The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC)


Abstract
:Simple Summary
Abstract
1. Introduction
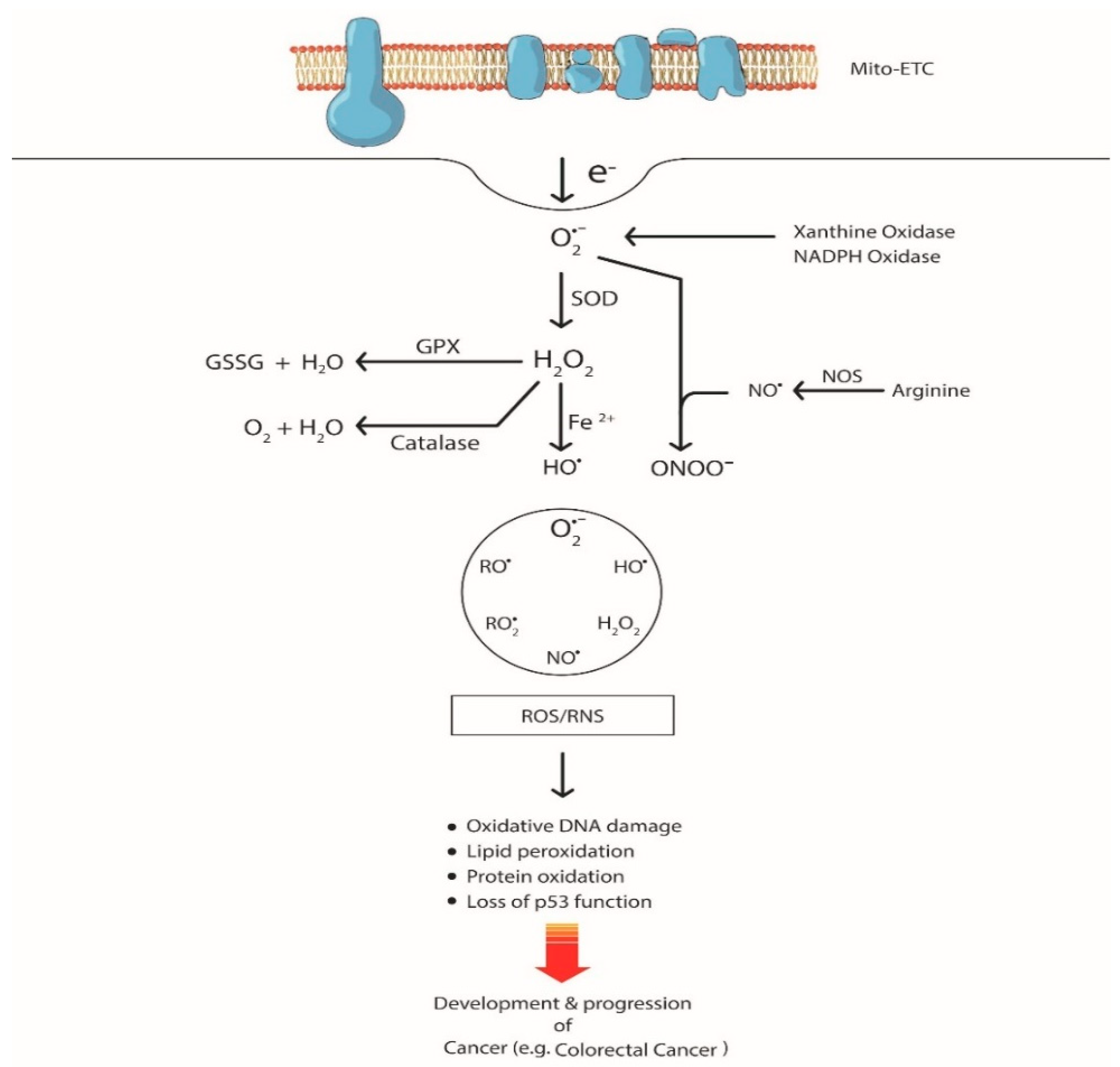
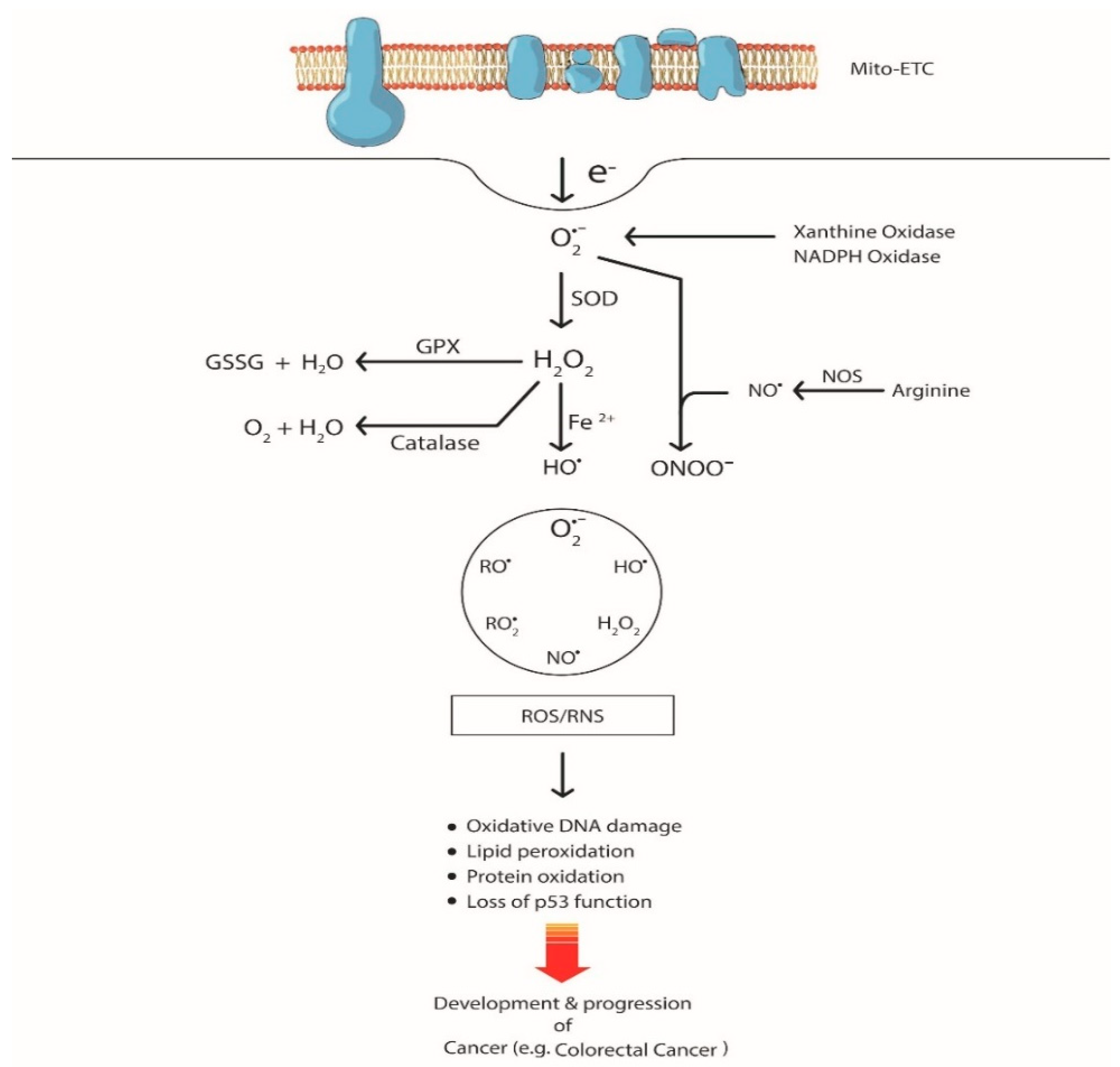
2. ROS-mediated Genetic Alterations in CRC
2.1. DNA Oxidation by ROS
2.2. Lipid Oxidation by ROS
2.3. Protein Oxidation by ROS
3. The Impact of Oxidative Stress-Induced Alteration of Signaling Pathways and Transcription Factors in CRC
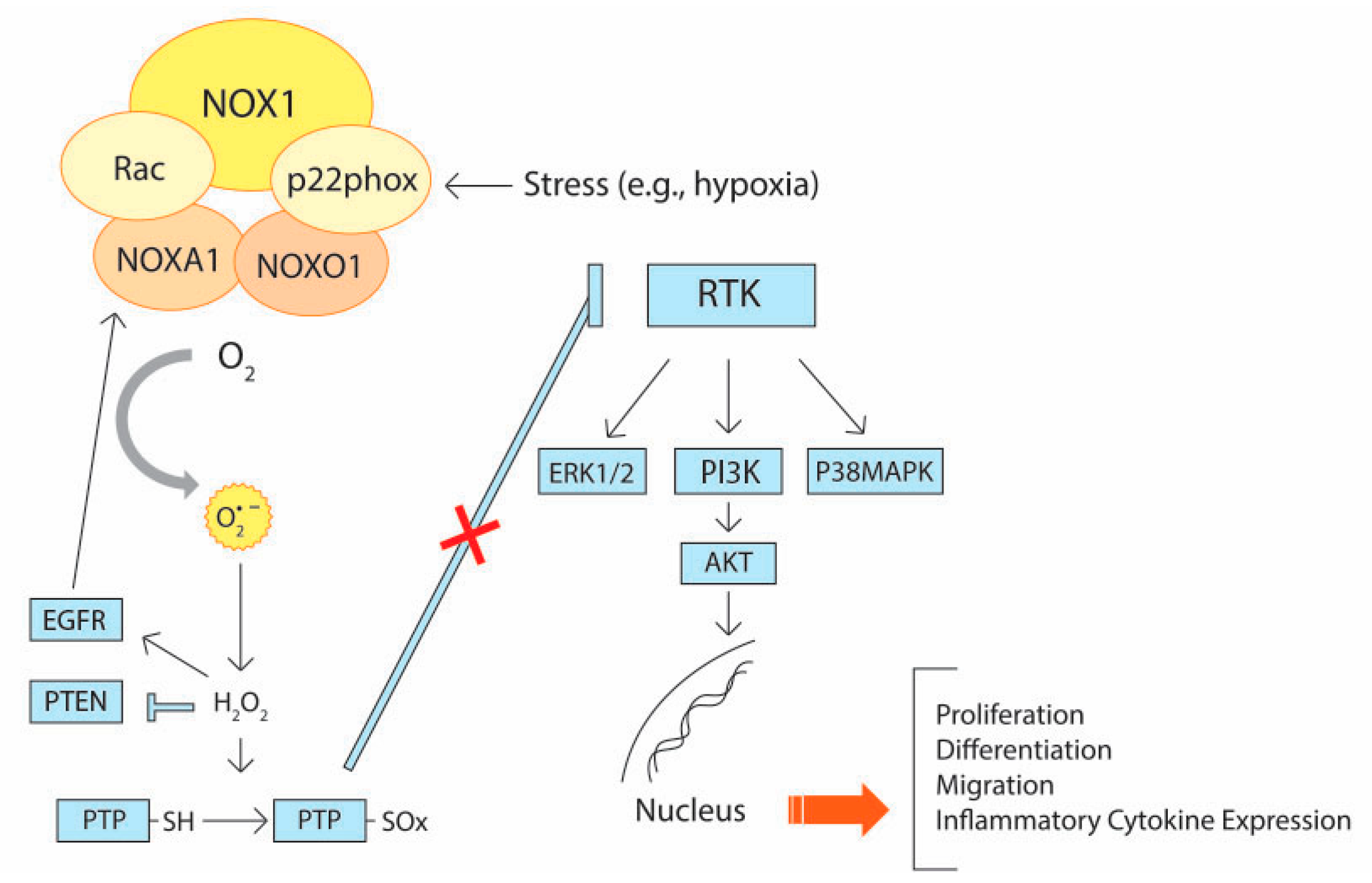
3.1. Signaling Pathways
3.2. Transcription Factors
4. The Role of Tumor Microenvironment (TME) in ROS Production and Their Pathophysiological Impact
5. The Role of ROS in Tumor Metastasis through Epithelial–Mesenchymal Transition (EMT)
6. Development of Drug Resistance Due to Redox Adaptation in CRC
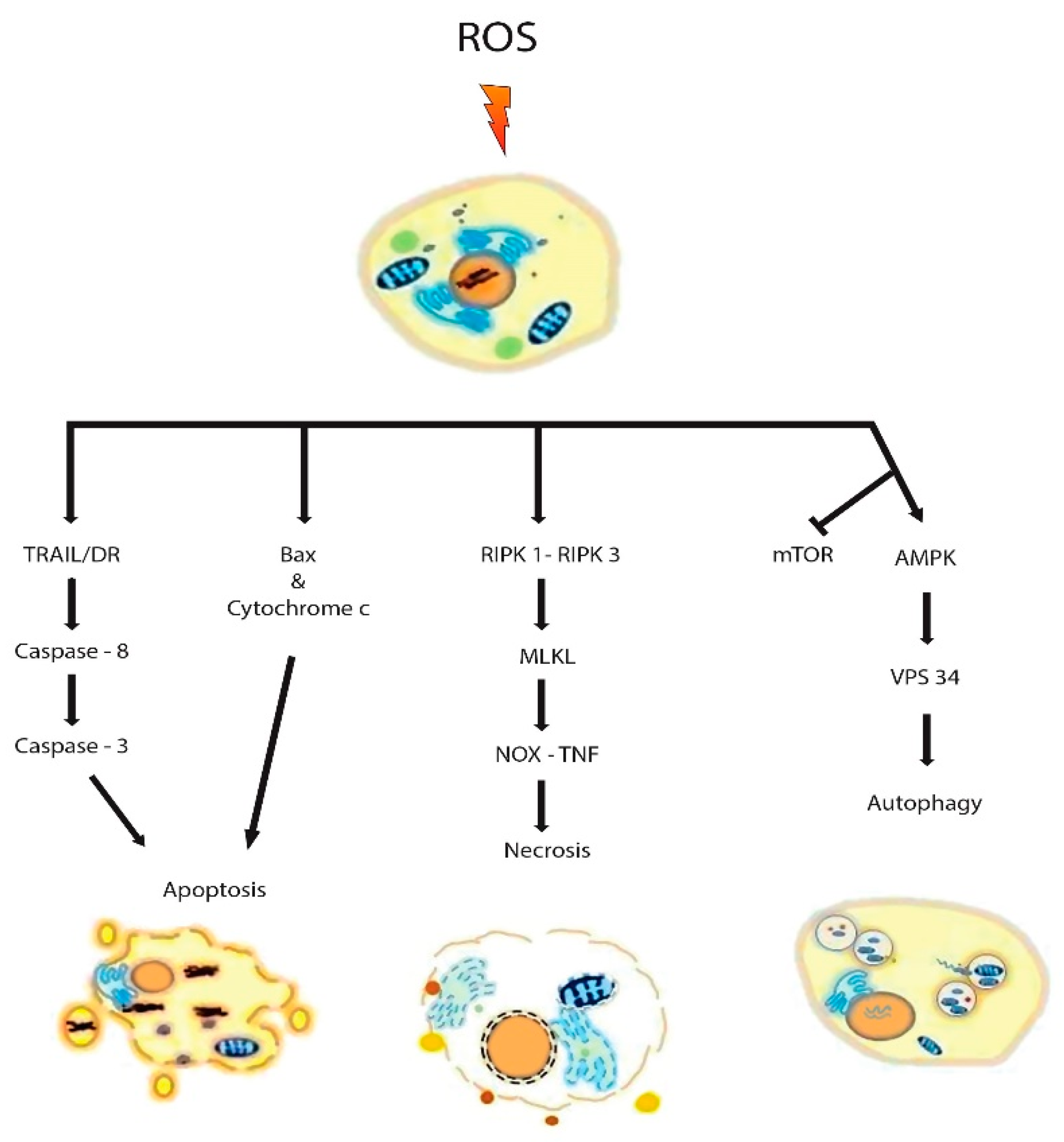
7. Counteractive Defense and Prevention of ROS from Occurring
8. Targeting Redox Alterations in CRC
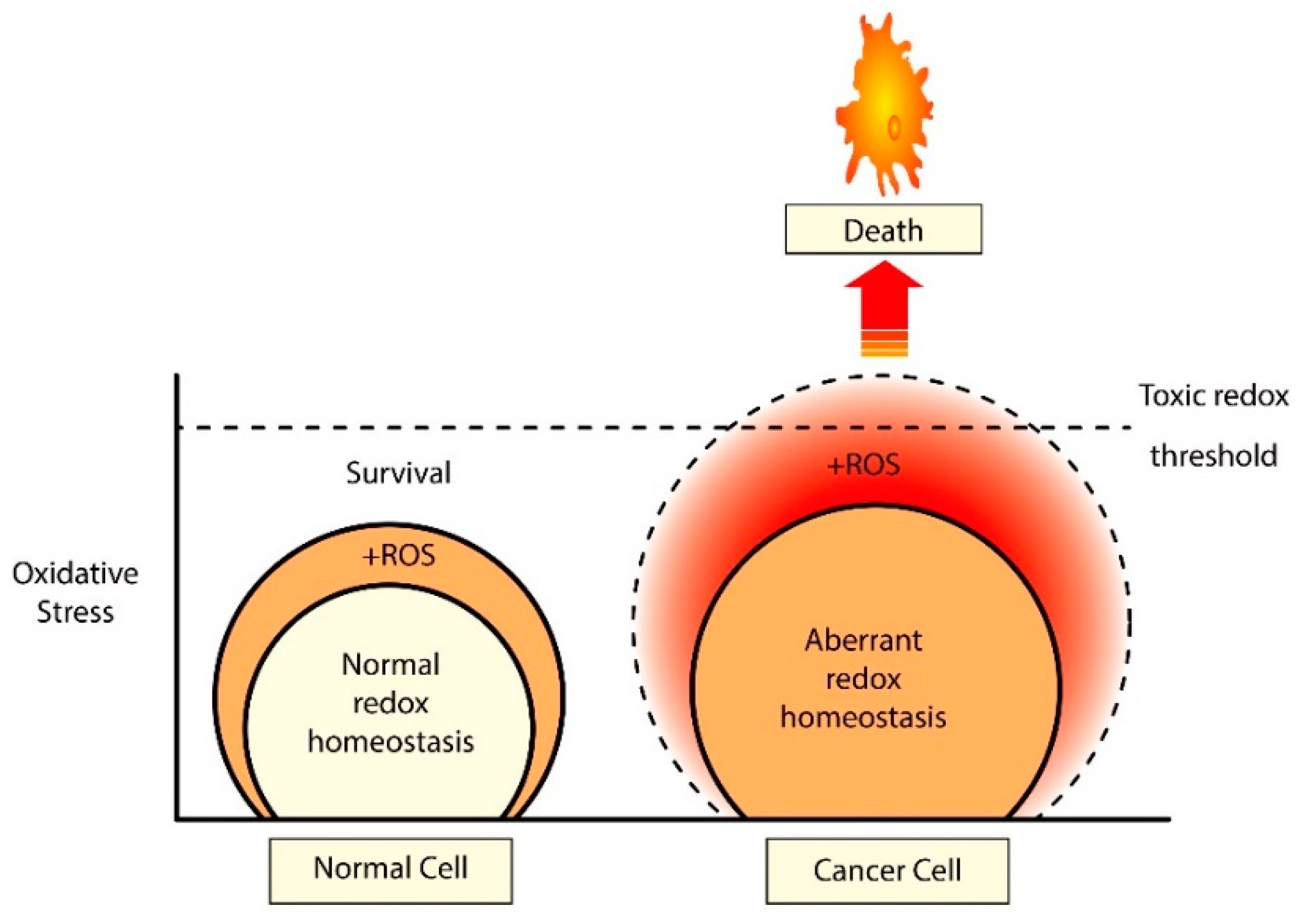
9. Concluding Statements and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 8-OHdG | 8-hydroxy-2′-deoxyguanosine |
| 8-oxodG | 8-oxo-7,8-dihydro-2′-deoxyguanosine |
| ACF | Aberrant crypt foci |
| AMPK | AMP-activated protein kinase |
| AOM | Azoxymethane |
| APC | Adenomatous polyposis coli |
| AR | Aldose reductase |
| ARE | Antioxidant responsive element |
| αSMA | α-smooth muscle actin |
| ATG4 | Autophagy related 4A cysteine peptidase |
| ATP | Adenosine triphosphate |
| Bcl-2 | B-cell lymphoma 2 |
| BER | Base excision repair |
| CAFs | Cancer-associated fibroblasts |
| CD | Crohn’s disease |
| COX-2 | Cyclooxygenase – 2 |
| CRC | Colorectal cancer |
| CSC | Cancer stem cells |
| dG | Deoxyguanosine |
| dGTP | Deoxyguanosine triphosphate |
| DR | Death receptor |
| DUOX | Dual oxidase |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| FAP | Fibroblast activation protein |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FLIP | Flice inhibitory protein |
| GIT | Gastrointestinal tract |
| GPx | Glutathione peroxidase |
| HDAC | Histone deacetylase |
| HDACIs | Histone deacetylase inhibitors |
| HIF-1α | Hypoxia inducible factor 1 alpha subunit |
| HNE | 4-hydroxy-2-nonenal |
| IBD | Inflammatory bowel diseases |
| JAK | Janus kinases |
| JNK | c-Jun NH(2)-terminal kinase |
| Keap1 | Kelch-like ECH-associated protein 1 |
| LCN-2 | Lipocalin-2 |
| LN | Lymphoid nodules |
| MAPK | Mitogen-activated protein kinase |
| MDA | Malondialdehyde |
| MDSC | Myeloid-derived suppressor cells |
| MMP | Matrix metalloproteinase |
| MMR | Mismatch repair |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase |
| MYH | MutY homologue |
| NAC | N-acetyl-l-cysteine |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NEMO | Nuclear factor-kappa B essential modulator |
| NER | Nucleotide excision repair |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NOD/SCID | Non-obese diabetic/Severe combined immunodeficient |
| NO-NSAIDs | Nitric oxide-releasing non-steroidal anti-inflammatory drugs |
| NOS | Nitric oxide synthase |
| NOX | Nicotinamide adenine dinucleotide phosphate oxidase |
| NOXA1 | NOX Activator 1 |
| NOXO1 | NOX Organizer 1 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| NRX | Nucleoredoxin |
| OGG1 | 8-oxoguanine DNA glycosylase 1 |
| PDGF | Platelet-derived growth factor |
| PDT | Photodynamic therapy |
| PG | Prostaglandin |
| PHD | Prolyl hydroxylase |
| PI3K | Phosphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| PMA | Phorbol 12-myristate 13-acetate |
| PPP | Pentose phosphate pathway |
| PS | Pterostilbene |
| PTEN | Phosphatase and tensin homolog |
| PTP | Protein tyrosine phosphatase |
| PUFAs | Polyunsaturated fatty acids |
| RIP | Receptor interacting protein |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| SNPs | Single-nucleotide polymorphisms |
| SOD | Superoxide dismutase |
| Sp | Specificity protein |
| STAT | Signal transducer and activator of transcription proteins |
| TAK1 | TGF-β1-TGF-β-activated kinase 1 |
| TAMs | Tumor-associated Macrophages |
| TGF-β | Transforming growth factor beta |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| TME | Tumor microenvironment |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Tregs | Regulatory T cells |
| UC | Ulcerative colitis |
| uPA | Urokinase-type plasminogen activator |
| VEGF | Vascular endothelial growth factor |
| VPS34 | Vacuolar protein sorting 34 |
| Wnt | Wingless-related integration site |
| XIAP | X-linked inhibitor of apoptosis protein |
| ZEB | Zinc finger-E-box-binding |
References
- Colorectal Cancer Statistics/World Cancer Research Fund. Available online: https://www.wcrf.org/dietandcancer/cancer-trends/colorectal-cancer-statistics (accessed on 6 March 2020).
- Colorectal Cancer Immunotherapy-Cancer Research Institute. Available online: https://www.cancerresearch.org/immunotherapy/cancer-types/colorectal-cancer (accessed on 6 March 2020).
- Tsikitis, V.L.; Larson, D.W.; Huebner, M.; Lohse, C.M.; Thompson, P.A. Predictors of recurrence free survival for patients with stage II and III colon cancer. BMC Cancer 2014, 14, 336. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Hayashi, N.; Sakamoto, Y.; Ohuchi, M.; Tokunagam, R.; Kurashige, J.; Hiyoshi, Y.; Baba, Y.; Iwagami, S.; Yoshida, N.; et al. Predictors of long-term survival in patients with stage IV colorectal cancer with multi-organ metastases: A single-center retrospective analysis. Int. J. Clin. Oncol. 2015, 20, 1140–1146. [Google Scholar] [CrossRef]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.J.; Tveit, K.M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Colorec. Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Hear. J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Kawanishi, S.; Hiraku, Y.; Pinlaor, S.; Ma, N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol. Chem. 2006, 387, 365–372. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef]
- Swartz, H.M.; Gutierrez, P.L. Free radical increases in cancer: Evidence that there is not a real increase. Science 1977, 198, 936–938. [Google Scholar] [CrossRef]
