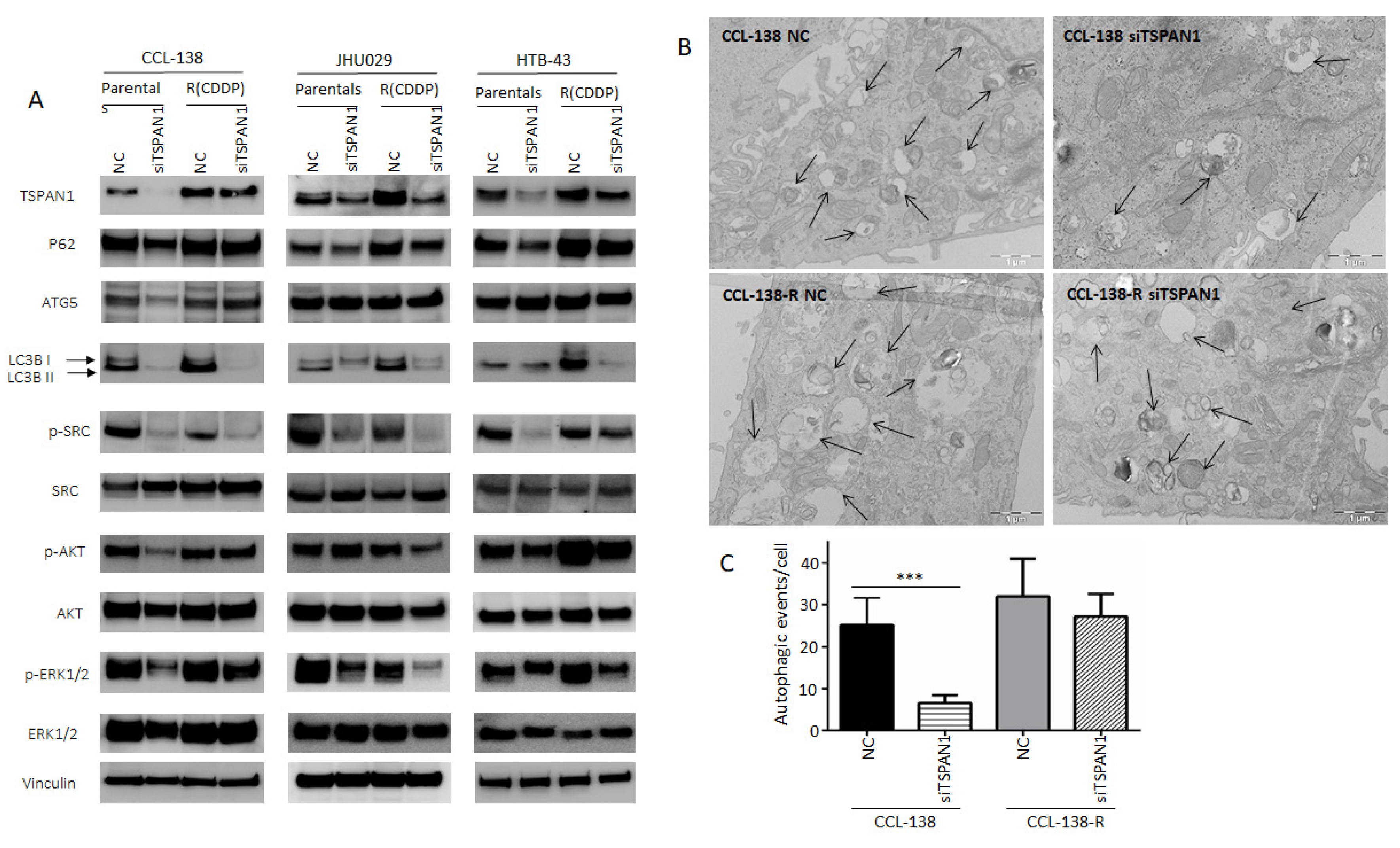
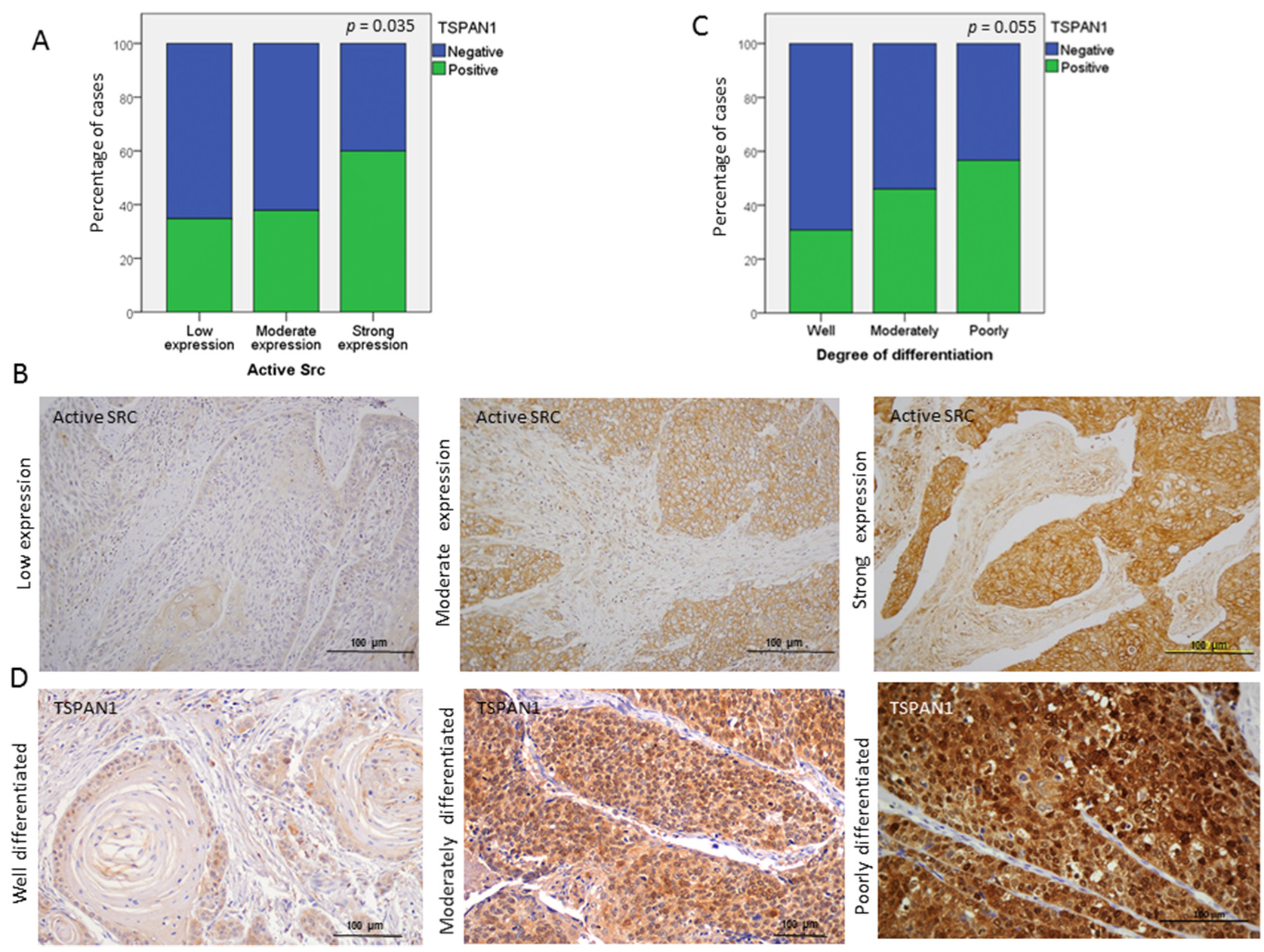
3.4. TSPAN1 Depletion Downregulates Several Signaling Cascades with SRC Kinase Signaling as a Central Node
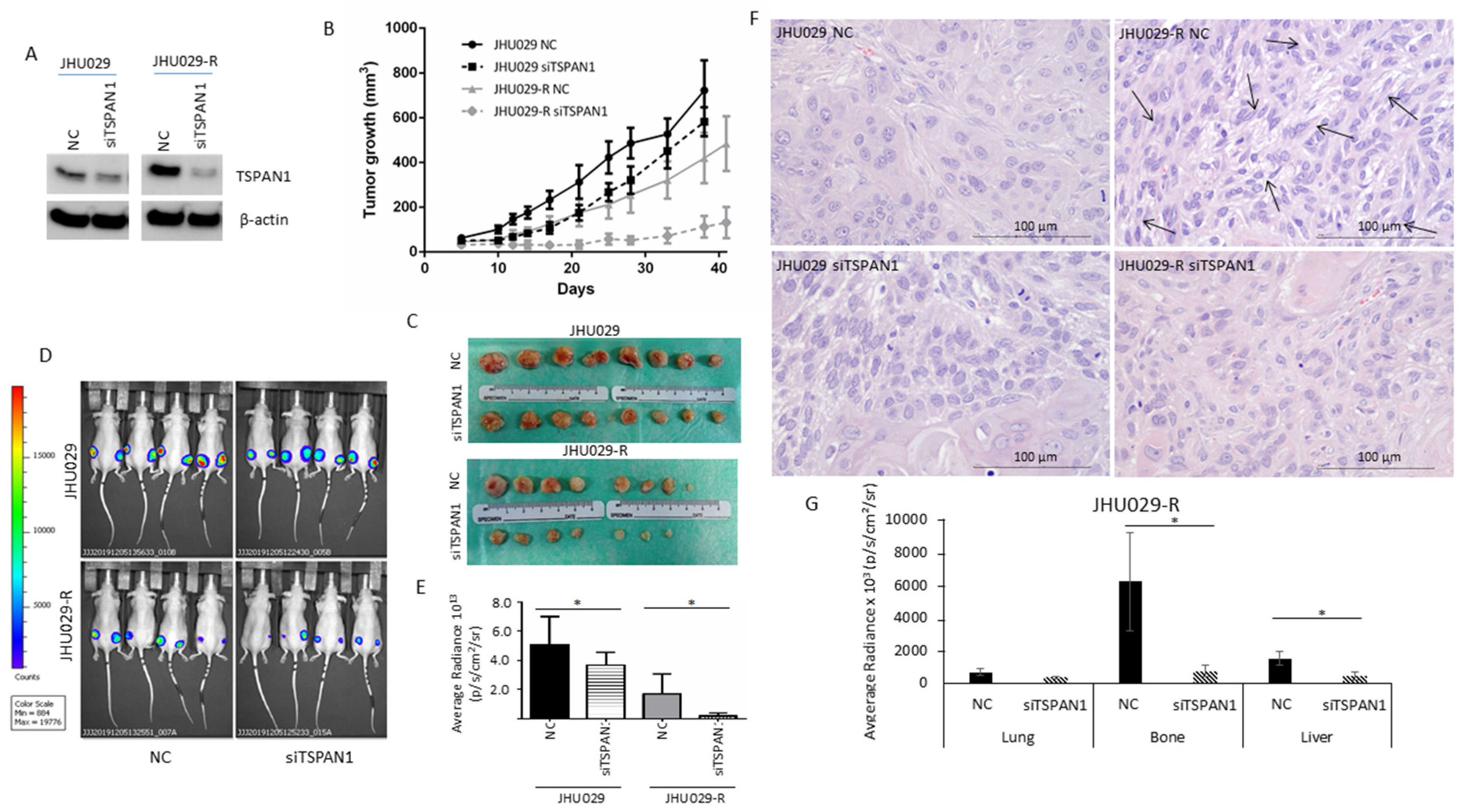
3.5. TSPAN1 Depletion Decreases Tumor Formation and Metastatic Capacity in Mice
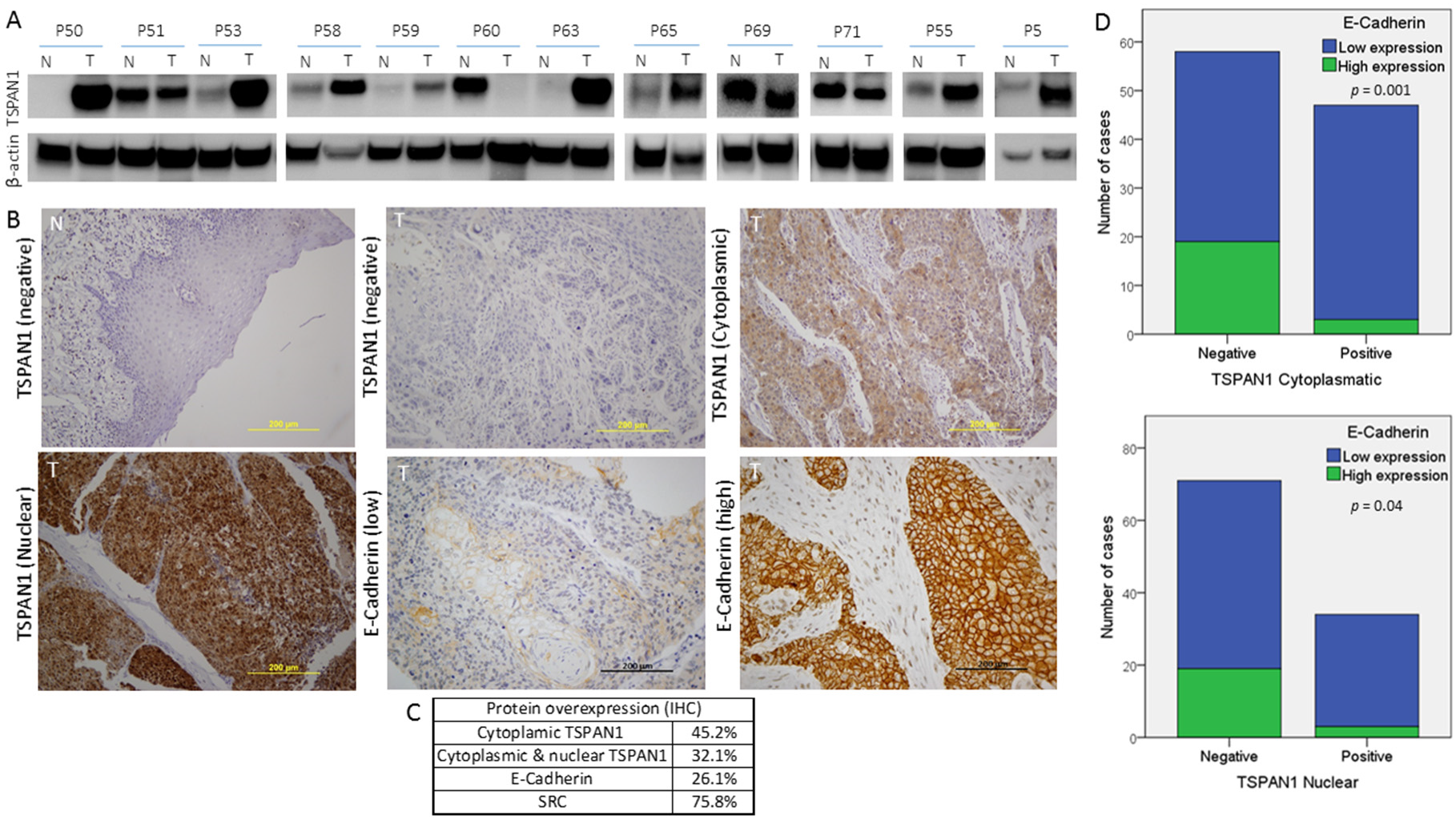
3.6. TSPAN1 Expression in Patient Samples. Correlations with Active SRC and EMT Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steuer, C.E.; El-Deiry, M.; Parks, J.R.; Higgins, K.A.; Saba, N.F. An update on larynx cancer. CA Cancer J. Clin. 2017, 67, 31–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porceddu, S.V.; Scotte, F.; Aapro, M.; Salmio, S.; Castro, A.; Launay-Vacher, V.; Licitra, L. Treating patients with locally advanced squamous cell carcinoma of the head and neck unsuitable to receive cisplatin-based therapy. Front. Oncol. 2019, 9, 1522. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Mayea, Y.; Mir, C.; Masson, F.; Paciucci, R.; Me, L.L. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin. Cancer Biol. 2019, 60, 166–180. [Google Scholar] [CrossRef]
- Cui, Q.; Wang, J.Q.; Assaraf, Y.G.; Ren, L.; Gupta, P.; Wei, L.; Ashby, C.R., Jr.; Yang, D.H.; Chen, Z.S. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist. Updat. 2018, 41, 1–25. [Google Scholar] [CrossRef]
- de Jong, M.C.; Pramana, J.; van der Wal, J.E.; Lacko, M.; Peutz-Kootstra, C.J.; de Jong, J.M.; Takes, R.P.; Kaanders, J.H.; van der Laan, B.F.; Wachters, J.; et al. CD44 expression predicts local recurrence after radiotherapy in larynx cancer. Clin. Cancer Res. 2010, 16, 5329–5338. [Google Scholar] [CrossRef] [Green Version]
