Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Spain: Clinical and Genetic Characterization

 , , , , add
Show full author list
, , , , add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Data Collection
2.2. Detection of Germline Mutations
2.3. Statistics
3. Results
3.1. Cutaneous Leiomyomas
3.2. Uterine Leiomyomas
3.3. Renal Cysts
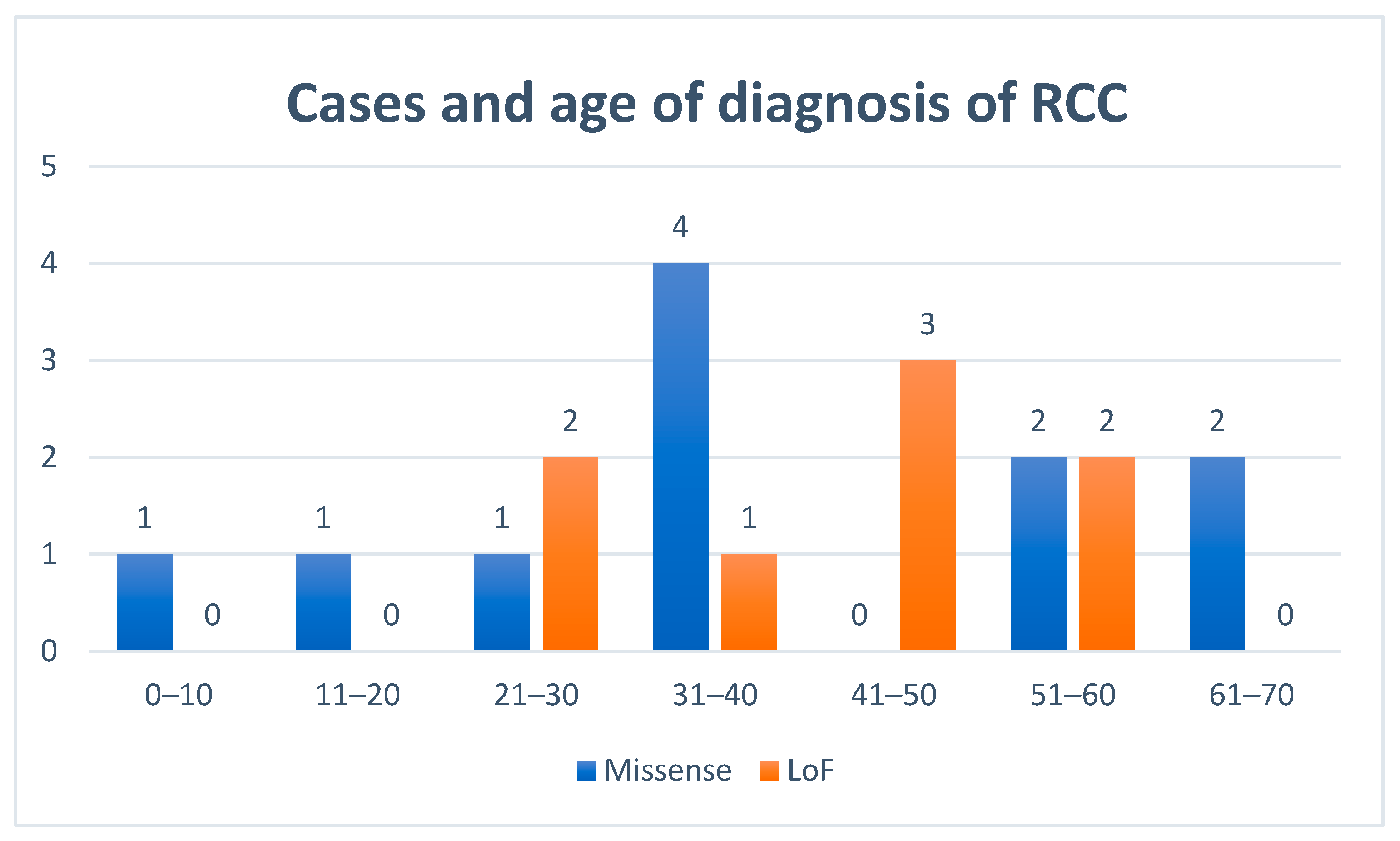
3.4. Renal Cell Cancers
3.5. Other Tumours
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reed, W.B.; Walker, R.; Horowitz, R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm. Venereol. 1973, 53, 409–416. [Google Scholar]
- Launonen, V.; Vierimaa, O.; Kiuru, M.; Isola, J.; Roth, S.; Pukkala, E.; Sistonen, P.; Herva, R.; Aaltonen, L.A. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 3387–3392. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 2002, 30, 406–410. [Google Scholar]
- Toro, J.R.; Nickerson, M.L.; Wei, M.H.; Warren, M.B.; Glenn, G.M.; Turner, M.L.; Stewart, L.; Duray, P.; Tourre, O.; Sharma, N.; et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am. J. Hum. Genet. 2003, 73, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Alam, N.A.; Barclay, E.; Rowan, A.J.; Tyrer, J.P.; Calonje, E.; Manek, S.; Kelsell, D.; Leigh, I.; Olpin, S.; Tomlinson, I.P. Clinical features of multiple cutaneous and uterine leiomyomatosis: An underdiagnosed tumor syndrome. Arch. Dermatol. 2005, 141, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.H.; Toure, O.; Glenn, G.M.; Pithukpakorn, M.; Neckers, L.; Stolle, C.; Choyke, P.; Grubb, R.; Middelton, L.; Turner, M.L.; et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet. 2006, 43, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Gardie, B.; Remenieras, A.; Kattygnarath, D.; Bombled, J.; Lefèvre, S.; Perrier-Trudova, V.; Rustin, P.; Barrois, M.; Slama, A.; Avril, M.F.; et al. French National Cancer Institute. “Inherited predisposition to kidney cancer” network. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J. Med. Genet. 2011, 48, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Ferlicot, S.; Guillaud-Bataille, M.; Le Teuff, G.; Genestie, C.; Deveaux, S.; Slama, A.; Poulalhon, N.; Escudier, B.; Albiges, L.; et al. Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin. Genet. 2017, 92, 606–615. [Google Scholar] [CrossRef]