- Martinez-Sanchez, G.; Giuliani, A. Cellular redox status regulates hypoxia inducible factor-1 activity. Role in tumour development. J. Exp. Clin. Cancer Res. 2007, 26, 39–50. [Google Scholar]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef] [Green Version]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Prot. Cell. 2014, 5, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Cheung, E.C.; Lee, P.; Ceteci, F.; Nixon, C.; Blyth, K.; Sansom, O.J.; Vousden, K.H. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Gen. Dev. 2016, 30, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Marumo, M.; Nakayama, J.; Matsumoto, M.; Yabe-Nishimura, C.; Kamata, T. The ROS-generating oxidase Nox1 is required for epithelial restitution following colitis. Exp. Anim. 2016, 65, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Moll, F.; Walter, M.; Rezende, F.; Helfinger, V.; Vasconez, E.; Oliveira, T.D.; Greten, F.R.; Olesch, C.; Weigert, A.; Radeke, H.H.; et al. NoxO1 Controls Proliferation of Colon Epithelial cells. Front. Immunol. 2018, 9, 973. [Google Scholar] [CrossRef] [Green Version]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013, 12, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Banfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, T.; Kohjima, M.; Kuwano, Y.; Mino, H.; Teshima-Kondo, S.; Takeya, R.; Tsunawaki, S.; Wada, A.; Sumimoto, H.; Rokutan, K. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am. J. Physiol. Cell Physiol. 2005, 288, C450–C457. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- O’Leary, D.P.; Bhatt, L.; Woolley, J.F.; Gough, D.R.; Wang, J.H.; Cotter, T.G.; Redmond, H.P. TLR-4 signalling accelerates colon cancer cell adhesion via NF-κB mediated transcriptional up-regulation of Nox-1. PLoS ONE 2012, 7, e44176. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.P.; Wang, X.; Gong, L.F.; Chen, W.J.; Hao, Z.; Feng, S.W.; Wu, Y.B.; Ye, T.; Cai, Y.K. Nox1 promotes colon cancer cell metastasis via activation of the ADAM17 pathway. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4474–4481. [Google Scholar] [PubMed]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohata, H.; Shiokawa, D.; Obata, Y.; Sato, A.; Sakai, H.; Fukami, M.; Hara, W.; Taniguchi, H.; Ono, M.; Nakagama, H.; et al. NOX1-dependent mTORC1 activation via S100A9 oxidation in cancer stem-like cells leads to colon cancer progression. Cell Rep. 2019, 28, 1282–1295.e8. [Google Scholar] [CrossRef] [Green Version]
- Makhezer, N.; Khemis, M.B.; Liu, D.; Khichane, Y.; Marzaioli, V.; Tlili, A.; Mojallali, M.; Pintard, C.; Letteron, P.; Hurtado-Nedelec, M.; et al. NOX1-derived ROS drive the expression of Lipocalin-2 in colonic epithelial cells in inflammatory conditions. Muc. Immun. 2019, 12, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Girgin, F.; Karaoglu, O.; Erkuş, M.; Tüzün, S.; Ozütemiz, O.; Dinçer, C.; Batur, Y.; Tanyalcin, T. Effects of trimetazidine on oxidant/antioxidant status in trinitrobenzenesulfonic acid-induced chronic colitis. J. Toxicol. Environ. Health Part A 2000, 59, 641–652. [Google Scholar]
- Moret-Tatay, I.; Iborra, M.; Cerrillo, E.; Tortosa, L.; Nos, P.; Beltrán, B. Possible biomarkers in blood for Crohn’s disease: Oxidative stress and microRNAs—Current evidences and further aspects to unravel. Oxid. Med. Cell. Long. 2015, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Piechota-Polanczyk, A.; Fichna, J. The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn Schmiedeberg’s Arch. Pharmacol. 2014, 387, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K. Does the interdependence between oxidative stress and inflammation explain the antioxidant paradox? Oxid. Med. Cell. Long. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hwang, I.; Kang, Y.N.; Choi, I.J.; Kim, D.K. Genetic characteristics of mitochondrial DNA was associated with colorectal carcinogenesis and its prognosis. PLoS ONE 2014, 10, e0118612. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbecks Arc. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Matosevic, P.; Klepac-Pulanic, T.; Kinda, E.; Augustin, G.; Brcic, I.; Jakic-Razumovic, J. Immunohistochemical expression of 8-oxo-7,8-dihydro-2′-deoxyguanosine in cytoplasm of tumour and adjacent normal mucosa cells in patients with colorectal cancer. World J. Surg. Oncol. 2015, 13, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, P.T.; Evans, M.D.; Cooke, M.S. Salvage of oxidized guanine derivatives in the (2′-deoxy)ribonucleotide pool as source of mutations in DNA. Mut. Res. 2010, 703, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, W.; Knaapen, M.W.; De Meyer, G.R.; Herman, A.G.; Kockx, M.M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 2002, 106, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Shinmura, K.; Yamada, H.; Tsuneyoshi, T.; Sugimura, H. OGG1, MYH and MTH1 gene variants identified in gastric cancer patients exhibiting both 8-hydroxy-2 ′ -deoxyguanosine accumulation and low inflammatory cell infiltration in their gastric mucosa. J. Genet. 2008, 87, 181–186. [Google Scholar] [CrossRef]
- Bravard, A.; Vacher, M.; Moritz, E.; Vaslin, L.; Hall, J.; Epe, B.; Radicella, J.P. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009, 69, 3642–3649. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef] [PubMed]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mut. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef] [PubMed]
- Van der Logt, E.M.; Roelofs, H.M.; Wobbes, T.; Nagengast, F.M.; Peters, W.H. High oxygen radical production in patients with sporadic colorectal cancer. Free Radic. Biol. Med. 2005, 39, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Guz, J.; Foksinski, M.; Siomek, A.; Gackowski, D.; Rozalski, R.; Dziaman, T.; Szpila, A.; Olinski, R. The relationship between 8-oxo-7,8-dihydro-2′-deoxyguanosine level and extent of cytosine methylation in leukocytes DNA of healthy subjects and in patients with colon adenomas and carcinomas. Mut. Res. 2008, 640, 170–173. [Google Scholar] [CrossRef]
- Rainis, T.; Maor, I.; Lanir, A.; Shnizer, S.; Lavy, A. Enhanced oxidative stress and leucocyte activation in neoplastic tissues of the colon. Diges. Dis. Sci. 2007, 52, 526–530. [Google Scholar] [CrossRef]
- Haklar, G.; Sayin-Ozveri, E.; Yüksel, M.; Aktan, A.O.; Yalçin, A.S. Different kinds of reactive oxygen and nitrogen species were detected in colon and breast tumors. Cancer Lett. 2001, 165, 219–224. [Google Scholar] [CrossRef]
- Perse, M. Oxidative stress in the pathogenesis of colorectal cancer: Cause or consequence? Biomed. Res. Int. 2013, 2013, 725710. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mut. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Skrzydlewska, E.; Sulkowski, S.; Koda, M.; Zalewski, B.; Kanczuga-Koda, L.; Sulkowska, M. Lipid peroxidation and antioxidant status in colorectal cancer. Worl. J. Gastro. 2005, 11, 403–406. [Google Scholar] [CrossRef]
- Uchida, K. HNE as an inducer of COX-2. Free Rad. Biol. Med. 2017, 111, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Yong, I.C.; Dubois, R.N. NSAIDs and cancer prevention: Targets downstream of COX-2. Ann. Rev. Med. 2007, 58, 239–252. [Google Scholar]
