Cancer Extracellular Matrix Proteins Regulate Tumour Immunity


Abstract
:Simple Summary
Abstract
1. Introduction
2. Extracellular Matrix Organisation in Healthy Tissue
2.1. Pericellular Membrane (Glycocalyx)
2.2. Basement Membrane
2.3. Interstitial ECM
3. Extracellular Matrix Organisation in Cancer
3.1. Peri-Cellular Membrane
3.2. Basement Membrane
3.3. Interstitial ECM
4. ECM Regulation of Immune Cell Migration
4.1. Structural ECM Proteins
4.2. Non-structural ECM Proteins
5. ECM Regulation of Immune Cell Function
5.1. Myeloid Cells
5.2. T-Lymphocytes, Natural Killer and Dendritic Cells
6. ECM Physics and Tumour Lymphatic Physiology
6.1. Targeting the ECM to Enhance Tumour Immunity
6.2. Renin-Angiotensin Inhibition
6.3. Focal-Adhesion Kinase Inhibition
6.4. Hyaluronan Depletion
6.5. Fibroblast Depletion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gruosso, T.; Zuo, D.; Omeroglu, A.; Meterissian, S.; Guiot, M.-C.; Salazar, A.; Park, M.; Levine, H. Infiltration of CD8+ T cells into tumor cell clusters in triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3678–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaday, G.G.; Lider, O. Extracellular matrix moieties, cytokines, and enzymes: dynamic effects on immune cell behavior and inflammation. J. Leukoc. Biol. 2000, 67, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Cha, J.; Jang, M.; Kim, P. Hyaluronic acid-based extracellular matrix triggers spontaneous M2-like polarity of monocyte/macrophage. Biomater. Sci. 2019, 7, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.T.; Ganguly, S.; Wang, T.L.; Anderson, C.W.; Sadtler, K.; Narain, R.; Cherry, C.; Parrillo, A.J.; Park, B.V.; Wang, G.; et al. A biologic scaffold-associated type 2 immune microenvironment inhibits tumor formation and synergizes with checkpoint immunotherapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Dziki, J.L.; Wang, D.S.; Pineda, C.; Sicari, B.M.; Rausch, T.; Badylak, S.F. Solubilized extracellular matrix bioscaffolds derived from diverse source tissues differentially influence macrophage phenotype. J. Biomed. Mater. Res. A 2017, 105, 138–147. [Google Scholar] [CrossRef]
- Mirochnik, Y.; Kwiatek, A.; Volpert, O. Thrombospondin and apoptosis: Molecular mechanisms and use for design of complementation treatments. Curr. Drug. Targets 2008, 9, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, F.; Slatter, D.A. Collagen cross-linking: Insights on the evolution of metazoan extracellular matrix. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 10. [Google Scholar] [CrossRef]
- Shurer, C.R.; Colville, M.J.; Gupta, V.K.; Head, S.E.; Kai, F.; Lakins, J.N.; Paszek, M.J. Genetically Encoded Toolbox for Glycocalyx Engineering: Tunable Control of Cell Adhesion, Survival, and Cancer Cell Behaviors. ACS Biomater. Sci. Eng. 2018, 4, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Kimata, K.; Itano, N. Key Roles of Hyaluronan and Its CD44 Receptor in the Stemness and Survival of Cancer Stem Cells. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Kuo, J.C.-H.; Gandhi, J.G.; Zia, R.N.; Paszek, M.J. Physical biology of the cancer cell glycocalyx. Nat. Phys. 2018, 14, 658–669. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Tzu, J.; Marinkovich, M.P. Bridging structure with function: Structural, regulatory, and developmental role of laminins. Int. J. Biochem. Cell Biol. 2008, 40, 199–214. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.H. The Glomerular Basement Membrane. Exp. Cell. Res. 2012, 318, 973–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Chaudhuri, O. Beyond proteases: Basement membrane mechanics and cancer invasion. J. Cell Biol. 2019, 218, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenstrup, R.J.; Smith, S.M.; Florer, J.B.; Zhang, G.; Beason, D.P.; Seegmiller, R.E.; Soslowsky, L.J.; Birk, D.E. Regulation of Collagen Fibril Nucleation and Initial Fibril Assembly Involves Coordinate Interactions with Collagens V and XI in Developing Tendon. J. Biol. Chem. 2011, 286, 20455–20465. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.J.; Mallory, C.; McDougal, O.M.; Oxford, J.T. Proteomic analysis of Col11a1-associated protein complexes. Proteomics 2011, 11, 4660–4676. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.M.; Collins, J.W.; Walma, D.A.C.; Doyle, A.D.; Morales, S.G.; Lu, J.; Matsumoto, K.; Nazari, S.S.; Sekiguchi, R.; Shinsato, Y.; et al. Extracellular matrix dynamics in cell migration, invasion and tissue morphogenesis. Int. J. Exp. Pathol. 2019, 100, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Eastwood, M.; Mudera, V.C.; Mcgrouther, D.A.; Brown, R.A. Effect of precise mechanical loading on fibroblast populated collagen lattices: Morphological changes. Cell Motil. 1998, 40, 13–21. [Google Scholar] [CrossRef]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Phan, S.H. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol. Res. 2016, 108, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Bird, D.; Baker, A.-M.; Barker, H.E.; Ho, M.W.-Y.; Lang, G.; Erler, J.T. LOX-Mediated Collagen Crosslinking Is Responsible for Fibrosis-Enhanced Metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, J.R.; Benson, A.B.; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. eLife 2014, 3, e01308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, J.D.; Myers, S.A.; Naba, A.; Abbruzzese, G.; Lamar, J.M.; Carr, S.A.; Hynes, R.O. Proteomic Profiling of the ECM of Xenograft Breast Cancer Metastases in Different Organs Reveals Distinct Metastatic Niches. Cancer Res. 2020, 80, 1475–1485. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Lim, S.Y.; Yuzhalin, A.; Lucotti, S.; Vermeer, J.A.F.; Jones, K.; Chen, J.; Muschel, R.J. Tumour-Derived Laminin α5 (LAMA5) Promotes Colorectal Liver Metastasis Growth, Branching Angiogenesis and Notch Pathway Inhibition. Cancers (Basel) 2019, 11, 630. [Google Scholar] [CrossRef] [Green Version]
- Deligne, C.; Murdamoothoo, D.; Gammage, A.N.; Gschwandtner, M.; Erne, W.; Loustau, T.; Marzeda, A.M.; Carapito, R.; Paul, N.; Velazquez-Quesada, I.; et al. Matrix-Targeting Immunotherapy Controls Tumor Growth and Spread by Switching Macrophage Phenotype. Cancer Immunol. Res. 2020, 8, 368–382. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [Green Version]
- Yuzhalin, A.E.; Lim, S.Y.; Kutikhin, A.G.; Gordon-Weeks, A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta. Rev. Cancer 2018, 1870, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; DuFort, C.C.; Rossier, O.; Bainer, R.; Mouw, J.K.; Godula, K.; Hudak, J.E.; Lakins, J.N.; Wijekoon, A.C.; Cassereau, L.; et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 2014, 511, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Wu, Q.; Sun, A.; Liu, X.; Fan, Y.; Deng, X. Cancer Cell Glycocalyx and Its Significance in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.-V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [Green Version]
- Kuppen, P.J.K.; van der Eb, M.M.; Jonges, L.E.; Hagenaars, M.; Hokland, M.E.; Nannmark, U.; Goldfarb, R.H.; Basse, P.H.; Fleuren, G.J.; Hoeben, R.C.; et al. Tumor structure and extracellular matrix as a possible barrier for therapeutic approaches using immune cells or adenoviruses in colorectal cancer. Histochem. Cell Biol. 2001, 115, 67–72. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive Stroma in Human Prostate Cancer: Induction of Myofibroblast Phenotype and Extracellular Matrix Remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, O.; Koshy, S.T.; Branco da Cunha, C.; Shin, J.-W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef] [Green Version]
- Kehlet, S.N.; Sanz-Pamplona, R.; Brix, S.; Leeming, D.J.; Karsdal, M.A.; Moreno, V. Excessive collagen turnover products are released during colorectal cancer progression and elevated in serum from metastatic colorectal cancer patients. Sci. Rep. 2016, 6, 30599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Feng, B.; Dong, T.; Yan, G.; Tan, B.; Shen, H.; Huang, A.; Zhang, X.; Zhang, M.; Yang, P.; et al. Up-regulation of type I collagen during tumorigenesis of colorectal cancer revealed by quantitative proteomic analysis. J. Proteom. 2013, 94, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Mehta, G.; Keikhosravi, A.; Liu, Y.; Lemancik, S.; Ricke, W.A.; Weber, S.M.; Kao, W.J.; et al. Comparison of Picrosirius Red Staining With Second Harmonic Generation Imaging for the Quantification of Clinically Relevant Collagen Fiber Features in Histopathology Samples. J. Histochem. Cytochem. 