- Lehtonen, H.J.; Kiuru, M.; Ylisaukko-Oja, S.K.; Salovaara, R.; Herva, R.; Koivisto, P.A.; Vierimaa, O.; Aittomäki, K.; Pukkala, E.; Launonen, V.; et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J. Med. Genet. 2006, 43, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Castro-Vega, L.J.; Buffet, A.; De Cubas, A.A.; Cascón, A.; Menara, M.; Khalifa, E.; Amar, L.; Azriel, S.; Bourdeau, I.; Chabre, O.; et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum. Mol. Genet. 2014, 23, 2440–2446. [Google Scholar] [CrossRef] [Green Version]
- Allegri, G.; Fernandes, M.J.; Scalco, F.B.; Correia, P.; Simoni, R.E.; Llerena, J.C., Jr.; de Oliveira, M.L. Fumaric aciduria: An overview and the first Brazilian case report. J. Inherit. Metab. Dis. 2010, 33, 411–419. [Google Scholar] [CrossRef]
- Orphanet Report Series—Rare diseases. Prevalence of Rare Diseases by Decreasing Prevalence, Incidence or Number of Published Cases. January 2020—n 2. Available online: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_decreasing_prevalence_or_cases.pdf (accessed on 31 August 2020).
- Badeloe, S.; van Geel, M.; van Steensel, M.A.; Bastida, J.; Ferrando, J.; Steijlen, P.M.; Frank, J.; Poblete-Gutiérrez, P. Diffuse and segmental variants of cutaneous leiomyomatosis: Novel mutations in the fumarate hydratase gene and review of the literature. Exp. Dermatol. 2006, 15, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, H.J.; Blanco, I.; Piulats, J.M.; Herva, R.; Launonen, V.; Aaltonen, L.A. Conventional renal cancer in a patient with fumarate hydratase mutation. Hum. Pathol. 2007, 38, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Oxford Bioinformatics, bty897. 30 October 2018. Available online: https://varsome.com/ (accessed on 22 July 2020).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar (accessed on 22 July 2020).