- Azimzade, Y.; Saberi, A.A.; Sahimi, M. Role of the interplay between the internal and external conditions in invasive behavior of tumors. Sci. Rep. 2018, 8, 5968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Atallah, N.M.; Pascuzzi, P.E.; Gupta, S.; Liu, X. Inhibition of the Wnt/beta-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar]
- Zhao, Z.L.; Zhang, L.; Huang, C.F.; Ma, S.R.; Bu, L.L.; Liu, J.F.; Yu, G.T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.H.; Wu, A.T.H.; Cheng, T.S.; Lin, K.T.; Lai, C.J.; Hsieh, H.W.; Chang, P.M.; Wu, C.W.; Huang, C.F.; Chen, K.Y. In silico identification of thiostrepton as an inhibitor of cancer stem cell growth and an enhancer for chemotherapy in non-small-cell lung cancer. J. Cell. Mol. Med. 2019, 23, 8184–8195. [Google Scholar] [CrossRef] [Green Version]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Abad, E.; Garcia-Mayea, Y.; Mir, C.; Sebastian, D.; Zorzano, A.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A.; Lleonart, M.E. Common metabolic pathways implicated in resistance to chemotherapy point to a key mitochondrial role in breast cancer. Mol. Cell. Proteom. 2019, 18, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Kuciauskas, D.; Dreize, N.; Ger, M.; Kaupinis, A.; Zemaitis, K.; Stankevicius, V.; Suziedelis, K.; Cicenas, J.; Graves, L.M.; Valius, M. Proteomic analysis of breast cancer resistance to the anticancer drug RH1 reveals the importance of cancer stem cells. Cancers 2019, 11, 972. [Google Scholar] [CrossRef] [Green Version]
- Maecker, H.T.; Todd, S.C.; Levy, S. The tetraspanin superfamily: Molecular facilitators. FASEB J. 1997, 11, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Maisonial-Besset, A.; Witkowski, T.; Navarro-Teulon, I.; Berthier-Vergnes, O.; Fois, G.; Zhu, Y.; Besse, S.; Bawa, O.; Briat, A.; Quintana, M.; et al. Tetraspanin 8 (TSPAN 8) as a potential target for radio-immunotherapy of colorectal cancer. Oncotarget 2017, 8, 22034–22047. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Gao, Y.; Wang, Y.; Pan, J.; Sha, J.; Shao, X.; Kang, X.; Qin, J.; You, M.J.; Huang, Y.; et al. Decreased TSPAN1 promotes prostate cancer progression and is a marker for early biochemical recurrence after radical prostatectomy. Oncotarget 2016, 7, 63294–63305. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Qi, W.; Liu, S.; Sun, L.; Ding, A.; Yu, G.; Li, H.; Wang, Y.; Qiu, W.; Lv, J. TSPAN9 suppresses the chemosensitivity of gastric cancer to 5-fluorouracil by promoting autophagy. Cancer Cell. Int. 2020, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Qu, L.; Shou, C. Activation of EGFR-PI3K-AKT signaling is required for Mycoplasma hyorhinis-promoted gastric cancer cell migration. Cancer Cell. Int. 2014, 14, 135. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mayea, Y.; Mir, C.; Munoz, L.; Benavente, S.; Castellvi, J.; Temprana, J.; Maggio, V.; Lorente, J.; Paciucci, R.; LLeonart, M.E. Autophagy inhibition as a promising therapeutic target for laryngeal cancer. Carcinogenesis 2019, 40, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Hannus, M.; Beitzinger, M.; Engelmann, J.C.; Weickert, M.T.; Spang, R.; Hannus, S.; Meister, G. siPools: Highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014, 42, 8049–8061. [Google Scholar] [CrossRef] [Green Version]
- Feliciano, A.; Garcia-Mayea, Y.; Jubierre, L.; Mir, C.; Hummel, M.; Castellvi, J.; Hernandez-Losa, J.; Paciucci, R.; Sansano, I.; Sun, Y.; et al. miR-99a reveals two novel oncogenic proteins E2F2 and EMR2 and represses stemness in lung cancer. Cell. Death Dis. 2017, 8, e3141. [Google Scholar] [CrossRef]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Hermida-Prado, F.; Granda-Diaz, R.; Del-Rio-Ibisate, N.; Villaronga, M.A.; Allonca, E.; Garmendia, I.; Montuenga, L.M.; Rodriguez, R.; Vallina, A.; Alvarez-Marcos, C.; et