2016, 64, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadiarnykh, O.; LaComb, R.B.; Brewer, M.A.; Campagnola, P.J. Alterations of the extracellular matrix in ovarian cancer studied by Second Harmonic Generation imaging microscopy. BMC Cancer 2010, 10, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, K.; Tang, P.; Brown, E. Second harmonic generation reveals matrix alterations during breast tumor progression. J. Biomed. Opt. 2013, 18, 31106. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing Role of Contact Guidance in Intrastromal T-cell Trapping in Human Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Han, W.; Chen, S.; Yuan, W.; Fan, Q.; Tian, J.; Wang, X.; Chen, L.; Zhang, X.; Wei, W.; Liu, R.; et al. Oriented collagen fibers direct tumor cell intravasation. Proc. Natl. Acad. Sci. USA 2016, 113, 11208–11213. [Google Scholar] [CrossRef] [Green Version]
- Brabrand, A.; Kariuki, I.I.; Engstrøm, M.J.; Haugen, O.A.; Dyrnes, L.A.; Åsvold, B.O.; Lilledahl, M.B.; Bofin, A.M. Alterations in collagen fibre patterns in breast cancer. A premise for tumour invasiveness? APMIS 2015, 123, 1–8. [Google Scholar] [CrossRef]
- Wu, J.; Liang, C.; Chen, M.; Su, W. Association between tumor-stroma ratio and prognosis in solid tumor patients: A systematic review and meta-analysis. Oncotarget 2016, 7, 68954–68965. [Google Scholar] [CrossRef] [Green Version]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, L.; Xia, Y.; Discher, D.E.; Janmey, P.A. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgel, J.P.R.O.; Madhurapantula, R.S. A structural prospective for collagen receptors such as DDR and their binding of the collagen fibril. Biochimica. Et. Biophysica. Acta. Mol. Cell Res. 2019, 1866, 118478. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orend, G.; Chiquet-Ehrismann, R. Tenascin-C induced signaling in cancer. Cancer Lett. 2006, 244, 143–163. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican—A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Takahashi, H.; Lin, W.-W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.-L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.; Liphardt, J.; Hwang, E.; Weaver, V. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integr. Biol. (Camb.) 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Mrass, P.; Takano, H.; Ng, L.G.; Daxini, S.; Lasaro, M.O.; Iparraguirre, A.; Cavanagh, L.L.; von Andrian, U.H.; Ertl, H.C.J.; Haydon, P.G.; et al. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J. Exp. Med. 2006, 203, 2749–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eynden, G.G.V. den; Colpaert, C.G.; Couvelard, A.; Pezzella, F.; Dirix, L.Y.; Vermeulen, P.B.; Marck, E.A.V.; Hasebe, T. A fibrotic focus is a prognostic factor and a surrogate marker for hypoxia and (lymph)angiogenesis in breast cancer: review of the literature and proposal on the criteria of evaluation. Histopathology 2007, 51, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Rivas-Caicedo, A.; Bougherara, H.; Salmon, H.; Donnadieu, E. Positive and negative influence of the matrix architecture on antitumor immune surveillance. Cell. Mol. Life Sci. 2013, 70, 4431–4448. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sun, H.; Wang, X.; Zhang, Z.; Zhou, Y.; Anderson, C.; Ma, X.-J. Abstract 4965: Extracellular matrix gene expression and cytotoxic T lymphocyte infiltration in the tumor microenvironment in non-small cell lung cancer. Cancer Res. 2019, 79, 4965. [Google Scholar] [CrossRef]
- Huang, J.-Y.; Cheng, Y.-J.; Lin, Y.-P.; Lin, H.-C.; Su, C.-C.; Juliano, R.; Yang, B.-C. Extracellular Matrix of Glioblastoma Inhibits Polarization and Transmigration of T Cells: The Role of Tenascin-C in Immune Suppression. J. Immunol. 2010, 185, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.-L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. JITC 2019, 7, 68. [Google Scholar] [CrossRef] [Green Version]
- Hazelbag, S.; Gorter, A.; Kenter, G.G.; van den Broek, L.; Fleuren, G. Transforming growth factor-beta1 induces tumor stroma and reduces tumor infiltrate in cervical cancer. Hum. Pathol. 2002, 33, 1193–1199. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF-β attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Hallmann, R.; Zhang, X.; Di Russo, J.; Li, L.; Song, J.; Hannocks, M.-J.; Sorokin, L. The regulation of immune cell trafficking by the extracellular matrix. Curr. Opin. Cell Biol. 2015, 36, 54–61. [Google Scholar] [CrossRef]
