BH3 Mimetics for the Treatment of B-Cell Malignancies—Insights and Lessons from the Clinic




Abstract
Simple Summary
Abstract
1. Introduction
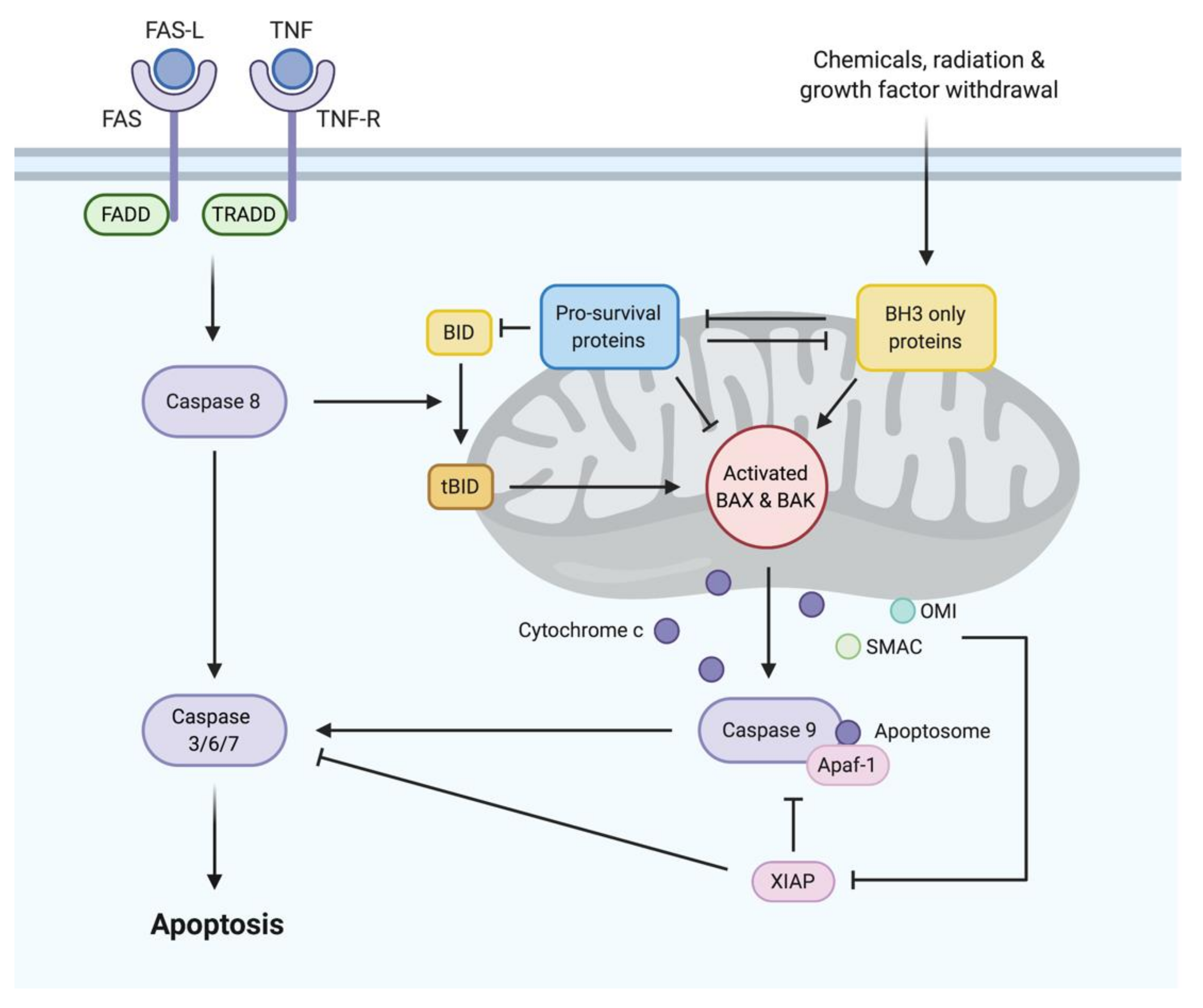
1.1. BCL2 Family Proteins: The Major Regulators of Apoptosis
1.2. Dysregulation of Apoptosis in B-Cell Malignancies
1.2.1. Overexpression of Pro-Survival Proteins
1.2.2. Loss of BH3-Only Protein Function
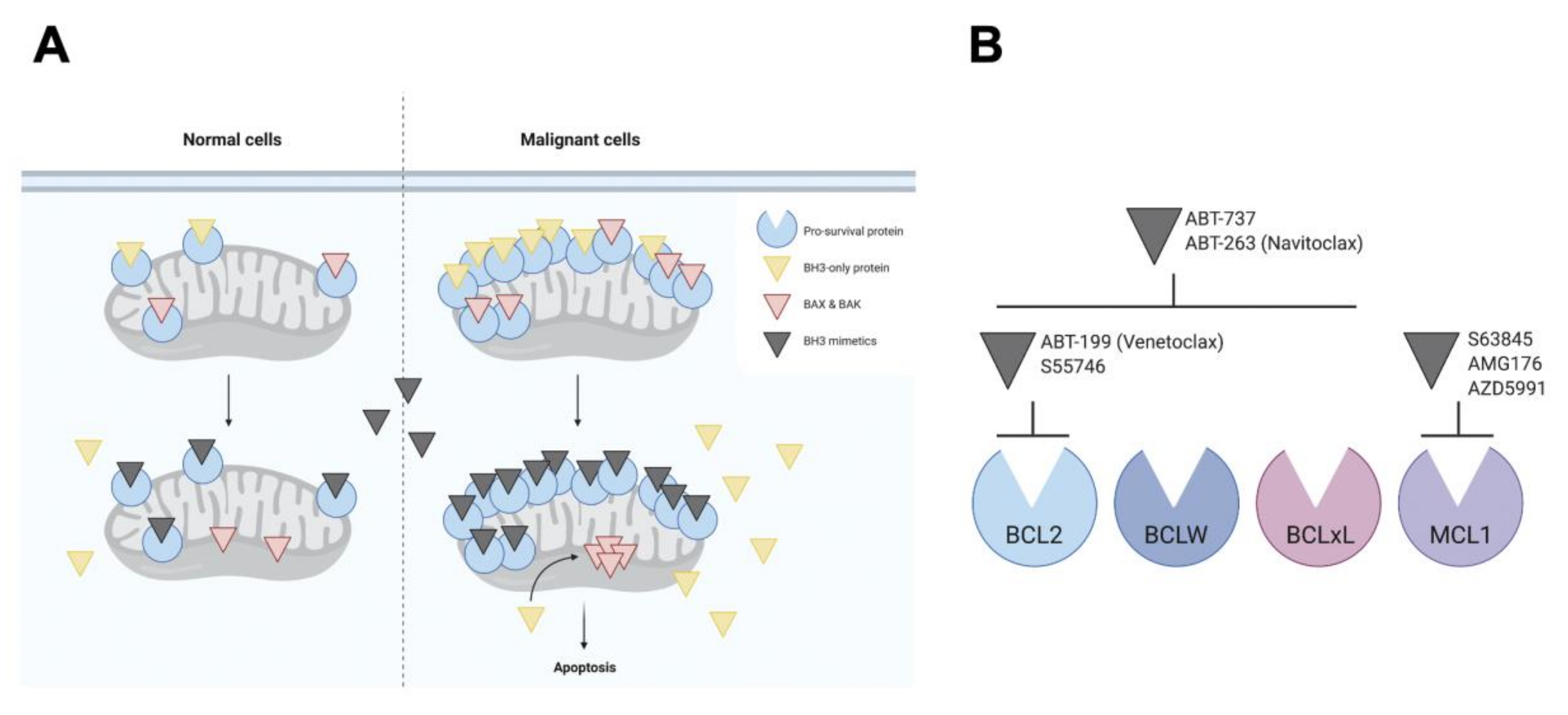
1.3. BH3 Mimetics
2. BCL2 Inhibitors
2.1. ABT-737 and ABT-263 (Navitoclax)
2.2. Venetoclax (ABT-199)
2.2.1. Venetoclax in CLL
2.2.2. Venetoclax in NHL
2.2.3. Venetoclax in Plasma Cell Dyscrasias
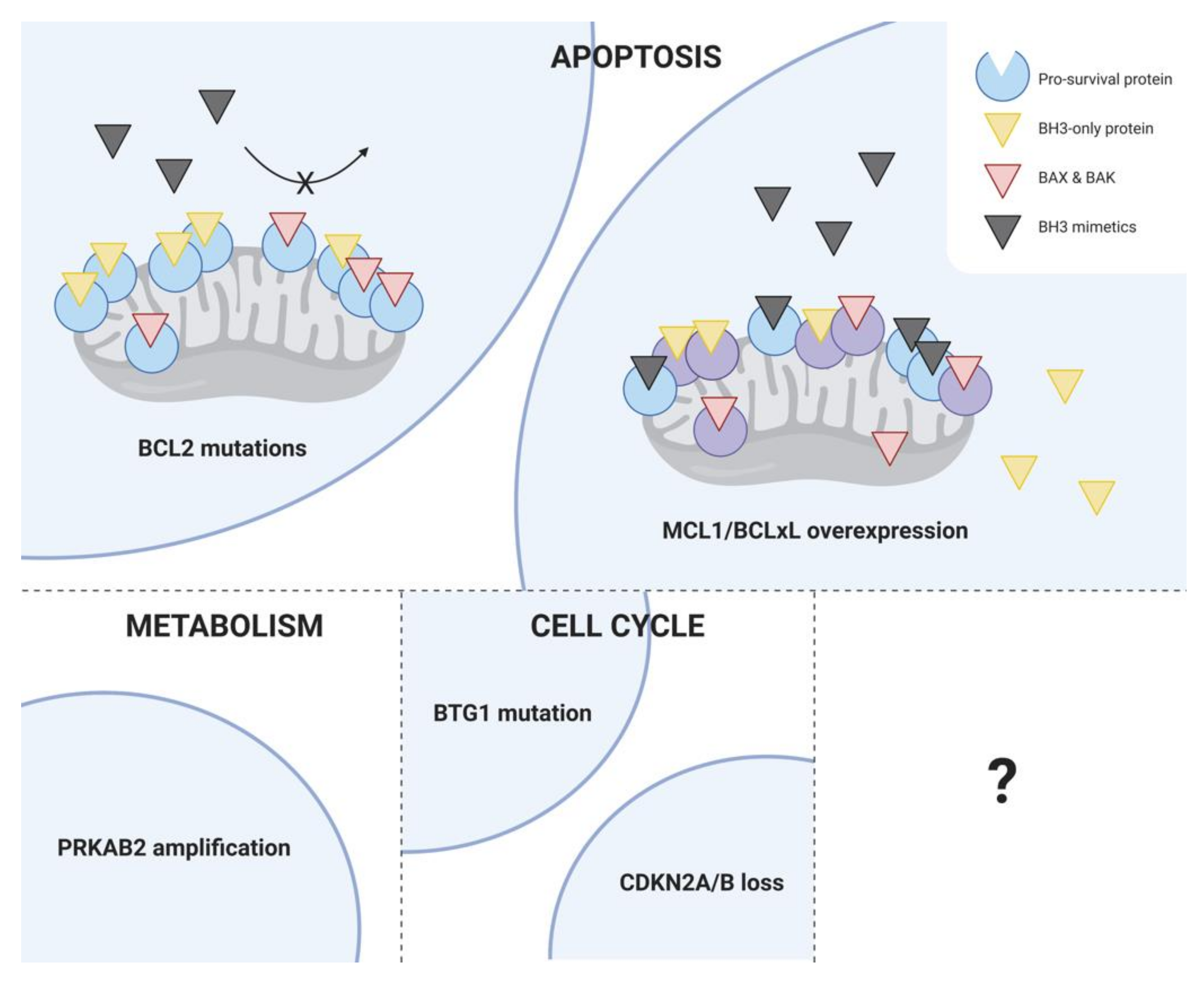
2.3. Resistance to BCL2 Inhibitors
2.3.1. Primary Resistance to Venetoclax
2.3.2. Acquired Resistance to Venetoclax
3. MCL1 Inhibitors
3.1. AZD5991
3.2. S63845 and S64315/MIK665
3.3. AMG-176 and AMG-397
4. BCLxL Inhibitors
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Diff. 2008, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Honarpour, N.; Gilbert, S.L.; Lahn, B.T.; Wang, X.; Herz, J. Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc. Natl. Acad. Sci. USA 2001, 98, 9683–9687. [Google Scholar] [CrossRef] [PubMed]
- Ke, F.F.S.; Vanyai, H.K.; Cowan, A.D.; Delbridge, A.R.D.; Whitehead, L.; Grabow, S.; Czabotar, P.E.; Voss, A.K.; Strasser, A. Embryogenesis and Adult Life in the Absence of Intrinsic Apoptosis Effectors BAX, BAK, and BOK. Cell 2018, 173, 1217–1230.e17. [Google Scholar] [CrossRef]
- Knudson, C.M.; Tung, K.S.; Tourtellotte, W.G.; Brown, G.A.; Korsmeyer, S.J. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 1995, 270, 96–99. [Google Scholar] [CrossRef]
- Eischen, C.M.; Roussel, M.F.; Korsmeyer, S.J.; Cleveland, J.L. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol. Cell Biol. 2001, 21, 7653–7662. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Dewson, G.; Kratina, T.; Sim, H.W.; Puthalakath, H.; Adams, J.M.; Colman, P.M.; Kluck, R.M. To Trigger Apoptosis, Bak Exposes Its BH3 Domain and Homodimerizes via BH3:Groove Interactions. Mol. Cell 2008, 30, 369–380. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Walle, L.V.; Gurp, M.V.; Loo, G.V.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: Clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985, 41, 899–906. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, T.J.; Deane, N.; Platt, F.M.; Nunez, G.; Jaeger, U.; McKearn, J.P.; Korsmeyer, S.J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 1989, 57, 79–88. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990, 348, 331–333. [Google Scholar] [CrossRef]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef]
- Ferrer, P.E.; Frederick, P.; Gulbis, J.M.; Dewson, G.; Kluck, R.M. Translocation of a Bak C-terminus mutant from cytosol to mitochondria to mediate cytochrome C release: Implications for Bak and Bax apoptotic function. PLoS ONE 2012, 7, e31510. [Google Scholar] [CrossRef]
- Griffiths, G.J.; Dubrez, L.; Morgan, C.P.; Jones, N.A.; Whitehouse, J.; Corfe, B.M.; Dive, C.; Hickman, J.A. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 1999, 144, 903–914. [Google Scholar] [CrossRef]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsmeyer, S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar] [CrossRef]
- Cheng, E.H.Y.A.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-XL Sequester BH3 Domain-Only Molecules Preventing BAX- and BAK-Mediated Mitochondrial Apoptosis. Mol. Cell 2001, 8, 705–711. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C.S. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001, 15, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Cartron, P.F.; Gallenne, T.; Bougras, G.; Gautier, F.; Manero, F.; Vusio, P.; Meflah, K.; Vallette, F.M.; Juin, P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol. Cell 2004, 16, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Gong, J.N.; Khong, T.; Segal, D.; Yao, Y.; Riffkin, C.D.; Garnier, J.M.; Khaw, S.L.; Lessene, G.; Spencer, A.; Herold, M.J.; et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: Pivotal role of MCL1. Blood 2016, 128, 1834–1844. [Google Scholar] [CrossRef]
- Kelly, G.L.; Grabow, S.; Glaser, S.P.; Fitzsimmons, L.; Aubrey, B.J.; Okamoto, T.; Valente, L.J.; Robati, M.; Tai, L.; Fairlie, W.D.; et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014, 28, 58–70. [Google Scholar] [CrossRef]
- Koss, B.; Morrison, J.; Perciavalle, R.M.; Singh, H.; Rehg, J.E.; Williams, R.T.; Opferman, J.T. Requirement for antiapoptotic MCL-1 in the survival of BCR-ABL B-lineage acute lymphoblastic leukemia. Blood 2013, 122, 1587–1598. [Google Scholar] [CrossRef]