al. The differential impact of SRC expression on the prognosis of patients with head and neck squamous cell carcinoma. Cancers 2019, 11, 1644. [Google Scholar] [CrossRef] [Green Version]
- Munkley, J.; McClurg, U.L.; Livermore, K.E.; Ehrmann, I.; Knight, B.; McCullagh, P.; McGrath, J.; Crundwell, M.; Harries, L.W.; Leung, H.Y.; et al. The cancer-associated cell migration protein TSPAN1 is under control of androgens and its upregulation increases prostate cancer cell migration. Sci. Rep. 2017, 7, 5249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, X.; Khan, A.A.; Li, H.; Tahir, M.; Yan, X.; Wang, J.; Huang, H. miR-216a-mediated upregulation of TSPAN1 contributes to pancreatic cancer progression via transcriptional regulation of ITGA2. Am. J. Cancer Res. 2020, 10, 1115–1129. [Google Scholar]
- Garcia-Mayea, Y.; LLeonart, M.E. Biomedical Research in Cancer Stem Cells; Vall d’Hebron Research Institute (VHIR): Barcelona, Spain, 2020; Personal observation. [Google Scholar]
- Bauman, J.E.; Duvvuri, U.; Gooding, W.E.; Rath, T.J.; Gross, N.D.; Song, J.; Jimeno, A.; Yarbrough, W.G.; Johnson, F.M.; Wang, L.; et al. Randomized, placebo-controlled window trial of EGFR, Src, or combined blockade in head and neck cancer. JCI Insight. 2017, 2, e90449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, C.I.; Chadaram, V.; Peterson, B.L.; Marcom, P.K.; Hopkins, J.; Kimmick, G.G.; Favaro, J.; Hamilton, E.; Welch, R.A.; Bacus, S.; et al. Phase II trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of Src inhibition to escalate dosing. Clin. Cancer Res. 2011, 17, 6061–6070. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liang, Y.; Yang, G.; Lan, Y.; Han, J.; Wang, J.; Yin, D.; Song, R.; Zheng, T.; Zhang, S.; et al. Tetraspanin 1 promotes epithelial-to-mesenchymal transition and metastasis of cholangiocarcinoma via PI3K/AKT signaling. J. Exp. Clin. Cancer Res. 2018, 37, 300. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Scholz, C.J.; Sauer, G.; Deissler, H. Glycosylation of tetraspanin Tspan-1 at four distinct sites promotes its transition through the endoplasmic reticulum. Protein Pept. Lett. 2009, 16, 1244–1248. [Google Scholar] [CrossRef]
- Gu, T.; Chen, W.; Chen, L.; Wang, G.; Li, T.; Zhu, Y.; Gao, X. Expression and function of tetraspanin 1 in esophageal carcinoma. Oncol. Lett. 2017, 14, 6815–6822. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Zhang, R.; Piao, H.; Li, X.; Sheng, W.; Zhou, J.; Dong, M.; Zhang, X.; Yan, X.; Shang, W.; et al. Silencing Tspan1 inhibits migration and invasion, and induces the apoptosis of human pancreatic cancer cells. Mol. Med. Rep. 2018, 18, 3280–3288. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.J.; Wang, J.L.; Chen, L. Inhibition of skin squamous cell carcinoma proliferation and promote apoptosis by dual silencing of NET-1 and survivin. Oncol. Rep. 2015, 34, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.Q.; Lei, X.F.; Yao, J.L.; Wang, Y.J.; Zhang, W. Tetraspanin 1 is involved in survival, proliferation and carcinogenesis of pancreatic cancer. Oncol. Rep. 2015, 34, 3068–3076. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.; Takimoto, T.; Hemler, M.E. Integrin-independent support of cancer drug resistance by tetraspanin CD151. Cell Mol. Life Sci. 2019, 76, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.D.; Martinez, C.H.; Vasquez, C.; Hebron, K.E.; Jones-Paris, C.; Arnold, S.A.; Chan, S.M.; Chalasani, V.; Gomez-Lemus, J.A.; Williams, A.K.; et al. Integrin-free tetraspanin CD151 can inhibit