- Wolf, K.; te Lindert, M.; Krause, M.; Alexander, S.; te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lämmermann, T.; Bader, B.L.; Monkley, S.J.; Worbs, T.; Wedlich-Söldner, R.; Hirsch, K.; Keller, M.; Förster, R.; Critchley, D.R.; Fässler, R.; et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 2008, 453, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Entschladen, F.; Conrad, C.; Niggemann, B.; Zänker, K.S. CD4+ T lymphocytes migrating in three-dimensional collagen lattices lack focal adhesions and utilize beta1 integrin-independent strategies for polarization, interaction with collagen fibers and locomotion. Eur. J. Immunol. 1998, 28, 2331–2343. [Google Scholar] [CrossRef]
- Sadjadi, Z.; Zhao, R.; Hoth, M.; Qu, B.; Rieger, H. Migration of Cytotoxic T Lymphocytes in 3D Collagen Matrices. bioRxiv 2020, 2020.01.14.906016. [Google Scholar] [CrossRef]
- Pruitt, H.C.; Lewis, D.; Ciccaglione, M.; Connor, S.; Smith, Q.; Hickey, J.W.; Schneck, J.P.; Gerecht, S. Collagen fiber structure guides 3D motility of cytotoxic T lymphocytes. Matrix Biol. 2020, 85–86, 147–159. [Google Scholar] [CrossRef]
- Bajénoff, M.; Egen, J.G.; Koo, L.Y.; Laugier, J.P.; Brau, F.; Glaichenhaus, N.; Germain, R.N. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity 2006, 25, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Boissonnas, A.; Fetler, L.; Zeelenberg, I.S.; Hugues, S.; Amigorena, S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007, 204, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Chetoui, N.; El Azreq, M.-A.; Boisvert, M.; Bergeron, M.-È.; Aoudjit, F. Discoidin domain receptor 1 expression in activated T cells is regulated by the ERK MAP kinase signaling pathway. J. Cell Biochem. 2011, 112, 3666–3674. [Google Scholar] [CrossRef]
- Hachehouche, L.N.; Chetoui, N.; Aoudjit, F. Implication of discoidin domain receptor 1 in T cell migration in three-dimensional collagen. Mol. Immunol. 2010, 47, 1866–1869. [Google Scholar] [CrossRef]
- Kamohara, H.; Yamashiro, S.; Galligan, C.; Yoshimura, T. Discoidin domain receptor 1 isoform-a (DDR1alpha) promotes migration of leukocytes in three-dimensional collagen lattices. FASEB J. 2001, 15, 2724–2726. [Google Scholar] [CrossRef]
- Afonso, P.V.; McCann, C.P.; Kapnick, S.M.; Parent, C.A. Discoidin domain receptor 2 regulates neutrophil chemotaxis in 3D collagen matrices. Blood 2013, 121, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Maller, O.; Drain, A.P.; Barrett, A.S.; Borgquist, S.; Ruffell, B.; Thanh, P.T.; Gruosso, T.; Kuasne, H.; Lakins, J.N.; Acerbi, I.; et al. Inflammation promotes tumor aggression by stimulating stromal cell-dependent collagen crosslinking and stromal stiffening. bioRxiv 2020, 2020.02.13.948141. [Google Scholar] [CrossRef]
- Thomas, A.H.; Edelman, E.R.; Stultz, C.M. Collagen fragments modulate innate immunity. Exp. Biol. Med. (Maywood) 2007, 232, 406–411. [Google Scholar] [PubMed]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: the promise of matrix-derived immune biomarkers. JITC 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Lyons, T.; Monks, J.; Lucia, M.S.; Wilson, R.S.; Hines, L.; Man, Y.; Borges, V.; Schedin, P. Alternatively Activated Macrophages and Collagen Remodeling Characterize the Postpartum Involuting Mammary Gland across Species. Am. J. Pathol. 2010, 176, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Leifler, K.S.; Svensson, S.; Abrahamsson, A.; Bendrik, C.; Robertson, J.; Gauldie, J.; Olsson, A.-K.; Dabrosin, C. Inflammation Induced by MMP-9 Enhances Tumor Regression of Experimental Breast Cancer. J. Immunol. 2013, 190, 4420–4430. [Google Scholar] [CrossRef]
- Liang, J.; Liu, X.; Xie, Q.; Chen, G.; Li, X.; Jia, Y.; Yin, B.; Qu, X.; Li, Y. Endostatin enhances antitumor effect of tumor antigen-pulsed dendritic cell therapy in mouse xenograft model of lung carcinoma. Chin. J. Cancer Res. 2016, 28, 452–460. [Google Scholar] [CrossRef]
- Li, M.; Huang, X.; Zhu, Z.; Wong, M.; Watkins, S.; Zhao, Q.; Herberman, R.; Gorelik, E. Immune Response Against 3LL Lewis Lung Carcinoma Potentiates the Therapeutic Efficacy of Endostatin. J. Immunother. 2001, 24, 472–481. [Google Scholar] [CrossRef]