- Pérez, S.; Taléns-Visconti, R.; Rius-Pérez, S.; Finamor, I.; Sastre, J. Redox signaling in the gastrointestinal tract. Free Rad. Biol. Med. 2017, 104, 75–103. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Cetrangolo, G.P.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Gen 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Yang, H.Y.; Chay, K.O.; Kwon, J.; Kwon, S.O.; Park, Y.K.; Lee, T.H. Comparative proteomic analysis of cysteine oxidation in colorectal cancer patients. Mol. Cell. 2013, 35, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.A.; Mallis, R.J. Aging and oxidation of reactive protein sulfhydryls. Exp. Geron. 2001, 36, 1519–1526. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Jung, Y.; Na, S.; Jeong, J.; Lee, E.; Kim, M.S.; Choi, S.; Shin, D.H.; Paek, E.; Lee, H.Y.; et al. Novel oxidative modifications in redox-active cysteine residues. Mol. Cell. Proteom. 2011, 10, M110.000513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Wang, G.; Li, W.; Hu, X.; Huang, Q.; Xu, K.; Lou, W.; Wu, J.; Liang, C.; Lou, Q.; et al. Activation of the JAK-STAT3 pathway is associated with the growth of colorectal carcinoma cells. Oncol. Rep. 2013, 31, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebio, A.; Kahn, M.; Lenz, H.J. The potential of targeting Wnt/β-catenin in colon cancer. Exp. Opin. Therap. Targ. 2014, 18, 611–615. [Google Scholar] [CrossRef]
- Pandurangan, A.K. Potential targets for prevention of colorectal cancer: A focus on PI3K/Akt/mTOR and Wnt pathways. Asian Pac. J. Cancer Preven. 2013, 14, 2201–2205. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Yuan, X.; Song, J.; Chen, Y.; Tan, X.; Li, Q. Association analyses of the JAK/STAT signaling pathway with the progression and prognosis of colon cancer. Oncol. Lett. 2019, 17, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Shaw, P.E. A STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem. Biophys. Res. Commun. 2004, 322, 1005–1011. [Google Scholar] [CrossRef]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’orco, D.; Mariotto, S. S-glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [Green Version]
- Ma, S. Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J. Gastroent. 2004, 10, 1569–1573. [Google Scholar] [CrossRef]
- Park, S.K.; Dahmer, M.K.; Quasney, M.W. MAPK and JAK-STAT signaling pathways are involved in the oxidative stress-induced decrease in expression of surfactant protein genes. Cell. Physiol. Biochem. 2012, 30, 334–346. [Google Scholar] [CrossRef]
- Truong, T.H.; Carroll, K.S. Redox regulation of epidermal growth factor receptor signaling through cysteine oxidation. Biochemistry 2012, 51, 9954–9965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.F.; Terada, L.S. Ras and Nox: Linked signaling networks? Free Rad. Biol. Med. 2009, 47, 1276–1281. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, X.; Zhang, C.; Zhu, H.; Xu, Q.; Bu, Y.; Lei, Y. Redox imbalance in the development of colorectal cancer. J. Canc. 2017, 8, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Kajla, S.; Mondol, A.S.; Nagasawa, A.; Zhang, Y.; Kato, M.; Matsuno, K.; Yabe-Nishimura, C.; Kamata, T. A crucial role for Nox 1 in redox-dependent regulation of Wnt-β-catenin signaling. FASEB J. 2012, 26, 2049–2059. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, A.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.F.; Chen, J.Z. Obesity, the PI3K/Akt signal pathway and colon cancer. Obes. Rev. 2009, 10, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Zuo, Z.X.; Liu, Z.X.; Wu, Q.N.; Mo, H.Y.; Wang, Z.X.; Wang, D.S.; Pu, H.Y.; et al. Modulation of redox homeostasis by inhibition of MTHFD2 in colorectal cancer: Mechanisms and therapeutic implications. J. Natl. Cancer Inst. 2019, 111, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Huang, K.; Gao, C.; Lau, Q.C.; Pan, H.; Xie, K.; Li, J.; Liu, R.; Zhang, T.; Xie, N.; et al. Proteomics identification of ITGB3 as a key regulator in reactive oxygen species-induced migration and invasion of colorectal cancer cells. Mol. Cell. Proteom. 2011, 10, M110.005397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamou, S.; Shimizu, N. Hydrogen peroxide preferentially enhances the tyrosine phosphorylation of epidermal growth factor receptor. FEBS Lett. 1995, 357, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular basis for redox activation of epidermal growth factor receptor kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Heppner, D.E.; Hristova, M.; Dustin, C.M.; Danyal, K.; Habibovic, A.; van der Vliet, A. The NADPH oxidases DUOX1 and NOX2 play distinct roles in redox regulation of epidermal growth factor receptor signaling. J. Biol. Chem. 2016, 291, 23282–23293. [Google Scholar] [CrossRef] [Green Version]
- Miladi-Abdennadher, I.; Abdelmaksoud-Dammak, R.; Ayed-Guerfali, D.B.; Ayadi, L.; Khabir, A.; Amouri, A.; Frikha, F.; Tahri, N.; Ellouz, S.; Frikha, M. Expression of COX-2 and E-cadherin in Tunisian patients with colorectal adenocarcinoma. Acta Histochem. 2012, 114, 577–581. [Google Scholar] [CrossRef]
- Soumaoro, L.T.; Uetake, H.; Higuchi, T.; Takagi, Y.; Enomoto, M.; Sugihara, K. Cyclooxygenase-2 expression: A significant prognostic indicator for patients with colorectal cancer. Clin. Cancer. Res. 2004, 10, 8465–8471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wu, Y.D.; Li, P.; Tu, J.; Niu, Y.L.; Xu, C.M.; Zhang, S.T. Effects of cyclooxygenase-2 on human esophageal squamous cell carcinoma. World J. Gastroent. 2011, 17, 4572–4580. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; Wang, X.Y.; Chang, J.L.; Xie, W.B.; Liu, T.F.; Zhang, Q.L.; Deng, Y.J.; Ding, Y.Q. The establishment of supramolecular immunobead real-time PCR and the identification of Cox-2 as a metastasis-related marker in colorectal carcinoma. Oncol. Rep. 2012, 28, 977–984. [Google Scholar] [PubMed] [Green Version]
- Tesei, A.; Rosetti, M.; Ulivi, P.; Fabbri, F.; Medri, L.; Vannini, I.; Bolla, M.; Amadori, D.; Zoli, W. Study of molecular mechanisms of pro-apoptotic activity of NCX 4040, a novel nitric oxide releasing aspirin, in colon cancer cell lines. J. Transl. Med. 2007, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar] [CrossRef] [Green Version]
- Pals, J.; Attene-Ramos, M.S.; Xia, M.; Wagner, E.D.; Plewa, M.J. Human cell toxicogenomic analysis linking reactive oxygen species to the toxicity of monohaloacetic acid drinking water disinfection byproducts. Environ. Sci. Technol. 2013, 47, 12514–12523. [Google Scholar] [CrossRef] [Green Version]
- Tammali, R.; Ramana, K.V.; Srivastava, S.K. Aldose reductase regulates TNF-alpha-induced PGE2 production in human colon cancer cells. Cancer Lett. 2007, 252, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Park, I.J.; Hwang, J.T.; Kim, Y.M.; Ha, J.; Park, O.J. Differential modulation of AMPK signaling pathways by low or high levels of exogenous reactive oxygen species in colon cancer cells. Ann. NY Acad. Sci. 2006, 1091, 102–109. [Google Scholar] [CrossRef]
- Chiou, Y.S.; Tsai, M.L.; Nagabhushanam, K.; Wang, Y.J.; Wu, C.H.; Ho, C.T.; Pan, M.H. Pterostilbene is more potent than resveratrol in preventing azoxymethane (AOM)-induced colon tumorigenesis via activation of the NF-E2-related factor 2 (Nrf2)-mediated antioxidant signaling pathway. J. Agric. Food. Chem. 2011, 59, 2725–2733. [Google Scholar] [CrossRef]