- Bayley, J.P.; Launonen, V.; Tomlinson, I.P. The FH mutation database: An online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med. Genet. 2008, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Dik, E.; Naamati, A.; Asraf, H.; Lehming, N.; Pines, O. Human Fumarate Hydratase Is Dual Localized by an Alternative Transcription Initiation Mechanism. Traffic 2016, 17, 720–732. [Google Scholar] [CrossRef]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Bulku, A.; Weaver, T.M.; Berkmen, M.B. Biochemical Characterization of Two Clinically-Relevant Human Fumarase Variants Defective for Oligomerization. Open Biochem. J. 2018, 12, 1–15. [Google Scholar] [CrossRef]
- Sanz-Ortega, J.; Vocke, C.; Stratton, P.; Linehan, W.M.; Merino, M.J. Morphologic and molecular characteristics of uterine leiomyomas in hereditary leiomyomatosis and renal cancer (HLRCC) syndrome. Am. J. Surg. Pathol. 2013, 37, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Rouault, T.A. Molecular pathways: Fumarate hydratase-deficient kidney cancer—Targeting the Warburg effect in cancer. Clin. Cancer Res. 2013, 19, 3345–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Su, H.; Soga, T.; Kranc, K.R.; Pollard, P.J. Prolyl hydroxylase domain enzymes: Important regulators of cancer metabolism. Hypoxia 2014, 2, 127–142. [Google Scholar] [PubMed] [Green Version]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, I.S.; Maksim, N.J.; Amouzougan, E.A.; Gallion, B.W.; Raviele, A.L.; Ooi, A. Sustained NRF2 activation in hereditary leiomyomatosis and renal cell cancer (HLRCC) and in hereditary tyrosinemia type 1 (HT1). Biochem. Soc. Trans. 2015, 43, 650–656. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Forde, C.; Lim, D.H.K.; Alwan, Y.; Burghel, G.; Butland, L.; Cleaver, R.; Dixit, A.; Evans, D.G.; Hanson, H.; Lalloo, F.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur. Urol. Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Tetzlaff, M.; Hick, R.; Duvic, M. Reed syndrome presenting with leiomyosarcoma. JAAD Case Rep. 2015, 1, 150–152. [Google Scholar] [CrossRef] [Green Version]
- Stewart, L.; Glenn, G.M.; Stratton, P.; Goldstein, A.M.; Merino, M.J.; Tucker, M.A.; Linehan, W.M.; Toro, J.R. Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch. Dermatol. 2008, 14, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Okolo, S. Incidence, aetiology and epidemiology of uterine fibroids. Best Pract. Res. Clin. Obstet. Gynaecol. 2008, 22, 571–588. [Google Scholar] [CrossRef]
- Walker, C.L.; Stewart, E.A. Uterine fibroids: The elephant in the room. Science 2005, 308, 1589–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tal, R.; Segars, J.H. The role of angiogenic factors in fibroid pathogenesis: Potential implications for future therapy. Hum. Reprod. Update 2014, 20, 194–216. [Google Scholar] [CrossRef] [Green Version]
- Pollard, P.; Wortham, N.; Barclay, E.; Alam, A.; Elia, G.; Manek, S.; Poulsom, R.; Tomlinson, I. Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J. Pathol. 2005, 205, 41–49. [Google Scholar] [CrossRef]