- Stroka, K.M.; Aranda-Espinoza, H. Neutrophils display biphasic relationship between migration and substrate stiffness. Cell Motility 2009, 66, 328–341. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.N.; Lim, S.Y.; Yuzhalin, A.E.; Jones, K.; Markelc, B.; Kim, K.J.; Buzzelli, J.N.; Fokas, E.; Cao, Y.; Smart, S.; et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology 2017, 65, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Madhurapantula, R.S.; Kalyanasundaram, A.; Sabharwal, T.; Antipova, O.; Bishnoi, S.W.; Orgel, J.P.R.O. Ultrastructural Location and Interactions of the Immunoglobulin Receptor Binding Sequence within Fibrillar Type I Collagen. Int. J. Mol. Sci. 2020, 21, 4166. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Ecker, B.L.; Douglass, S.M.; Kugel, C.H.; Webster, M.R.; Almeida, F.V.; Somasundaram, R.; Hayden, J.; Ban, E.; Ahmadzadeh, H.; et al. Remodeling of the Collagen Matrix in Aging Skin Promotes Melanoma Metastasis and Affects Immune Cell Motility. Cancer Discov. 2019, 9, 64–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Manso, G.; Galli, S.; Ridnour, L.A.; Tsokos, M.; Wink, D.A.; Roberts, D.D. Thrombospondin-1 promotes tumor macrophage recruitment and enhances tumor cell cytotoxicity of differentiated U937 cells. Cancer Res. 2008, 68, 7090–7099. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-Y.; Yin, K.-M.; Bai, Y.-H.; Zhang, Z.-G.; Di, W.; Zhang, S. CTHRC1 promotes M2-like macrophage recruitment and myometrial invasion in endometrial carcinoma by integrin-Akt signaling pathway. Clin. Exp. Metastasis 2019, 36, 351–363. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Prada, F.; Salvatierra, E.; Bravo, A.I.; Lutzky, V.P.; Carbone, C.; Pitossi, F.J.; Chuluyan, H.E.; Podhajcer, O.L. Secreted protein acidic and rich in cysteine produced by human melanoma cells modulates polymorphonuclear leukocyte recruitment and antitumor cytotoxic capacity. Cancer Res. 2005, 65, 5123–5132. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Hey, D.R.V.D.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3–Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef]
- Rodríguez-Baena, F.J.; Redondo-García, S.; Peris-Torres, C.; Martino-Echarri, E.; Fernández-Rodríguez, R.; del Carmen Plaza-Calonge, M.; Anderson, P.; Rodríguez-Manzaneque, J.C. ADAMTS1 protease is required for a balanced immune cell repertoire and tumour inflammatory response. Sci. Rep. 2018, 8, 13103. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, R.; Rodríguez-Baena, F.J.; Martino-Echarri, E.; Peris-Torres, C.; del Carmen Plaza-Calonge, M.; Rodríguez-Manzaneque, J.C. Stroma-derived but not tumor ADAMTS1 is a main driver of tumor growth and metastasis. Oncotarget 2016, 7, 34507–34519. [Google Scholar] [CrossRef]
- Franitza, S.; Hershkoviz, R.; Kam, N.; Lichtenstein, N.; Vaday, G.G.; Alon, R.; Lider, O. TNF-alpha associated with extracellular matrix fibronectin provides a stop signal for chemotactically migrating T cells. J. Immunol. 2000, 165, 2738–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, F.; Rochotte, J.; Colacios, C.; Montfort, A.; Tilkin-Mariamé, A.-F.; Touriol, C.; Rochaix, P.; Lajoie-Mazenc, I.; Andrieu-Abadie, N.; Levade, T.; et al. Blocking Tumor Necrosis Factor α Enhances CD8 T-cell–Dependent Immunity in Experimental Melanoma. Cancer Res. 2015, 75, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson-Haidaris, P.J.; Rybarczyk, B. Tumors and fibrinogen. The role of fibrinogen as an extracellular matrix protein. Ann. N. Y. Acad. Sci. 2001, 936, 406–425. [Google Scholar] [CrossRef]
- Zhang, P.; Ozdemir, T.; Chung, C.-Y.; Robertson, G.P.; Dong, C. Sequential binding of αVβ3 and ICAM-1 determines fibrin-mediated melanoma capture and stable adhesion to CD11b/CD18 on neutrophils. J. Immunol. 2011, 186, 242–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, T.; Zhang, P.; Fu, C.; Dong, C. Fibrin serves as a divalent ligand that regulates neutrophil-mediated melanoma cells adhesion to endothelium under shear conditions. Am. J. Physiol. Cell Physiol. 2012, 302, C1189–C1201. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.-T.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef]
- Ochieng, J.; Leite-Browning, M.L.; Warfield, P. Regulation of Cellular Adhesion to Extracellular Matrix Proteins by Galectin-3. Biochem. Biophys. Res. Commun. 1998, 246, 788–791. [Google Scholar] [CrossRef]