- Slomp, A.; Peperzak, V. Role and Regulation of Pro-survival BCL-2 Proteins in Multiple Myeloma. Front. Oncol. 2018, 8, 533. [Google Scholar] [CrossRef]
- Shah, V.; Sherborne, A.L.; Walker, B.A.; Johnson, D.C.; Boyle, E.M.; Ellis, S.; Begum, D.B.; Proszek, P.Z.; Jones, J.R.; Pawlyn, C.; et al. Prediction of outcome in newly diagnosed myeloma: A meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2017, 32, 102–110. [Google Scholar] [CrossRef]
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dörken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2012, 27, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Xerri, L.; Parc, P.; Brousset, P.; Schlaifer, D.; Hassoun, J.; Reed, J.C.; Krajewski, S.; Birnbaum, D. Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br. J. Haematol. 1996, 92, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, H.; Karnan, S.; Suzuki, R.; Matsuo, K.; Zhang, X.; Ota, A.; Morishima, Y.; Nakamura, S.; Seto, M. Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005, 24, 1348–1358. [Google Scholar] [CrossRef]
- Mestre-Escorihuela, C.; Rubio-Moscardo, F.; Richter, J.A.; Siebert, R.; Climent, J.; Fresquet, V.; Beltran, E.; Agirre, X.; Marugan, I.; Marín, M.; et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood 2006, 109, 271–280. [Google Scholar] [CrossRef]
- Bachmann, P.S.; Piazza, R.G.; Janes, M.E.; Wong, N.C.; Davies, C.; Mogavero, A.; Bhadri, V.A.; Szymanska, B.; Geninson, G.; Magistroni, V.; et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood 2010, 116, 3013–3022. [Google Scholar] [CrossRef]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 Mutation and Survival in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Investig. 2007, 117, 112–121. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Khaw, S.L.; Rayeroux, K.C.; Chew, E.; Lee, E.F.; Fairlie, W.D.; Grigg, A.P.; Seymour, J.F.; Szer, J.; Huang, D.C.; et al. The BH3 mimetic compound, ABT-737, synergizes with a range of cytotoxic chemotherapy agents in chronic lymphocytic leukemia. Leukemia 2009, 23, 2034–2041. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vogler, M.; Dinsdale, D.; Dyer, M.J.; Cohen, G.M. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br. J. Haematol. 2013, 163, 139–142. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Bottcher, S.; Letestu, R.; Villamor, N.; Fazi, C.; Kartsios, H.; de Tute, R.M.; Shingles, J.; Ritgen, M.; Moreno, C.; et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia 2013, 27, 142–149. [Google Scholar] [CrossRef]
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.-M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Jones, J.A.; Mato, A.R.; Wierda, W.G.; Davids, M.S.; Choi, M.; Cheson, B.D.; Furman, R.R.; Lamanna, N.; Barr, P.M.; Zhou, L.; et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: An interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018, 19, 65–75. [Google Scholar] [CrossRef]
- Coutre, S.; Choi, M.; Furman, R.R.; Eradat, H.; Heffner, L.; Jones, J.A.; Chyla, B.; Zhou, L.; Agarwal, S.; Waskiewicz, T.; et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood 2018, 131, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Hallek, M.; Wierda, W.; Roberts, A.W.; Stilgenbauer, S.; Jones, J.A.; Gerecitano, J.F.; Kim, S.Y.; Potluri, J.; Busman, T.; et al. Comprehensive Safety Analysis of Venetoclax Monotherapy for Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2018, 24, 4371–4379. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [CrossRef]
- Sampath, D.; Herter, S.; Herting, F.; Ingalla, E.; Nannini, M.; Bacac, M.; Fairbrother, W.J.; Klein, C. Combination of the glycoengineered Type II CD20 antibody obinutuzumab (GA101) and The novel Bcl-2 selective Inhibitor GDC-0199 Results in superior In Vitro and In Vivo Anti-tumor activity in models Of B-Cell Malignancies. Blood 2013, 122, 4412. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef]
- Kater, A.P.; Kersting, S.; van Norden, Y.; Dubois, J.; Dobber, J.A.; Mellink, C.H.; Evers, L.M.; Croon-de Boer, F.; Schreurs, J.; van der Spek, E.; et al. Obinutuzumab pretreatment abrogates tumor lysis risk while maintaining undetectable MRD for venetoclax + obinutuzumab in CLL. Blood Adv. 2018, 2, 3566–3571. [Google Scholar] [CrossRef]
- Herishanu, Y.; Pérez-Galán, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-κB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574. [Google Scholar] [CrossRef]