- Bonsib, S.M. Renal cystic diseases and renal neoplasms: A mini-review. Clin. J. Am. Soc. Nephrol. 2009, 4, 1998–2007. [Google Scholar] [CrossRef] [Green Version]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Ristau, B.T.; Kamat, S.N.; Tarin, T.V. Abnormal Cystic Tumor in a Patient with Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome: Evidence of a Precursor Lesion? Case Rep. Urol. 2015, 2015, 303872. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.S.; Linehan, W.M. Genetic predisposition to kidney cancer. Semin. Oncol. 2016, 43, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef]
- Grubb, R.L.; Franks, M.E.; Toro, J.; Middelton, L.; Choyke, L.; Fowler, S.; Torres-Cabala, C.; Glenn, G.M.; Choyke, P.; Merino, M.J.; et al. Hereditary leiomyomatosis and renal cell cancer: A syndrome associated with an aggressive form of inherited renal cancer. J. Urol. 2007, 177, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.J.; Torres-Cabala, C.; Pinto, P.; Linehan, W.M. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am. J. Surg. Pathol. 2007, 31, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Menko, F.H.; Maher, E.R.; Schmidt, L.S.; Middelton, L.A.; Aittomäki, K.; Tomlinson, I.; Richard, S.; Linehan, W.M. Hereditary leiomyomatosis and renal cell cancer (HLRCC): Renal cancer risk, surveillance and treatment. Fam. Cancer 2014, 13, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Trpkov, K.; Hes, O.; Agaimy, A.; Bonert, M.; Martinek, P.; Magi-Galluzzi, C.; Kristiansen, G.; Lüders, C.; Nesi, G.; Compérat, E.; et al. Fumarate hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am. J. Surg. Pathol. 2016, 40, 865–875. [Google Scholar] [CrossRef]
- Scelo, G.; Larose, T.L. Epidemiology and Risk Factors for Kidney Cancer. J. Clin. Oncol. 2018, 36, 3574–3581. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Oudard, S.; De Fromont, M.; Chevreau, C.; Gravis, G.; Zanetta, S.; Theodore, C.; Jimenez, M.; Sevin, E.; Laguerre, B.; et al. First-line treatment with sunitinib for type 1 and type 2 locally advanced or metastatic papillary renal cell carcinoma: A phase II study (SUPAP) by the French Genitourinary Group (GETUG). Ann. Oncol. 2015, 26, 1123–1128. [Google Scholar] [CrossRef]
- Srinivasan, R.; Gurram, S.; Al Harthy, M.; Singer, E.A.; Sidana, A.; Shuch, B.M.; Ball, M.W.; Friend, J.C.; Mac, L.; Purcell, E.; et al. Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. J. Clin. Oncol. 2020, 38 (Suppl. S15), 5004. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Hereditary+Leiomyomatosis+and+Renal+Cell+Cancer&draw=1&rank=1#rowId0 (accessed on 13 September 2020).
- Suarez Rodriguez, C.; Larkin, J.M.G.; Patel, P.; Pérez Valderrama, B.; Rodriguez-Vida, A.; Glen, H.; Thistlethwaite, F.; Ralph, C.; Srinivasan, G.; Mendez Vidal, M.J.; et al. Overall survival results for durvalumab and savolitinib in metastatic papillary renal cancer. J. Clin. Oncol. 2020, 38 (Suppl. S6), 619. [Google Scholar] [CrossRef]
- Matyakhina, L.; Freedman, R.J.; Bourdeau, I.; Wei, M.H.; Stergiopoulos, S.G.; Chidakel, A.; Walther, M.; Abu-Asab, M.; Tsokos, M.; Keil, M.; et al. Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: A clinical and molecular genetic investigation. J. Clin. Endocrinol. Metab. 2005, 90, 3773–3779. [Google Scholar] [CrossRef] [Green Version]