- RABINOVICH, G.A.; ARIEL, A.; HERSHKOVIZ, R.; HIRABAYASHI, J.; KASAI, K.-I.; LIDER, O. Specific inhibition of T-cell adhesion to extracellular matrix and proinflammatory cytokine secretion by human recombinant galectin-1. Immunology 1999, 97, 100–106. [Google Scholar] [CrossRef]
- Nambiar, D.K.; Aguilera, T.; Cao, H.; Kwok, S.; Kong, C.; Bloomstein, J.; Wang, Z.; Rangan, V.S.; Jiang, D.; Eyben, R. von; et al. Galectin-1–driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J. Clin. Investig. 2019, 129, 5553–5567. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Alonso, M.; Hirsch, T.; Wildmann, C.; van der Bruggen, P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat. Commun. 2017, 8, 793. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil (TAN) Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedemann, M.; Kalbitzer, L.; Franz, S.; Moeller, S.; Schnabelrauch, M.; Simon, J.-C.; Pompe, T.; Franke, K. Instructing Human Macrophage Polarization by Stiffness and Glycosaminoglycan Functionalization in 3D Collagen Networks. Adv. Healthc. Mater. 2017, 6, 1600967. [Google Scholar] [CrossRef] [PubMed]
- Soares da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katja, F.; Liv, K.; Sandra, F. Tilo Pompe Modulating plasticity of primary human macrophages by extracellular matrix signals of biomimetic 3D matrices. Front. Bioeng. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Kajahn, J.; Franz, S.; Rueckert, E.; Forstreuter, I.; Hintze, V.; Moeller, S.; Simon, J.C. Artificial extracellular matrices composed of collagen I and high sulfated hyaluronan modulate monocyte to macrophage differentiation under conditions of sterile inflammation. Biomatter 2012, 2, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.Y.; Keating, M.T.; Smith, T.D.; Meli, V.S.; Botvinick, E.L.; Liu, W.F. Matrix crosslinking enhances macrophage adhesion, migration, and inflammatory activation. APL Bioeng. 2019, 3, 016103. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Li, J.; Wei, Y.; Gao, W.; Fu, X.; Wang, Y. Substrate stiffness affects the immunosuppressive and trophic function of hMSCs via modulating cytoskeletal polymerization and tension. Biomater. Sci. 2019, 7, 5292–5300. [Google Scholar] [CrossRef]
- Choi, J.S.; Harley, B.A.C. Marrow-inspired matrix cues rapidly affect early fate decisions of hematopoietic stem and progenitor cells. Sci. Adv. 2017, 3, e1600455. [Google Scholar] [CrossRef] [Green Version]
- Huleihel, L.; Dziki, J.L.; Bartolacci, J.G.; Rausch, T.; Scarritt, M.E.; Cramer, M.C.; Vorobyov, T.; LoPresti, S.T.; Swineheart, I.T.; White, L.J.; et al. Macrophage phenotype in response to ECM bioscaffolds. Semin. Immunol. 2017, 29, 2–13. [Google Scholar] [CrossRef]
- Sicari, B.M.; Dziki, J.L.; Siu, B.F.; Medberry, C.J.; Dearth, C.L.; Badylak, S.F. The promotion of a constructive macrophage phenotype by solubilized extracellular matrix. Biomaterials 2014, 35, 8605–8612. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yang, Z.; Jin, Y.; Qi, X.; Chu, J.; Deng, X. ADM Scaffolds Generate a Pro-regenerative Microenvironment During Full-Thickness Cutaneous Wound Healing Through M2 Macrophage Polarization via Lamtor1. Front. Physiol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.M.H.; Kuczek, D.E.; Kalvisa, A.; Siersbæk, M.S.; Thorseth, M.-L.; Johansen, A.Z.; Carretta, M.; Grøntved, L.; Vang, O.; Madsen, D.H. Collagen Density Modulates the Immunosuppressive Functions of Tumor-Associated Macrophages. bioRxiv 2019, 513986. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.-Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- García-Mendoza, M.G.; Inman, D.R.; Ponik, S.M.; Jeffery, J.J.; Sheerar, D.S.; Van Doorn, R.R.; Keely, P.J. Neutrophils drive accelerated tumor progression in the collagen-dense mammary tumor microenvironment. Breast Cancer Res. 2016, 18, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettritz, R.; Xu, Y.-X.; Kerren, T.; Quass, P.; Klein, J.; Luft, F.C.; Haller, H. Extracellular matrix regulates apoptosis in human neutrophils. Kidney Int. 1999, 55, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Srimal, S.; Farber, C.; Sanchez, E.; Kabbash, L.; Asch, A.; Gailit, J.; Wright, S.D. Cytokine-induced respiratory burst of human neutrophils: Dependence on extracellular matrix proteins and CD11/CD18 integrins. J. Cell Biol. 1989, 109, 1341–1349. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.F. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J. Clin. Investig. 1987, 80, 1550–1560. [Google Scholar] [CrossRef]