- Liu, Y.; Kulesz-Martin, M. p53 protein at the hub of cellular DNA damage response pathways through sequence-specific and non-sequence-specific DNA binding. Carcinogenesis 2001, 22, 851–860. [Google Scholar] [CrossRef]
- Eaton, P.; Jones, M.E.; McGregor, E.; Dunn, M.J.; Leeds, N.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Pratt, J.R.; Shattock, M.J. Reversible cysteine-targeted oxidation of proteins during renal oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S290–S296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainwater, R.; Parks, D.; Anderson, M.E.; Tegtmeyer, P.; Mann, K. Role of cysteine residues in regulation of p53 function. Mol. Cell. Biol. 1995, 15, 3892–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainaut, P.; Mann, K. Zinc binding and redox control of p53 structure and function. Antioxid. Red. Signal. 2001, 3, 611–623. [Google Scholar] [CrossRef]
- Wu, H.H.; Yuan, Y.C.; Momand, J.; Sherman, M. Direct redox modulation of p53 protein: Potential sources of redox control and potential outcomes. Gen. Ther. Mol. Biol. 1999, 4, 119–132. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; White, E. p53-dependent apoptosis pathways. Adv. Cancer Res. 2001, 82, 55–84. [Google Scholar]
- Giatromanolaki, A.; Sivridis, E.; Stathopoulos, G.; Fountzilas, G.; Kalofonos, H.; Tsamandas, A.; Vrettou, E.; Scopa, C.; Polychronidis, A.; Simopoulos, K.; et al. Bax protein expression in colorectal cancer: Association with p53, bcl-2 and patterns of relapse. AntiCancer Res. 2001, 21, 253–259. [Google Scholar]
- Richie-Jp, J.; Komninou, D. Induction of colon tumorigenesis by glutathione depletion in p53-knock-out mice. Int. J. Oncol. 2007, 30, 1539–1543. [Google Scholar]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Gen. Develop. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Boldin-Adamsky, S.; Thimmulappa, R.K.; Rath, S.K.; Ashush, H.; Coulter, J.; Blackford, A.; Goodman, S.N.; Bunz, F.; Watson, W.H. RNAi-mediated silencing of nuclear factor erythroid-2–related factor 2 gene expression in non–small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008, 68, 7975–7984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viennois, E.; Chen, F.; Merlin, D. NF-κB pathway in colitis-associated cancers. Transl. Gastro. Cancer 2013, 2, 21–29. [Google Scholar]
- Berardi, R.; Maccaroni, E.; Mandolesi, A.; Mantello, G.; Onofri, A.; Biscotti, T.; Pierantoni, C.; Siquini, W.; Marmorale, C.; Guerrieri, M.; et al. Nuclear factor-κB predicts outcome in locally advanced rectal cancer patients receiving neoadjuvant radio-chemotherapy. Diges. Liv. Dis. 2012, 44, 617–622. [Google Scholar] [CrossRef]
- Herscovitch, M.; Comb, W.; Ennis, T.; Coleman, K.; Yong, S.; Armstead, B.; Kalaitzidis, D.; Chandani, S.; Gilmore, T.D. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem. Biophys. Res. Commun. 2008, 367, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Kil, I.S.; Kim, S.Y.; Park, J.W. Glutathionylation regulates IkappaB. Biochem. Biophys. Res. Commun. 2008, 373, 169–173. [Google Scholar] [CrossRef]
- Li, H.; Rokavec, M.; Jiang, L.; Horst, D.; Hermeking, H. Antagonistic effects of p53 and HIF1A on microrna-34a regulation of ppp1r11 and stat3 and hypoxia-induced epithelial to mesenchymal transition in colorectal cancer cells. Gastroenterology 2017, 153, 505–520. [Google Scholar] [CrossRef]
- Xiang, J.; Sun, H.; Su, L.; Liu, L.; Shan, J.; Shen, J.; Yang, Z.; Chen, J.; Zhong, X.; Ávila, M.A. Myocyte enhancer factor 2D promotes colorectal cancer angiogenesis downstream of hypoxia-inducible factor 1alpha. Cancer Lett. 2017, 400, 117–126. [Google Scholar] [CrossRef]
- Srinivasan, S.; Chitalia, V.; Meyer, R.D.; Hartsough, E.; Mehta, M.; Harrold, I.; Anderson, N.; Feng, H.; Smith, L.E.; Jiang, Y. Hypoxia-induced expression of phosducin-like 3 regulates expression of VEGFR-2 and promotes angiogenesis. Angiogenesis 2015, 18, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, H.P.; Jin, Y.; Choi, A.M.; Ryter, S.W. Beclin 1 deficiency is associated with increased hypoxia-induced angiogenesis. Autophagy 2011, 7, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fu, L.; Li, J.B.; Qin, Y.; Zeng, T.T.; Zhou, J.; Zeng, Z.L.; Chen, J.; Cao, T.T.; Ban, X. Increased expression of EIF5A2, via hypoxia or gene amplification, contributes to metastasis and angiogenesis of esophageal squamous cell carcinoma. Gastroenterology 2014, 146, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.; Paraskeva, E.; Baxevanidou, K.; Simos, G.; Papamichali, R.; Papacharalambous, C.; Samara, M.; Koukoulis, G. HIF-1α in colorectal carcinoma: Review of the literature. J. Buon. 2015, 20, 680–689. [Google Scholar] [PubMed]
- Schmitz, K.J.; Müller, C.I.; Reis, H.; Alakus, H.; Winde, G.; Baba, H.A.; Wohlschlaeger, J.; Jasani, B.; Fandrey, J.; Schmid, K.W. Combined analysis of hypoxia-inducible factor 1 alpha and metallothionein indicates an aggressive subtype of colorectal carcinoma. Int. J. Colorec. Dis. 2009, 24, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Yasinska, I.M.; Sumbayev, V.V. S -nitrosation of Cys-800 of HIF-1α protein activates its interaction with p300 and stimulates its transcriptional activity. Febs. Lett. 2003, 549, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhu, L.; Fang, J.; Ge, Z.; Li, X. LRG1 modulates epithelial-mesenchymal transition and angiogenesis in colorectal cancer via HIF-1α activation. J. Exp. Clin. Cancer Res. 2016, 35, 29. [Google Scholar] [CrossRef] [Green Version]
- Peddareddigari, V.G.; Wang, D.; DuBois, R.N. The tumor microenvironment in colorectal carcinogenesis. Cancer Microenviron. 2010, 3, 149–166. [Google Scholar] [CrossRef] [Green Version]
- Wigerup, C.; Pahlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Simiantonaki, N.; Taxeidis, M.; Jayasinghe, C.; Kurzik-Dumke, U.; Kirkpatrick, C.J. Hypoxia-inducible factor-1 alpha expression increases during colorectal carcinogenesis and tumor progression. BMC Cancer 2008, 8, 320. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Met. Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007, 12, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, J.J.; Land, S.C. A non-hypoxic, ROS-sensitive pathway mediates TNF-α-dependent regulation of HIF-1α. FEBS Lett. 2001, 505, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. Biom. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [Green Version]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Red. Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Barman, S.; Yu, Y.; Haigh, S.; Wang, Y.; Dou, H.; Bagi, Z.; Han, W.; Su, Y.; Fulton, D.J.R. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Rad. Biol. Med. 2014, 73, 201–213. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.S.K.; Tan, M.J.; Sng, M.K.; Teo, Z.; Phua, T.; Choo, C.C.; LI, L.; Zhu, P.; Tan, N.S. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017, 8, e2562. [Google Scholar] [CrossRef]