tumor cell motility upon clustering and is a clinical indicator of prostate cancer progression. Cancer Res. 2014, 74, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.; Akbar, A.; Ng, N.N.; Concepcion, W.; Thakor, A.S. Mesenchymal stem cells confer chemoresistance in breast cancer via a CD9 dependent mechanism. Oncotarget 2019, 10, 3435–3450. [Google Scholar] [CrossRef] [Green Version]
- Kohmo, S.; Kijima, T.; Otani, Y.; Mori, M.; Minami, T.; Takahashi, R.; Nagatomo, I.; Takeda, Y.; Kida, H.; Goya, S.; et al. Cell surface tetraspanin CD9 mediates chemoresistance in small cell lung cancer. Cancer Res. 2010, 70, 8025–8035. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Gires, O.; Zhu, L.; Liu, J.; Li, J.; Yang, H.; Ju, G.; Huang, J.; Ge, W.; Chen, Y.; et al. TSPAN8 promotes cancer cell stemness via activation of sonic Hedgehog signaling. Nat. Commun. 2019, 10, 2863. [Google Scholar] [CrossRef]
- Huang, Y.; Sook-Kim, M.; Ratovitski, E. Midkine promotes tetraspanin-integrin interaction and induces FAK-Stat1alpha pathway contributing to migration/invasiveness of human head and neck squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2008, 377, 474–478. [Google Scholar] [CrossRef]
- de Bree, R.; Wolf, G.T.; de Keizer, B.; Nixon, I.J.; Hartl, D.M.; Forastiere, A.A.; Haigentz, M., Jr.; Rinaldo, A.; Rodrigo, J.P.; Saba, N.F.; et al. Response assessment after induction chemotherapy for head and neck squamous cell carcinoma: From physical examination to modern imaging techniques and beyond. Head Neck 2017, 39, 2329–2349. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef]
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to mesenchymal transition: A mechanism that fuels cancer radio/chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef] [Green Version]