- Mockridge, C.I.; Potter, K.N.; Wheatley, I.; Neville, L.A.; Packham, G.; Stevenson, F.K. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood 2007, 109, 4424–4431. [Google Scholar] [CrossRef]
- Duhren-von Minden, M.; Ubelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Kohler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Deng, J.; Wiestner, A.; Lannutti, B.J.; Wang, L.; Wu, C.J.; Wilson, W.H.; Brown, J.R.; Letai, A. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood 2012, 120, 3501–3509. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.M.; Balakrishnan, K.; Douglas, M.; Tibbitts, T.; Xu, E.Y.; Kutok, J.L.; Ayers, M.; Sarkar, A.; Guerrieri, R.; Wierda, W.G.; et al. Duvelisib treatment is associated with altered expression of apoptotic regulators that helps in sensitization of chronic lymphocytic leukemia cells to venetoclax (ABT-199). Leukemia 2017, 31, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Gomez, F.; Lamothe, B.; Woyach, J.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Gandhi, V. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2015, 21, 3705–3715. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef]
- Hillmen, P.; Rawstron, A.C.; Brock, K.; Munoz-Vicente, S.; Yates, F.J.; Bishop, R.; Boucher, R.; MacDonald, D.; Fegan, C.; McCaig, A.; et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J. Clin. Oncol. 2019, 37, 2722–2729. [Google Scholar] [CrossRef]
- Rogers, K.A.; Huang, Y.; Ruppert, A.S.; Awan, F.T.; Heerema, N.A.; Hoffman, C.; Lozanski, G.; Maddocks, K.J.; Moran, M.E.; Reid, M.A.; et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood 2018, 132, 1568–1572. [Google Scholar] [CrossRef]
- Rogers, K.A.; Huang, Y.; Stark, A.; Awan, F.T.; Maddocks, K.J.; Woyach, J.A.; Whitlow, T.; Byrd, J.C.; Jones, J.A. Initial Results of the Phase 2 Treatment Naive Cohort in a Phase 1b/2 Study of Obinutuzumab, Ibrutinib, and Venetoclax in Chronic Lymphocytic Leukemia. Blood 2017, 130 (Suppl. 1), 431. [Google Scholar]
- Rogers, K.A.; Huang, Y.; Ruppert, A.S.; Awan, F.T.; Hoffman, C.; Maddocks, K.J.; Reid, M.; Woyach, J.A.; Whitlow, T.; Jones, J.A.; et al. Phase 2 Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naive and Relapsed/Refractory Chronic Lymphocytic Leukemia. Blood 2018, 132 (Suppl. 1), 693. [Google Scholar] [CrossRef]