- Sangaletti, S.; Talarico, G.; Chiodoni, C.; Cappetti, B.; Botti, L.; Portararo, P.; Gulino, A.; Consonni, F.M.; Sica, A.; Randon, G.; et al. SPARC Is a New Myeloid-Derived Suppressor Cell Marker Licensing Suppressive Activities. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Nardin, A.; Wong, W.-C.; Tow, C.; Molina, T.J.; Tissier, F.; Audebourg, A.; Garcette, M.; Caignard, A.; Avril, M.-F.; Abastado, J.-P.; et al. Dacarbazine promotes stromal remodeling and lymphocyte infiltration in cutaneous melanoma lesions. J. Invest. Dermatol. 2011, 131, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Papadas, A.; Flietner, E.; Morrow, Z.; Wiesner, J.; Cicala, A.; Pagenkopf, A.; Hope, C.L.; Rajagopalan, A.; Wen, Z.; Emmerich, P.; et al. Versican Proteolytic Fragments (Matrikines) Regulate the Intratumoral Dendritic Cell Milieu In Vivo: Implications for in Situ Tumor Vaccination. Blood 2019, 134, 1210. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediators Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E.; Urbonas, T.; Silva, M.A.; Muschel, R.J.; Gordon-Weeks, A.N. A core matrisome gene signature predicts cancer outcome. Br. J. Cancer 2018, 118, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 2018, 128, 5549–5560. [Google Scholar] [CrossRef]
- Jachetti, E.; Caputo, S.; Mazzoleni, S.; Brambillasca, C.S.; Parigi, S.M.; Grioni, M.; Piras, I.S.; Restuccia, U.; Calcinotto, A.; Freschi, M.; et al. Tenascin-C Protects Cancer Stem–like Cells from Immune Surveillance by Arresting T-cell Activation. Cancer Res. 2015, 75, 2095–2108. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Guo, H.; Geng, J.; Zheng, X.; Wei, H.; Sun, R.; Tian, Z. Tumor-released Galectin-3, a Soluble Inhibitory Ligand of Human NKp30, Plays an Important Role in Tumor Escape from NK Cell Attack. J. Biol. Chem. 2014, 289, 33311–33319. [Google Scholar] [CrossRef] [Green Version]
- Golden-Mason, L.; McMahan, R.H.; Strong, M.; Reisdorph, R.; Mahaffey, S.; Palmer, B.E.; Cheng, L.; Kulesza, C.; Hirashima, M.; Niki, T.; et al. Galectin-9 Functionally Impairs Natural Killer Cells in Humans and Mice. J. Virol 2013, 87, 4835–4845. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, J.; Ma, C.; Gao, W.; Song, B.; Xue, H.; Chen, W.; Chen, X.; Zhang, Y.; Shao, Q.; et al. Reduced Expression of Galectin-9 Contributes to a Poor Outcome in Colon Cancer by Inhibiting NK Cell Chemotaxis Partially through the Rho/ROCK1 Signaling Pathway. PLoS ONE 2016, 11, e0152599. [Google Scholar] [CrossRef] [Green Version]
- Bordry, N.; Broggi, M.A.S.; de Jonge, K.; Schaeuble, K.; Gannon, P.O.; Foukas, P.G.; Danenberg, E.; Romano, E.; Baumgaertner, P.; Fankhauser, M.; et al. Lymphatic vessel density is associated with CD8+ T cell infiltration and immunosuppressive factors in human melanoma. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Keskin, D.; Kalluri, R. Interaction between the extracellular matrix and lymphatics—Consequences for lymphangiogenesis and lymphatic function. Matrix Biol. 2010, 29, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianopoulos, T.; Martin, J.D.; Snuderl, M.; Mpekris, F.; Jain, S.R.; Jain, R.K. Co-evolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013, 73, 3833–3841. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Bindea, G.; Kirilovsky, A.; Angell, H.K.; Obenauf, A.C.; Tosolini, M.; Church, S.E.; Maby, P.; Vasaturo, A.; Angelova, M.; et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med. 2016, 8, 327ra26. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.W.; Wagner, M.; Fankhauser, M.; Steinskog, E.S.; Broggi, M.A.; Spranger, S.; Gajewski, T.F.; Alitalo, K.; Eikesdal, H.P.; Wiig, H.; et al. Lymphatic vessels regulate immune microenvironments in human and murine melanoma. J. Clin. Investig. 2016, 126, 3389–3402. [Google Scholar] [CrossRef]