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and mutagenesis. Cancer Cell 2017, 32, 727–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell. 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Molon, B.; Ugel, S.; Pozzo, F.D.; Soldani, C.; Zilio, S.; Avella, D.; Palma, A.D.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef]
- Kraaij, M.D.; Savage, N.D.L.; Kooij, S.W.V.D.; Koekkoek, K.; Wang, J.; Berg, J.M.V.D.; Ottenhoff, T.H.M.; Kuijpers, T.W.; Holmdahl, R.; Kooten, C.V.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef] [Green Version]
- Enukidze, M.G.; Machavariani, M.G.; Intskirveli, N.A.; Bezhitashvili, N.D.; Sanikidze, T.V. Cell death in Jurkat cells induced by oxygen/nitrogen stress. Georg. Med. News. 2009, 167, 109–113. [Google Scholar]
- Hervera, A.; Virgiliis, F.D.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.T.; Santos, C.X.C.; et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 307–319. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Gen. Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Cannito, S.; Novo, E.; Bonzo, L.V.D.; Busletta, C.; Colombatto, S.; Parola, M. Epithelial-mesenchymal transition: From molecular mechanisms, redox regulation to implications in human health and disease. Antioxid. Red. Signal. 2010, 12, 1383–1430. [Google Scholar] [CrossRef]
- Stemmer, V.; Craene, B.D.; Berx, G.; Behrens, J. Snail promotes Wnt target gene expression and interacts with β-catenin. Oncogene 2008, 27, 5075–5080. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.; Li, D.D.; Yang, C.L.; Peng, R.Q.; Guo, Y.Q.; Zhang, X.S.; Zhu, X.F. Reactive oxygen species mediate oxaliplatin-induced epithelial-mesenchymal transition and invasive potential in colon cancer. Tum. Biol. 2016, 37, 8413–8423. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Sig. Trans. Targ. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, T.D.; Garbati, M.R. Inhibition of NF-κB signaling as a strategy in disease therapy. Curr. Top. Microbiol. Immunol. 2011, 349, 245–263. [Google Scholar]
- Kamiya, T.; Goto, A.; Kurokawa, E.; Hara, H.; Adachi, T. Cross talk mechanism among EMT, ROS, and histone acetylation in phorbol ester-treated human breast cancer MCF-7 cells. Oxid. Med. Cell Longev. 2016, 2016, 1284372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.H.; Dier, U.; Melendez, J.A.; Hempel, N. Regulation of MMP-1 expression in response to hypoxia is dependent on the intracellular redox status of metastatic bladder cancer cells. Biochim. Biophys. Acta 2015, 1852, 2593–2602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.R.I.; Tan, C.; Teo, Z.; Tay, C.Y.; Phua, T.; Wu, Y.L.; Cai, P.Q.; Tan, L.P.; Chen, X.; Zhu, P.; et al. Loss of TAK1 increases cell traction force in a ROS-dependent manner to drive epithelial–mesenchymal transition of cancer cells. Cell Death Dis. 2013, 4, e848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuno, Y.; Kiwamoto, T.; Morishima, Y.; Ishii, Y.; Hizawa, N. ROS-Nrf2 pathway mediates the development of TGF-β1-induced epithelial-mesenchymal transition through the interaction with Notch signaling. Am. J. Respir. Crit. Care Med. 2018, 197, A3805. [Google Scholar] [CrossRef]
- Demelash, A.; Pfannenstiel, L.W.; Liu, L.; Gastman, B.R. Mcl-1 regulates reactive oxygen species via NOX4 during chemotherapy-induced senescence. Oncotarget 2017, 8, 28154–28168. [Google Scholar] [CrossRef] [Green Version]
- Carew, J.S.; Zhou, Y.; Albitar, M.; Carew, J.D.; Keating, M.J.; Huang, P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: Clinical significance and therapeutic implications. Leukemia 2003, 17, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Turkington, R.C.; Longley, D.B.; Allen, W.L.; Stevenson, L.; McLaughlin, K.; Dunne, P.D.; Blayney, J.K.; Salto-Tellez, M.; Van Schaeybroeck, S.; Johnston, P.G. Fibroblast growth factor receptor 4 (FGFR4): A targetable regulator of drug resistance in colorectal cancer. Cell Death Dis. 2014, 5, e1046. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.H.; Rojanasakul, Y. The Fas death signaling pathway connecting reactive oxygen species generation and FLICE inhibitory protein down-regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef] [Green Version]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9, e106485. [Google Scholar] [CrossRef]
- Chian, S.; Li, Y.Y.; Wang, X.J.; Tang, X.W. Luteolin sensitizes two oxaliplatin-resistant colorectal cancer cell lines to chemotherapeutic drugs via inhibition of the Nrf2 pathway. Asian Pac. J. Cancer Prev. 2014, 15, 2911–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Shilnikova, K.; Kang, H.K.; Yi, J.M.; Boulanger, M.; Paolillo, R.; Bossis, G.; Yoon, S.Y.; et al. DUOX2-mediated production of reactive oxygen species induces epithelial mesenchymal transition in 5-fluorouracil resistant human colon cancer cells. Red. Biol. 2018, 17, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dashwood, W.M.; Nian, H.; Löhr, C.V.; Fischer, K.A.; Tsuchiya, N.; Nakagama, H.; Ashktorab, H.; Dashwood, R.H. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Int. J. Cancer 2011, 128, 2581–2590. [Google Scholar] [CrossRef]
- Lin, S.; Li, Y.; Zamyatnin, A.A., Jr.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Welbourn, B.; Wolff, R.K.; Corcoran, C. Oxidative balance and colon and rectal cancer: Interaction of lifestyle factors and genes. Mut. Res. 2012, 734, 30–40. [Google Scholar] [CrossRef] [Green Version]
- M’eplan, C.; Hughes, D.J.; Pardini, B.; Naccarati, A.; Soucek, P.; Vodickova, L.; Hlavatá, I.; Vrána, D.; Vodicka, P.; Hesketh, J.E. Genetic variants in selenoprotein genes increase risk of colorectal cancer. Carcinogenesis 2010, 31, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, A.; Kim, D.H.; Relton, C.; Ahn, Y.O.; Hesketh, J. Polymorphisms in the selenoprotein S and 15-kDa selenoprotein genes are associated with altered susceptibility to colorectal cancer. Genes Nutr. 2010, 5, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; Lundgreen, A.; Wolff, R.K. MAP kinase genes and colon and rectal cancer. Carcinogenesis 2012, 33, 2398–2408. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; Lundgreen, A.; Welbourn, B.; Corcoran, C.; Wolff, R.K. Genetic variation in selenoprotein genes, lifestyle, and risk of colon and rectal cancer. PLoS ONE 2012, 7, e37312. [Google Scholar] [CrossRef]
- Chang, D.; Wang, F.; Zhao, Y.S.; Pan, H.Z. Evaluation of oxidative stress in colorectal cancer patients. Biomed. Environ. Sci. 2008, 21, 286–289. [Google Scholar] [CrossRef]