- Hammarstedt, L.; Muth, A.; Wängberg, B.; Björneld, L.; Sigurjónsdóttir, H.A.; Götherström, G.; Almqvist, E.; Widell, H.; Carlsson, S.; Ander, S.; et al. Adrenal lesion frequency: A prospective, cross-sectional CT study in a defined region, including systematic re-evaluation. Acta Radiol. 2010, 51, 1149–1156. [Google Scholar] [CrossRef]
- Lattouf, J.B.; Pautler, S.E.; Reaume, M.N.; Kim, R.H.; Care, M.; Green, J.; So, A.; Violette, P.D.; Saliba, I.; Major, P.; et al. Kidney Cancer Research Network of Canada. Structured assessment and followup for patients with hereditary kidney tumour syndromes. Can. Urol. Assoc. J. 2016, 10, E214–E222. [Google Scholar] [CrossRef] [Green Version]
- Graumann, O.; Osther, S.S.; Osther, P.J. Characterization of complex renal cysts: A critical evaluation of the Bosniak classification. Scand. J. Urol. Nephrol. 2011, 45, 84–90. [Google Scholar] [CrossRef]
- Linton, J.M.; Martin, G.R.; Reichardt, L.F. The ECM Protein Nephronectin Promotes Kidney Development via Integrin alpha8beta1-mediated Stimulation of Gdnf Expression. Development 2007, 134, 2501–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Ferreira, M.; Donati, G.; Marciano, D.K.; Linton, J.M.; Sato, Y.; Hartner, A.; Sekiguchi, K.; Reichardt, L.F.; Watt, F.M. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell 2011, 144, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Cook, J.D.; Walker, C.L. The Eker rat: Establishing a genetic paradigm linking renal cell carcinoma and uterine leiomyoma. Curr. Mol. Med. 2004, 4, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Valera, V.A.; Padilla-Nash, H.M.; Sourbier, C.; Vocke, C.D.; Vira, M.A.; bu-Asab, M.S.; Bratslavsky, G.; Tsokos, M.; Merino, M.J.; et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: In vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet. Cytogenet. 2010, 196, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Class | Variant Type | No. Families/ No. Individual (Male; Female) | CLM * | ULM * | RCy * | RCC * | Origin |
|---|---|---|---|---|---|---|---|---|
| No. affected patients/Total no | ||||||||
| Del FH | 5 | LoF | 2/6 (4; 2) | 2/6 | 1/2 | 2/6 | 0/6 | Spain |
| Del exon 2 | 5 | LoF | 1/1 (0; 1) | 1/1 | 0/1 | 0/1 | 1/1 | Spain |
| c.139C > T | 5 | LoF | 1/1 (0; 1) | 1/1 | 1/1 | ? | 0/1 | Spain |
| c.267 + 1_267 + 10del | 5 | LoF | 1/1 (0; 1) | 1/1 | 0/1 | 0/1 | 0/1 | Spain |
| c.301C > T | 5 | LoF | 1/1 (0; 1) | 1/1 | 1/1 | 1/1 | 0/1 | Spain |
| c.349G > C | 4 | Missense | 2/3 (1; 2) | 2/2 | 2/2 | ? | 0/3 | Spain |
| c.395delT | 5 | LoF | 1/16 (5; 11) | 1/15 | 6/10 | 0/14 | 2/16 | Spain |
| c.553delC | 5 | LoF | 1/1 (0; 1) | 1/1 | 1/1 | ? | ? | Spain |
| c.555 + 1G > A | 5 | LoF | 1/1 (0; 1) | 1/1 | 1/1 | 1/1 | 0/1 | Spain |
| c.563A > G | 4 | Missense | 1/2 (1; 1) | 1/2 | 0/1 | 0/1 | 0/1 | Spain |
| c.575C > T | 4 | Missense | 4/11 (4; 7) | 11/11 | 7/7 | 5/11 | 0/11 | Spain |
| c.697C > T | 4 | Missense | 5/10 (5; 5) | 5/9 | 4/4 | 3/6 | 0/6 | Spain |
| c.698G > A | 5 | Missense | 4/7 (2; 5) | 4/6 | 4/4 | 1/1 | 1/3 | Spain |
| c.703C > T | 4 | Missense | 1/1 (0; 1) | ? | ? | ? | 0/1 | Spain |
| c.845G > T | 4 | Missense | 3/6 (2; 4) | 5/5 | 3/4 | 3/4 | 1/5 | Spain |
| c.893del | 5 | LoF | 1/2 (0; 2) | 2/2 | 1/1 | 0/1 | 0/1 | Spain |