- Subrungruanga, I.; Thawornkunob, C.; Chawalitchewinkoon-Petmitrc, P.; Pairojkul, C.; Wongkham, S.; Petmitrb, S. Gene expression profiling of intrahepatic cholangiocarcinoma. Asian Pacific J. Cancer Prev. APJCP 2013, 14, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fei, B.; Wang, Q.; Song, M.; Yin, Y.; Zhang, B.; Ni, S.; Guo, W.; Bian, Z.; Quan, C.; et al. MicroRNA-638 inhibits cell proliferation, invasion and regulates cell cycle by targeting tetraspanin 1 in human colorectal carcinoma. Oncotarget 2014, 5, 12083–12096. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, J.; Chen, L.; Wang, G.; Qin, J.; Xu, Y.; Li, X. Expression and function of NET-1 in human skin squamous cell carcinoma. Arch. Dermatol. Res. 2014, 306, 385–397. [Google Scholar] [CrossRef] [Green Version]
- Scholz, C.J.; Kurzeder, C.; Koretz, K.; Windisch, J.; Kreienberg, R.; Sauer, G.; Deissler, H. Tspan-1 is a tetraspanin preferentially expressed by mucinous and endometrioid subtypes of human ovarian carcinomas. Cancer Lett. 2009, 275, 198–203. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Wang, G.L.; Wang, Y.; Zhu, Y.Y.; Zhu, J. Clinicopathological significance of overexpression of TSPAN1, Ki67 and CD34 in gastric carcinoma. Tumori 2008, 94, 531–538. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, T.; Nie, M.; Pang, T.; Zhang, X.; Shen, X.; Ma, L.; Bi, J.; Wei, G.; Fang, G.; et al. TSPAN1 functions as an oncogene in gastric cancer and is downregulated by miR-573. FEBS Lett. 2015, 589, 1988–1994. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.M.; Backsch, C.; Schneider, A.; Durst, M. HPV induced cervical carcinogenesis: Molecular basis and vaccine development. Zent. Gynakol. 2002, 124, 511–524. [Google Scholar]