- Barrett, C.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase Gpx3 in colitis-associated carcinoma. Cancer Res. 2012, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Demaison, L.; Sergiel, J.P.; Moreau, D.; Grynberg, A. Influence of the phospholipid n-6/n-3 polyunsaturated fatty acid ratio on the mitochondrial oxidative metabolism before and after myocardial ischemia. Bioch. Biophys. Acta 1994, 1227, 53–59. [Google Scholar] [CrossRef]
- Oudart, H.; Groscolas, R.; Calgari, C.; Nibbelink, M.; Leray, C.; Maho, Y.L.; Malan, A. Brown fat thermogenesis in rats fed high-fat diets enriched with n-3 polyunsaturated fatty acids. Int. J. Obes. 1997, 21, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Pehowich, D.J. Thyroid hormone status and membrane n-3 fatty acid content influence mitochondrial proton leak. Bioch. Biophys. Acta. 1999, 1411, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Weindruch, R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Gredilla, R.; Sanz, A.; Lopez-Torres, M.; Barja, G. Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 2001, 15, 1589–1591. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A. Mild uncoupling of mitochondria. Biosc. Rep. 1997, 17, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Guarente, L. SIR2: A potential target for calorie restriction mimetics. Trend. Mol. Med. 2007, 13, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ingram, D.K.; Zhu, M.; Mamczarz, J.; Zou, S.; Lane, M.A.; Roth, G.S.; DeCabo, R. Calorie restriction mimetics: An emerging research field. Ag. Cell. 2006, 5, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Pathways of oxidative damage. Ann. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef]
- Dickens, L.S.; Powley, I.R.; Hughes, M.A.; MacFarlane, M. The “complexities” of life and death: Death receptor signalling platforms. Exp. Cell Res. 2012, 318, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., 3rd; Wondrak, G.T. Experimental therapeutics: Targeting the redox Achilles heel of cancer. Curr. Opin. Invest. 2007, 8, 1022–1037. [Google Scholar]
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Su, R.Y.; Chi, K.H.; Huang, D.Y.; Tai, M.H.; Lin, W.W. 15-deoxy-Delta12,14-prostaglandin J2 up-regulates death receptor 5 gene expression in HCT116 cells: Involvement of reactive oxygen species and C/EBP homologous transcription factor gene transcription. Mol. Cancer Therap. 2008, 7, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Banerjee, V.; Czinn, S.; Blanchard, T. Increased reactive oxygen species levels cause ER stress and cytotoxicity in andrographolide treated colon cancer cells. Oncotarget 2017, 8, 26142–26153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Yang, P.; Li, Z.; Liu, W.; Amin, S.; Li, Z. Avenanthramide A triggers potent ROS-mediated anti-tumor effects in colorectal cancer by directly targeting DDX3. Cell Death Dis. 2019, 10, 593. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Kim, J.H.; Chung, Y.H.; Lee, S.H. Bakuchiol sensitizes cancer cells to TRAIL through ROS- and JNK-mediated upregulation of death receptors and downregulation of survival proteins. Biochem. Biophys. Res. Commun. 2016, 473, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Lei, P.; Pathi, S.; Safe, S. Betulinic acid inhibits colon cancer cell and tumor growth and induces proteasome-dependent and -independent downregulation of specificity proteins (Sp) transcription factors. BMC Cancer 2011, 11, 371. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Chu, B.Z.; Liu, F.; Li, B.; Gao, C.M.; Li, L.L.; Sun, Q.S.; Shen, Z.F.; Jiang, Y.Y. New benzimidazole acridine derivative induces human colon cancer cell apoptosis in vitro via the ROS-JNK signaling pathway. Acta Pharmacol. Sin. 2015, 36, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.M.; Chan, W.Y.; Yu, S.; Zhao, J.; Cheng, C.H. Bufalin induces autophagy-mediated cell death in human colon cancer cells through reactive oxygen species generation and JNK activation. Free Rad. Biol. Med. 2011, 51, 1365–1375. [Google Scholar] [CrossRef]
- Sung, B.; Prasad, S.; Ravindran, J.; Yadav, V.R.; Aggarwal, B.B. Capsazepine, a TRPV1 antagonist, sensitizes colorectal cancer cells to apoptosis by TRAIL through ROS-JNK-CHOP-mediated upregulation of death receptors. Free Rad. Biol. Med. 2012, 53, 1977–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.R.; Prasad, S.; Aggarwal, B.B. Cardamonin sensitizes tumour cells to TRAIL through ROS- and CHOP-mediated up-regulation of death receptors and down-regulation of survival proteins. Br. J. Pharmacol. 2012, 165, 741–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.Y.; Zhong, M.Z.; Yuan, G.J.; Hou, S.P.; Yin, L.L.; Jiang, H.; Yu, Z. Casticin, a flavonoid, potentiates TRAIL-induced apoptosis through modulation of anti-apoptotic proteins and death receptor 5 in colon cancer cells. Oncol. Rep. 2013, 29, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, H.S.; Liu, J.Y.; Lu, H.F.; Chiang, H.S.; Lin, C.H.; Chen, A.; Lin, Y.F.; Chung, J.G. Casticin induced apoptotic cell death and altered associated gene expression in human colon cancer colo 205 cells. Environ. Toxicol. 2017, 32, 2041–2052. [Google Scholar] [CrossRef]
- Enayat, S.; Şeyma, C.M.; Taşkoparan, B.; Stefek, M.; Banerjee, S. CHNQ, a novel 2-Chloro-1,4-naphthoquinone derivative of quercetin, induces oxidative stress and autophagy both in vitro and in vivo. Arch. Biochem. Biophy. 2016, 596, 84–98. [Google Scholar] [CrossRef]
- Sheikh, B.Y.; Sarker, M.M.R.; Kamarudin, M.N.A.; Mohan, G. Antiproliferative and apoptosis inducing effects of citral via p53 and ROS-induced mitochondrial-mediated apoptosis in human colorectal HCT116 and HT29 cell lines. Biol. Pharmacother. 2017, 96, 834–846. [Google Scholar] [CrossRef]
- Singh, M.P.; Park, K.H.; Khaket, T.P.; Kang, S.C. CJK-7, a Novel Flavonoid from Paulownia tomentosa triggers cell death cascades in HCT-116 human colon carcinoma cells via redox signaling. AntiCancer Agents Med. Chem. 2018, 18, 428–437. [Google Scholar] [CrossRef]
- Pierre, A.S.; Minville-Walz, M.; Fèvre, C.; Hichami, A.; Gresti, J.; Pichon, L.; Bellenger, S.; Bellenger, J.; Ghiringhelli, F.; Narce, M.; et al. Trans-10, cis-12 conjugated linoleic acid induced cell death in human colon cancer cells through reactive oxygen species-mediated ER stress. Biochim. Biophys. Acta 2013, 1831, 759–768. [Google Scholar] [CrossRef]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Kim, H.S.; Hyun, J.W. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Kasinathan, A.; Ganesan, R.; Balasubramanian, A.; Bhaskaran, J.; Suresh, S.; Srinivasan, R.; Aravind, K.B.; Sivalingam, N. Curcumin induces apoptosis and cell cycle arrest via the activation of reactive oxygen species-independent mitochondrial apoptotic pathway in Smad4 and p53 mutated colon adenocarcinoma HT29 cells. Nutr. Res. 2018, 51, 67–81. [Google Scholar] [CrossRef]