- Frye, M.; Taddei, A.; Dierkes, C.; Martinez-Corral, I.; Fielden, M.; Ortsäter, H.; Kazenwadel, J.; Calado, D.P.; Ostergaard, P.; Salminen, M.; et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat. Commun. 2018, 9, 1511. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Hoersch, S.; Liu, H.; Carr, S.A.; Hynes, R.O. The matrisome: In silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 2012, 11, M111.014647. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, T.; Gavras, I. Renin–Angiotensin Inhibition in Combating Malignancy: A Review. Anticancer Res. 2019, 39, 4597–4602. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, X.; Lu, J.; Salfenmoser, M.; Wirsik, N.M.; Schleussner, N.; Imle, A.; Freire Valls, A.; Radhakrishnan, P.; Liang, J.; et al. Reduction of Liver Metastasis Stiffness Improves Response to Bevacizumab in Metastatic Colorectal Cancer. Cancer Cell 2020, 37. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.; Ewachiw, T.; Jung, C.; Huang, A.; Norberg, K.J.; Marchionni, L.; McMillan, R.; Penchev, V.; Rajeshkumar, N.V.; Maitra, A.; et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0180181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canel, M.; Taggart, D.; Sims, A.H.; Lonergan, D.W.; Waizenegger, I.C.; Serrels, A. T-cell co-stimulation in combination with targeting FAK drives enhanced anti-tumor immunity. eLife 2020, 9, e48092. [Google Scholar] [CrossRef]
- Mohan, N.; Hosain, S.; Zhao, J.; Shen, Y.; Luo, X.; Jiang, J.; Endo, Y.; Wu, W.J. Atezolizumab potentiates Tcell-mediated cytotoxicity and coordinates with FAK to suppress cell invasion and motility in PD-L1+ triple negative breast cancer cells. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa With Nab-Paclitaxel Plus Gemcitabine for Patients With Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. JCO 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Guan, X.; Chen, J.; Hu, Y.; Lin, L.; Sun, P.; Tian, H.; Chen, X. Highly enhanced cancer immunotherapy by combining nanovaccine with hyaluronidase. Biomaterials 2018, 171, 198–206. [Google Scholar] [CrossRef]
- Singha, N.C.; Nekoroski, T.; Zhao, C.; Symons, R.; Jiang, P.; Frost, G.I.; Huang, Z.; Shepard, H.M. Tumor-Associated Hyaluronan Limits Efficacy of Monoclonal Antibody Therapy. Mol. Cancer Ther. 2015, 14, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Wang, L.-C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ertl, H.C.J. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget 2016, 7, 23282–23299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narra, K.; Mullins, S.R.; Lee, H.-O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers (Basel) 2020, 12, 1307. [Google Scholar] [CrossRef]
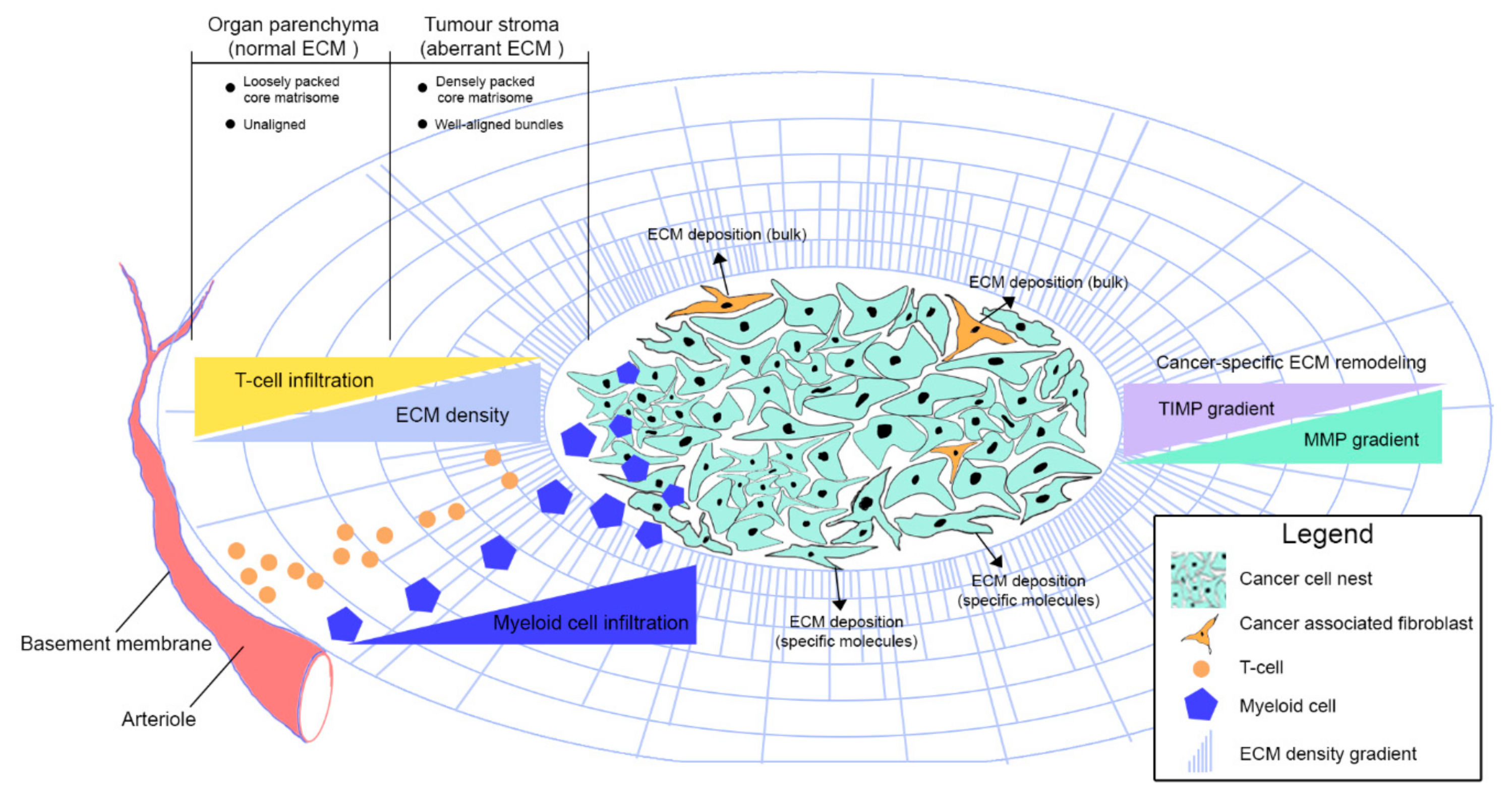

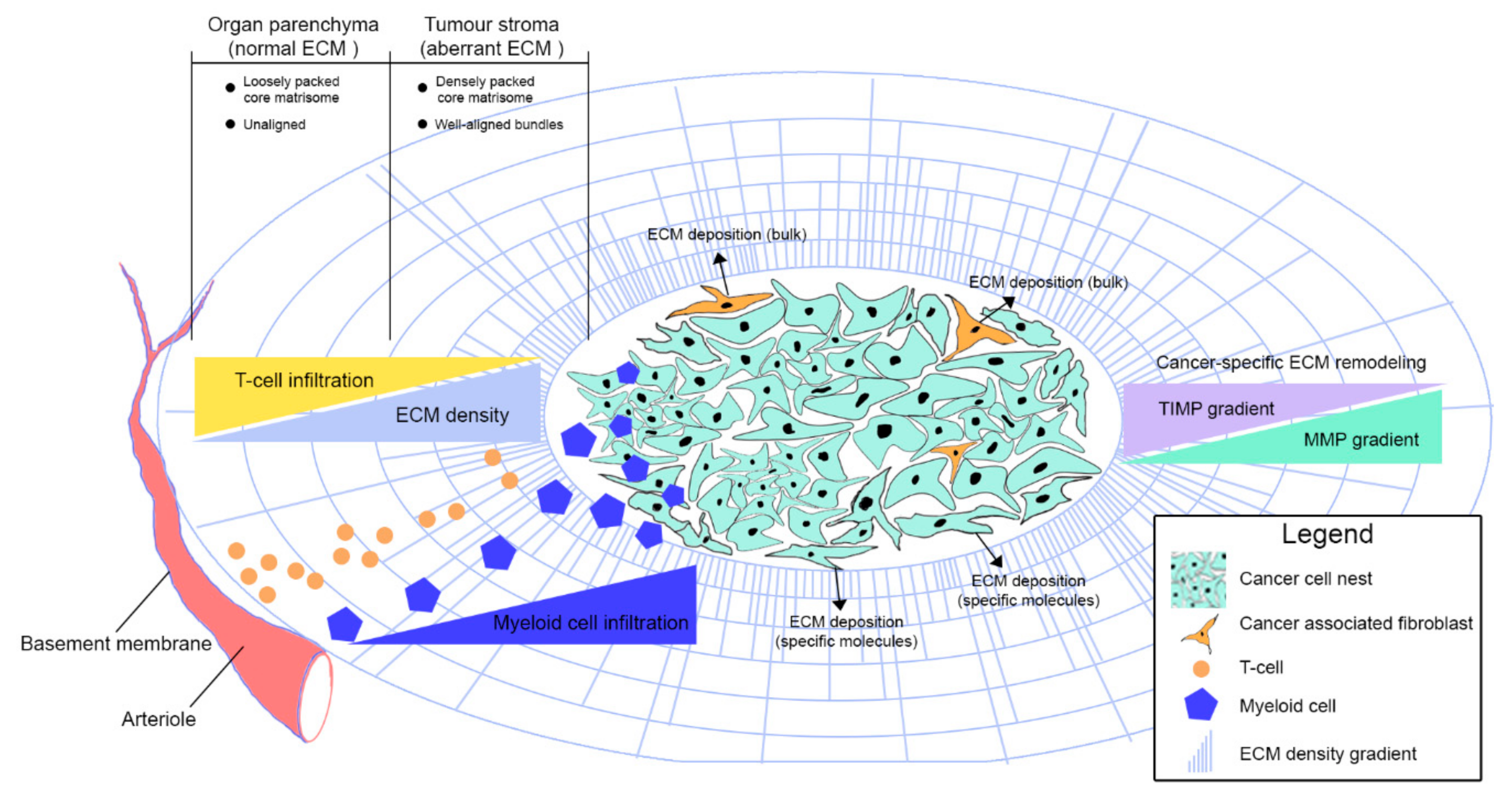{kind=link}
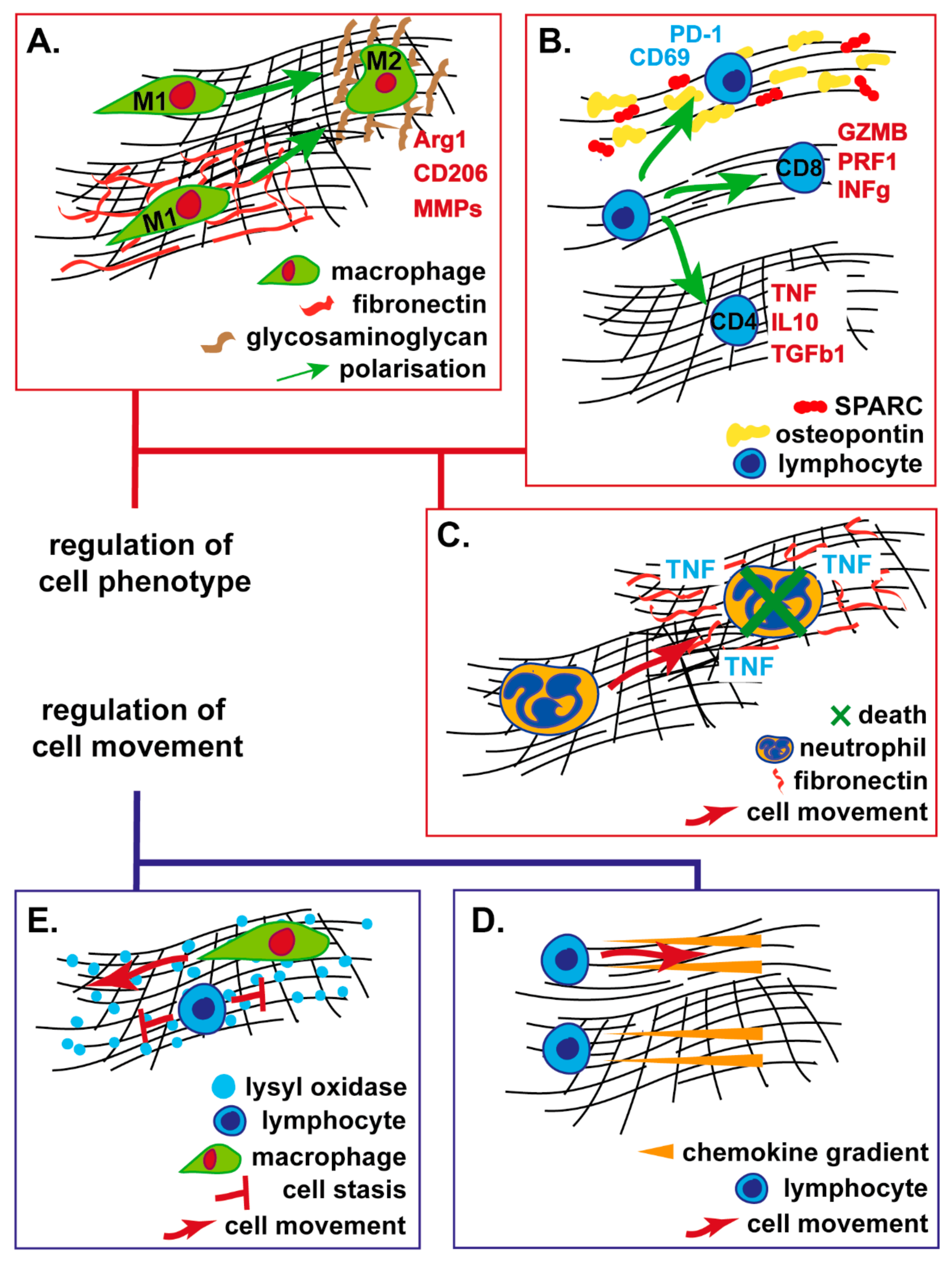
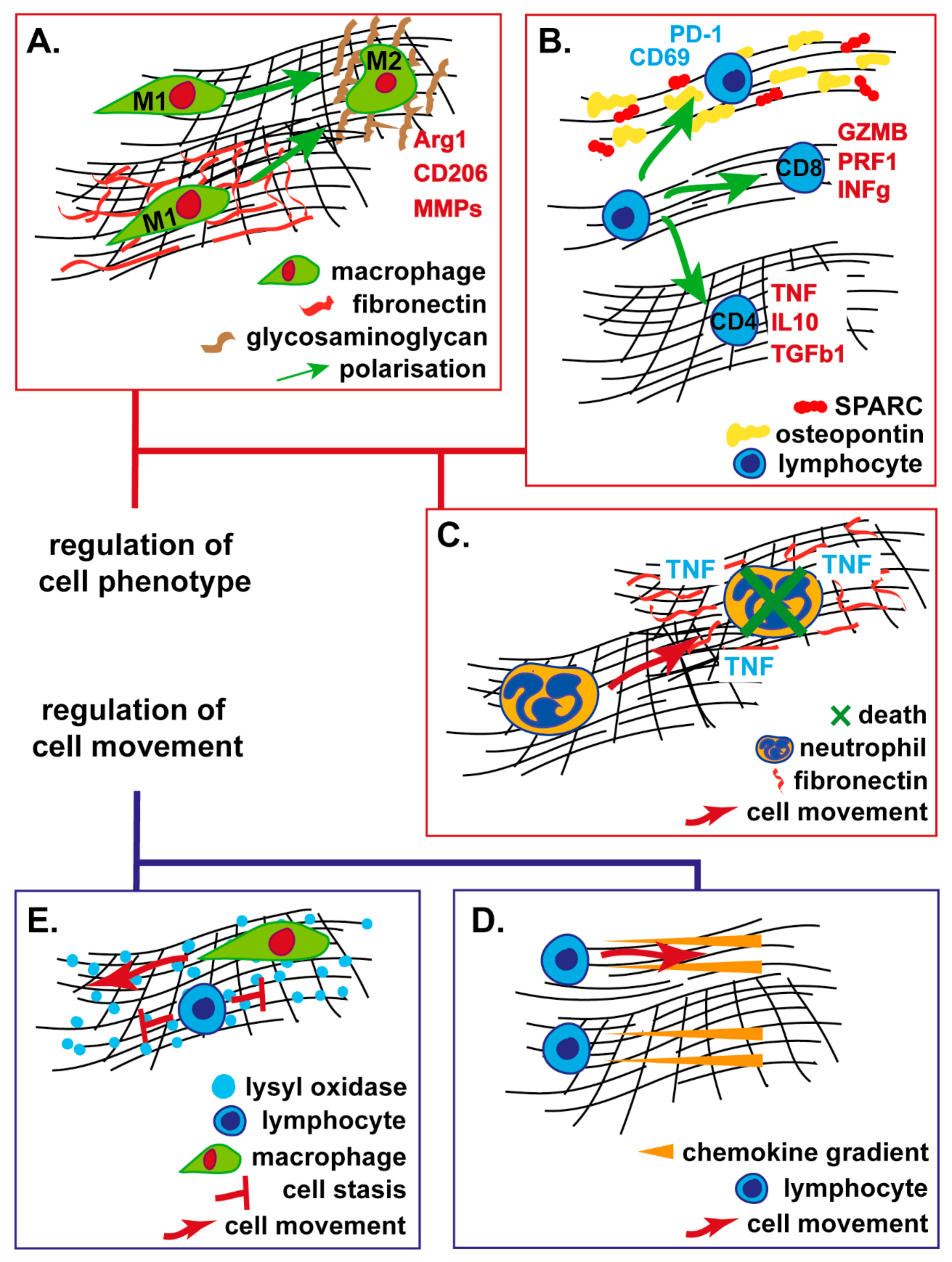{kind=link}
| Target | Cancer | Drug | Additional Treatments | Setting | Trial Number | Phase | Primary Outcome/Aim |
|---|---|---|---|---|---|---|---|
| FAK | Pancreas | Defactinib | PD-1 (Pembrolizumab) | Neoadjuvant and adjuvant | NCT03727880 | 2 | Pathological response |
| Lung, mesothelioma, pancreas | Defactinib | PD-1 (Pembrolizumab) | Palliative | NCT02758587 | 1 and 2 | safety | |
| Advanced solid cancer | Defactinib | PD-1 (Pembrolizumab), Gemcitabine | Palliative | NCT02546531 | 1 | Dose escalation | |
| RAS | Pancreas | losartan | PD-1 (Nivolumab), FOLFIRINOX, SBRT | Neoadjuvant | NCT03563248 | 2 | R0 resection rate |
| Hyaluronin | Stomach, lung | PEGPH20 (PEGylated recombinant human hyaluronidase | PD-1 (Pembrolizumab) | Palliative | NCT02563548 | 1 | Safety, dose escalation |
| Metastatic pancreas | PEGPH20 | PD-1 (Pembrolizumab) | Palliative | NCT03634332 | 1 | Progression-free survival |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gordon-Weeks, A.; Yuzhalin, A.E. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers 2020, 12, 3331. https://doi.org/10.3390/cancers12113331
Gordon-Weeks A, Yuzhalin AE. Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers. 2020; 12(11):3331. https://doi.org/10.3390/cancers12113331
Chicago/Turabian StyleGordon-Weeks, Alex, and Arseniy E. Yuzhalin. 2020. "Cancer Extracellular Matrix Proteins Regulate Tumour Immunity" Cancers 12, no. 11: 3331. https://doi.org/10.3390/cancers12113331
APA StyleGordon-Weeks, A., & Yuzhalin, A. E. (2020). Cancer Extracellular Matrix Proteins Regulate Tumour Immunity. Cancers, 12(11), 3331. https://doi.org/10.3390/cancers12113331