| c.905-2A > G | 5 | LoF | 1/1 (1; 0) | 1/1 | - | 1/1 | 0/1 | Spain |
| c.965T > G | 4 | Missense | 1/1 (0; 1) | 1/1 | 1/1 | 0/1 | 0/1 | Spain |
| c.974delG | 5 | LoF | 1/3 (1; 2) | 1/1 | ? | 0/1 | 1/1 | Spain |
| Del exon 8 | 5 | LoF | 3/6 (1; 5) | 4/5 | 4/4 | 3/6 | 1/6 | Spain |
| c.1112A > G | 4 | Missense | 1/1 (1; 0) | 1/1 | - | 0/1 | 0/1 | Perú |
| c.1118A > G | 5 | Missense | 31/104 (51; 53) | 64/99 | 50/51 | 36/85 | 10/101 | Spain |
| c.1126delC | 5 | LoF | 1/6 (4; 2) | 5/6 | 2/2 | 0/6 | 0/6 | Spain |
| c.1189G > A | 4 | Missense | 2/2 (0; 2) | 1/2 | 2/2 | 0/2 | 0/2 | Colombia |
| c.1217A > C | 4 | Missense | 1/1 (1; 0) | 1/1 | - | 0/1 | 0/1 | Cuba |
| c.1240A > G | 4 | Missense | 1/1 (0; 1) | 0/1 | 1/1 | 1/1 | 1/1 | Belarus |
| Del exon 9 | 5 | LoF | 1/1 (0; 1) | ? | 1/1 | ? | 1/1 | Spain |
| 74/197 (84; 113) | 118/182 (64.8%) | 93/103 (90.3%) | 57/153 (37.3%) | 19/175 (10.9%) | ||||
| Loss of Function | 17/47 (16; 31) | 21/41 (51.2%) | 18/25 (72.0%) | 8/38 (24.8%) | 6/42 (14.3%) | |||
| Missense | 57/150 (68; 82) | 97/141 (68.8%) | 75/78 (96.1%) | 49/115 (42.6%) | 13/133 (9.8%) | |||
| Clinical Manifestations | Missense No. Affected/Total (%) | LoF No. Affected/Total (%) | OR (95%CI) | p-Value * |
|---|---|---|---|---|
| CLM | 97/141 (68.8) | 21/41 (51.2) | 0.47 (0.23–0.96) | 0.038 |
| ULM | 75/78 (96.1) | 18/25 (72.0) | 0.10 (0.02–0.43) | 0.001 |
| RCy | 49/115 (42.6) | 8/38 (21.0) | 0.35 (0.15–0.85) | 0.017 |
| RCC | 13/133 (9.7) | 6/42 (14.2) | 1.53 (0,55–4,34) | 0.412 |
| Variable | RCy | |
|---|---|---|
| OR (95%CI) | p-value * | |
| Variant type | ||
| Missense | Ref. | |
| Lof | 0.23 (0.08–0.65) | 0.005 |
| Tobacco | ||
| No | Ref. | |
| Yes | 2.05 (0.96–4.39) | 0.065 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Heras, A.B.; Castillejo, A.; García-Díaz, J.D.; Robledo, M.; Teulé, A.; Sánchez, R.; Zúñiga, Á.; Lastra, E.; Durán, M.; Llort, G.; et al. Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Spain: Clinical and Genetic Characterization. Cancers 2020, 12, 3277. https://doi.org/10.3390/cancers12113277
Sánchez-Heras AB, Castillejo A, García-Díaz JD, Robledo M, Teulé A, Sánchez R, Zúñiga Á, Lastra E, Durán M, Llort G, et al. Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Spain: Clinical and Genetic Characterization. Cancers. 2020; 12(11):3277. https://doi.org/10.3390/cancers12113277
Chicago/Turabian StyleSánchez-Heras, A. Beatriz, Adela Castillejo, Juan D. García-Díaz, Mercedes Robledo, Alexandre Teulé, Rosario Sánchez, Ángel Zúñiga, Enrique Lastra, Mercedes Durán, Gemma Llort, and et al. 2020. "Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Spain: Clinical and Genetic Characterization" Cancers 12, no. 11: 3277. https://doi.org/10.3390/cancers12113277
APA StyleSánchez-Heras, A. B., Castillejo, A., García-Díaz, J. D., Robledo, M., Teulé, A., Sánchez, R., Zúñiga, Á., Lastra, E., Durán, M., Llort, G., Yagüe, C., Ramon y Cajal, T., López San Martin, C., López-Fernández, A., Balmaña, J., Robles, L., Mesa-Latorre, J. M., Chirivella, I., Fonfria, M., ... Soto, J. L. (2020). Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome in Spain: Clinical and Genetic Characterization. Cancers, 12(11), 3277. https://doi.org/10.3390/cancers12113277