- Wang, L.; Hu, T.; Shen, J.; Zhang, L.; Chan, R.L.; Lu, L.; Li, M.; Cho, C.H.; Wu, W.K. Dihydrotanshinone I induced apoptosis and autophagy through caspase dependent pathway in colon cancer. Phytomedicine 2015, 22, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhao, Y.; Ma, C.Y.; Xu, X.M.; Zhang, Y.Q.; Wang, C.G.; Jin, J.; Shen, X.; Gao, J.L.; Li, N.; et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br. J. Pharmacol. 2015, 172, 3929–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yang, W.; Zeng, H.; Hu, C.; Zhang, Y.; Ding, N.; Fan, G.; Shao, L.; Kuang, B. Droxinostat sensitizes human colon cancer cells to apoptotic cell death via induction of oxidative stress. Cell Mol. Biol. Lett. 2018, 23, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.F.; Su, Y.Z.; Tseng, Y.H.; Wang, S.Y.; Wang, H.M.; Chueh, P.J. Flavokawain B, a novel chalcone from Alpinia pricei Hayata with potent apoptotic activity: Involvement of ROS and GADD153 upstream of mitochondria-dependent apoptosis in HCT116 cells. Free Rad. Biol. Med. 2010, 49, 214–226. [Google Scholar] [CrossRef]
- Pathi, S.S.; Jutooru, I.; Chadalapaka, G.; Sreevalsan, S.; Anand, S.; Thatcher, G.R.; Safe, S. GT-094, a NO-NSAID, inhibits colon cancer cell growth by activation of a reactive oxygen species-microRNA-27a: ZBTB10-specificity protein pathway. Mol. Cancer Res. 2011, 9, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Lee, Y.M.; Park, S.R.; Kim, D.H.; Lim, B.O. Anticancer activity of hispidin via reactive oxygen species-mediated apoptosis in colon cancer cells. AntiCancer Res. 2014, 34, 4087–4093. [Google Scholar]
- Bhardwaj, M.; Kim, N.H.; Paul, S.; Jakhar, R.; Han, J.; Kang, S.C. 5- Hydroxy-7-methoxyflavone triggers mitochondrial-associated cell death via reactive oxygen species signaling in human colon carcinoma cells. PLoS ONE 2016, 11, e0154525. [Google Scholar] [CrossRef]
- Do, M.T.; Na, M.; Kim, H.G.; Khanal, T.; Choi, J.H.; Jin, S.W.; Oh, S.H.; Hwang, I.H.; Chung, Y.C.; Kim, H.S.; et al. Ilimaquinone induces death receptor expression and sensitizes human colon cancer cells to TRAIL-induced apoptosis through activation of ROS-ERK/p38 MAPK-CHOP signaling pathways. Food Chem. Toxicol. 2014, 71, 51–59. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wang, L.; Lee, S. Levistolide A induces apoptosis via ROS-mediated ER stress pathway in colon cancer cells. Cell Physiol. Biochem. 2017, 42, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wu, X.; Gao, H.; Yu, J.; Zhao, W.; Lu, J.J.; Wang, J.; Du, G.; Chen, X. Cytosolic calcium mediates RIP1/RIP3 complex-dependent necroptosis through JNK activation and mitochondrial ROS production in human colon cancer cells. Free Rad. Biol. Med. 2017, 108, 433–444. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Cacciola, N.A.; Martino, E.; Borrelli, F.; Fiorino, F.; Lombardi, A.; Neglia, G.; Balestrieri, M.L.; Campanile, G. ROS-mediated apoptotic cell death of human colon cancer LoVo cells by milk δ-Valerobetaine. Sci. Rep. 2020, 10, 8978. [Google Scholar] [CrossRef] [PubMed]
- Zeriouh, W.; Nani, A.; Belarbi, M.; Dumont, A.; de Rosny, C.; Aboura, I.; Ghanemi, F.Z.; Murtaza, B.; Patoli, D.; Thomas, C.; et al. Phenolic extract from oleaster (Olea europaea var. Sylvestris) leaves reduces colon cancer growth and induces caspase dependent apoptosis in colon cancer cells via the mitochondrial apoptotic pathway. PLoS ONE 2017, 12, e0170823. [Google Scholar] [CrossRef]
- Ma, Y.M.; Han, W.; Li, J.; Hu, L.H.; Zhou, Y.B. Physalin B not only inhibits the ubiquitin-proteasome pathway but also induces incomplete autophagic response in human colon cancer cells in vitro. Acta Pharmacol. Sin. 2015, 36, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Punganuru, S.R.; Srivenugopal, K.S. Piperlongumine exerts cytotoxic effects against cancer cells with mutant p53 proteins at least in part by restoring the biological functions of the tumor suppressor. Int. J. Oncol. 2016, 48, 1426–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, H.; Uehara, N.; Kimura, A.; Sasaki, T.; Yuri, T.; Yoshizawa, K.; Tsubura, A. Resveratrol induces apoptosis via ROS-triggered autophagy in human colon cancer cells. Int. J. Oncol. 2012, 40, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Han, M.H.; Kim, G.Y.; Yoo, Y.H.; Choi, Y.H. Sanguinarine induces apoptosis in human colorectal cancer HCT-116 cells through ROS-mediated Egr-1 activation and mitochondrial dysfunction. Toxicol. Lett. 2013, 220, 157–166. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, C.; Li, R.; Zhang, L.; Tian, J. TEOA, a triterpenoid from Actinidia eriantha, induces autophagy in SW620 cells via endoplasmic reticulum stress and ROS-dependent mitophagy. Arch. Pharm. Res. 2017, 40, 579–591. [Google Scholar] [CrossRef]
- Pathi, S.; Lei, P.; Sreevalsan, S.; Chadalapaka, G.; Jutooru, I.; Safe, S. Pharmacologic doses of ascorbic acid repress specificity protein (Sp) transcription factors and Sp-regulated genes in colon cancer cells. Nutr. Cancer 2011, 63, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, Z.; Wang, C.; Zhou, H.; Liu, W.; Kanchana, K.; Dai, X.; Zou, P.; Gu, J.; Cai, L.; et al. Curcumin derivative WZ35 efficiently suppresses colon cancer progression through inducing ROS production and ER stress-dependent apoptosis. Am. J. Cancer Res. 2017, 7, 275–288. [Google Scholar]
- Lkhagvasuren, K.; Kim, J.K. Ziyuglycoside II induces caspases-dependent and caspases-independent apoptosis in human colon cancer cells. Toxicol. Vitro 2019, 59, 255–262. [Google Scholar] [CrossRef]
- Prasad, S.; Yadav, V.R.; Ravindran, J.; Aggarwal, B.B. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res. 2011, 71, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Cheng, S.; Wu, W.; Wang, L.; Zhao, Y.; Shen, Y.; Janin, A.; Zhao, W.L. c-FLIP is involved in tumor progression of peripheral T-cell lymphoma and targeted by histone deacetylase inhibitors. J. Hematol. Oncol. 2014, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelrahim, M.; Safe, S. Cyclooxygenase-2 inhibitors decrease vascular endothelial growth factor expession in colon cancer cells by enhanced degradation of Sp1 and Sp4 proteins. Mol. Pharmacol. 2005, 68, 317–329. [Google Scholar] [CrossRef]
- Chadalapaka, G.; Jutooru, I.; Chintharlapalli, S.; Papineni, S.; Smith, R.; Li, X.; Safe, S. Curcumin decreases specificity protein expression in bladder cancer cells. Cancer Res. 2008, 68, 5345–5354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens-Talcott, S.U.; Chintharlapalli, S.; Li, X.; Safe, S. The oncogenic microRNA-27a targets genes that regulate specificity protein (Sp) transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 2007, 67, 11001–11011. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.C.; Wang, L.; Wei, D.; Gong, W.; Hassan, M.; Wu, T.T.; Mansfield, P.; Ajani, J.; Xie, K. Association between expression of transcription factor Sp1 and increased vascular endothelial growth factor expression, advanced stage, and poor survival in patients with resected gastric cancer. Clin. Cancer Res. 2004, 10, 4109–4117. [Google Scholar] [CrossRef] [Green Version]