- Wollscheid, V.; Kuhne-Heid, R.; Stein, I.; Jansen, L.; Kollner, S.; Schneider, A.; Durst, M. Identification of a new proliferation-associated protein NET-1/C4.8 characteristic for a subset of high-grade cervical intraepithelial neoplasia and cervical carcinomas. Int. J. Cancer 2002, 99, 771–775. [Google Scholar] [CrossRef]
- Nees, M.; van Wijngaarden, E.; Bakos, E.; Schneider, A.; Durst, M. Identification of novel molecular markers which correlate with HPV-induced tumor progression. Oncogene 1998, 16, 2447–2458. [Google Scholar] [CrossRef]
- Ke, L.; Xiang, Y.; Guo, X.; Lu, J.; Xia, W.; Yu, Y.; Peng, Y.; Wang, L.; Wang, G.; Ye, Y.; et al. c-Src activation promotes nasopharyngeal carcinoma metastasis by inducing the epithelial-mesenchymal transition via PI3K/Akt signaling pathway: A new and promising target for NPC. Oncotarget 2016, 7, 28340–28355. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Mayea, Y.; Mir, C.; Carballo, L.; Castellvi, J.; Temprana-Salvador, J.; Lorente, J.; Benavente, S.; García-Pedrero, J.M.; Allonca, E.; Rodrigo, J.P.; et al. TSPAN1: A Novel Protein Involved in Head and Neck Squamous Cell Carcinoma Chemoresistance. Cancers 2020, 12, 3269. https://doi.org/10.3390/cancers12113269
Garcia-Mayea Y, Mir C, Carballo L, Castellvi J, Temprana-Salvador J, Lorente J, Benavente S, García-Pedrero JM, Allonca E, Rodrigo JP, et al. TSPAN1: A Novel Protein Involved in Head and Neck Squamous Cell Carcinoma Chemoresistance. Cancers. 2020; 12(11):3269. https://doi.org/10.3390/cancers12113269
Chicago/Turabian StyleGarcia-Mayea, Yoelsis, Cristina Mir, Laia Carballo, Josep Castellvi, Jordi Temprana-Salvador, Juan Lorente, Sergi Benavente, Juana M. García-Pedrero, Eva Allonca, Juan P. Rodrigo, and et al. 2020. "TSPAN1: A Novel Protein Involved in Head and Neck Squamous Cell Carcinoma Chemoresistance" Cancers 12, no. 11: 3269. https://doi.org/10.3390/cancers12113269