- Abdelrahim, M.; Smith III, R.; Burghardt, R.; Safe, S. Role of Sp proteins in regulation of vascular endothelial growth factor expression and proliferation of pancreatic cancer cells. Cancer Res. 2004, 64, 6740–6749. [Google Scholar] [CrossRef] [Green Version]
- Jutooru, I.; Chadalapaka, G.; Lei, P.; Safe, S. Inhibition of NF-κB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein downregulation. J. Biol. Chem. 2010, 285, 25332–25344. [Google Scholar] [CrossRef] [Green Version]
- Koveitypour, Z.; Panahi, F.; Vakilian, M.; Peymani, M.; Forootan, F.S.; Esfahani, M.H.N.; Ghaedi, K. Signaling pathways involved in colorectal cancer progression. Cell Biosci. 2019, 9, 97. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chen, Y.; Guo, W.; Gao, Y.; Song, C.; Zhang, Q.; Zheng, N.; Han, X.; Guo, C. Ferrite-based nanoplatform design: An ablation mechanism study of solid tumor and NIR-triggered photothermal/photodynamic combination cancer therapy. Adv. Funct. Mater. 2018, 28, 1706827. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radics. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Tian, J.; He, W.; Guo, Z. H2O2-activatable and O2-evolving nanoparticles for highly efficient and selective photodynamic therapy against hypoxic tumor cells. J. Am. Chem. Soc. 2015, 137, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Rad. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Habtetsion, T.; Ding, Z.C.; Pi, W.; Li, T.; Lu, C.; Chen, T.; Xi, C.; Spartz, H.; Liu, K.; Hao, Z.; et al. Alteration of tumor metabolism by CD4+ T cells leads to TNF-α-dependent intensification of oxidative stress and tumor cell death. Cell Metab. 2018, 28, 228–242.e6. [Google Scholar] [CrossRef] [Green Version]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed catalase protects chimeric antigen receptor–redirected T cells as well as bystander cells from oxidative stress–induced loss of antitumor activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cell Lines | Major Outcomes | Mechanisms | References |
|---|---|---|---|---|
| 5-FU | HT-29 | ↑ Caspase-7, Src | ROS-dependent apoptosis | [205] |
| 15dPGJ (2) | HCT116, SW480 | ↑ CHOP, GRP78, XBP1 | ROS/TRAIL-dependent apoptosis | [206] |
| Andrographolide | T84, COLO205 | ↑ Nrf2, GPx, PrX-6, LPO, TRX; ↓ ΔΨm | ROS/ER/caspase dependent apoptosis | [207] |
| Avenanthramide A | HCT116, DLD-1 | ↑ Caspase-3, Cyto c | ROS-mediated apoptosis | [208] |
| Bakuchiol | HCT116, HT-29 | ↑ Caspase-3, -8, -9, PARP; ↓ Bcl-2, survivin, cFLIP, XIAP | ROS/DR-dependent apoptosis | [209] |
| Betulinic acid | RKO, SW480 | ↓ Sp, microRNA-27a, ZBTB10 gene | ROS-apoptosis | [210] |
| Benzimidazole acridine derivative | HCT116, SW480 | ↑ Caspase-3, -7, -8, -9, Bid, PARP; ↓ Bcl-2 | ROS/DR-dependent apoptosis | [211] |
| Bufalin | HT-29, Caco-2 | ↑ LC3-II, ATG5, Beclin-1 | ROS-autophagy | [212] |
| Capsazepine | HCT116, HT-29 | ↑ Caspase-8, -9, Bax; ↓ cFLIP, survivin | ROS/DR-dependent apoptosis | [213] |
| Cardamonin | HCT116 | ↑ Cleaved PARP, caspase-8, -9, -3, Bax; ↓ cIAP-1, cFLIP, XIAP, Bcl-2 | ROS /DR/TRAIL-dependent apoptosis | [214] |
| Casticin | HCT116, SW480, HT-29, COLO205 | ↓ Bcl-2, Bcl-xL, cFLIP, XIAP; ↑ CDKN1B gene, TRAP1 gene, G2/M phase arrest; ↓ ΔΨm, [Ca2+]i, MMP-2, RKAR2B gene, CaMK4 gene | ROS/DR-dependent apoptosis ROS/caspase-dependent apoptosis | [215] [216] |
| CHNQ | HCT116, HT-29 | ↑ LC3-II, puncta formation, acidic vesicle; ↓ AKT/PKB | ROS-autophagy | [217] |
| Citral | HCT116, HT-29 | ↑ phospho-p53, Bax, Cleaved caspase-3; ↓ Bcl-2, Bcl-xL | ROS-apoptosis | [218] |
| CJK-7 | HCT116 | ↑ p53, Puma, ATG5, Beclin-1, LC3-I/II; ↓ Bcl-2 | ROS-apoptosis and autophagy | [219] |
| CLA | SW480 | ↑ Phosphorylated eIF2α, Xbp1 mRNA, CHOP | ROS/ER/caspase dependent apoptosis | [220] |
| Compound K | HCT116 | ↑ Caspase-3, -9, LC3-II, flux ATG6, ATG7; ↓ Bcl-2 | ROS-apoptosis and autophagy | [221] |
| Curcumin | HT-29 | ↑ S & G2/M arrest, DNA fragmentation; ↓ ΔΨm | ROS-apoptosis | [222] |
| DHTS | HCT116 | ↑ Bax, Bcl-xl, caspase-3, Cyto c, AIF, LC3-II | ROS/caspase-apoptosis and autophagy | [223] |
| DMF | HCT116, CT26, HT-29 | ↓ GSH | ROS-mediated necroptosis | [224] |
| Droxinostat | HT-29 | ↑ Acetylated H3, H4, caspase-3, Bax, Puma; ↓ HDAC3, 6, Bcl-2, Bcl-xl | ROS-apoptosis | [225] |
| Flavokawain B | HCT116 | ↑ Cyto c, GADD153; ↓ Bcl-2 family members | ROS-apoptosis | [226] |
| GT-094 | RKO, SW480 | ↓ VEGF, MMP, c-Met, EGFR, Sp microRNA-27a | ROS-apoptosis | [227] |
| Hispidin | HCT116, CMT-93 | ↑ p53, Bax, caspase-3, -8; ↓ Bcl-2 | ROS/DR/caspase-dependent apoptosis | [228] |
| HMF | HCT116 | ↑ [Ca2+]i, Cyto c, BID, Bax; ↓ Bcl-2 | ROS/ER/caspase-dependent apoptosis | [229] |
| Ilimaquinone | HCT116 | ↑ Caspase-8, -3; ↓ Bcl-2, Bcl-xL | ROS/DR-dependent apoptosis | [230] |
| Levistolide A | HCT116 | ↑ Caspase-3, cleaved-PARP | ROS-apoptosis | [231] |
| MAM | HCT116, HT-29 | ↑ [Ca2+]i, RIP1/RIP3 | ROS-dependent necroptosis | [232] |
| Milk δ-Valerobetaine (δVB) | LoVo | ↑ Caspase-9, -3, Bax, Sirtuin6 | ROS-mediated apoptosis | [233] |
| PEOL | HCT116, HCT-8 | ↑ [Ca2+]i, Cyto c; ↓ ΔΨm | ROS/ER/caspase-dependent apoptosis | [234] |
| Physalin B | HCT116 | ↑ Cleaved-PARP, p62; ↓ Caspase-3, LC3-II | ROS-autophagy | [235] |
| Piperlongumine | HT-29, SW620 | ↑ Cleaved caspase-3, PARP, Bax | ROS-apoptosis | [236] |
| Resveratrol | HT-29, COLO201 | ↑ Caspase-8, -3, LC3-II | ROS-apoptosis and autophagy | [237] |
| Sanguinarine | HCT116 | ↑ Caspase-3, -9; ↓ Bcl-2, XIAP, cIAP-1 | ROS-apoptosis | [238] |
| TEOA | SW620 | ↑ p62, Cleaved-PARP, LC3-II | ROS/ER/caspase-dependent apoptosis | [239] |
| Vitamin C | RKO, SW480 | ↓ EGFR, VEGF, c-Met, VEGFR1, Sp | ROS-dependent apoptosis and necrosis | [240] |
| WZ35 | CT26 | ↑ Cleaved-PARP; ↓ CyclinB1, Cdc2, MDM-2 | ROS/ER/caspase 3-mediated apoptosis | [241] |
| Ziyuglycoside II | HCT116 | ↑ p53, cleaved-PARP, caspase-3, -7, -8, -caspase-9; ↓ Bcl-2 | ROS-apoptosis | [242] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basak, D.; Uddin, M.N.; Hancock, J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers 2020, 12, 3336. https://doi.org/10.3390/cancers12113336
Basak D, Uddin MN, Hancock J. The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers. 2020; 12(11):3336. https://doi.org/10.3390/cancers12113336
Chicago/Turabian StyleBasak, Debasish, Mohammad Nasir Uddin, and Jake Hancock. 2020. "The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC)" Cancers 12, no. 11: 3336. https://doi.org/10.3390/cancers12113336
APA StyleBasak, D., Uddin, M. N., & Hancock, J. (2020). The Role of Oxidative Stress and Its Counteractive Utility in Colorectal Cancer (CRC). Cancers, 12(11), 3336. https://doi.org/10.3390/cancers12113336