- Cramer, P.; von Tresckow, J.; Bahlo, J.; Robrecht, S.; Langerbeins, P.; Al-Sawaf, O.; Engelke, A.; Fink, A.M.; Fischer, K.; Tausch, E.; et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): Primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018, 19, 1215–1228. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Hillmen, P.; Seymour, J.F.; Coutre, S.; Jurczak, W.; Mulligan, S.P.; Schuh, A.; Assouline, S.; et al. Venetoclax for Patients With Chronic Lymphocytic Leukemia with 17p Deletion: Results From the Full Population of a Phase II Pivotal Trial. J. Clin. Oncol. 2018, 36, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Kater, A.P.; Seymour, J.F.; Hillmen, P.; Eichhorst, B.; Langerak, A.W.; Owen, C.; Verdugo, M.; Wu, J.; Punnoose, E.A.; Jiang, Y.; et al. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J. Clin. Oncol. 2019, 37, 269–277. [Google Scholar] [CrossRef]
- Vandenberg, C.J.; Cory, S. ABT-199, a new Bcl-2–specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood 2013, 121, 2285–2288. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Anderson, M.A.; Pott, C.; Agarwal, R.; Handunnetti, S.; Hicks, R.J.; Burbury, K.; Turner, G.; Di Iulio, J.; Bressel, M.; et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Zelenetz, A.D.; Salles, G.; Mason, K.D.; Casulo, C.; Le Gouill, S.; Sehn, L.H.; Tilly, H.; Cartron, G.; Chamuleau, M.E.D.; Goy, A.; et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: Results from the CAVALLI phase 1b trial. Blood 2019, 133, 1964–1976. [Google Scholar] [CrossRef]
- Gomez-Bougie, P.; Amiot, M. Apoptotic Machinery Diversity in Multiple Myeloma Molecular Subtypes. Front. Immunol. 2013, 4, 467. [Google Scholar] [CrossRef]
- Morales, A.A.; Kurtoglu, M.; Matulis, S.M.; Liu, J.; Siefker, D.; Gutman, D.M.; Kaufman, J.L.; Lee, K.P.; Lonial, S.; Boise, L.H. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1–expressing myeloma cells. Blood 2011, 118, 1329–1339. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maiga, S.; Bene, M.C.; Moreau, P.; Pellat-Deceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef]
- Matulis, S.M.; Gupta, V.A.; Nooka, A.K.; Hollen, H.V.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia 2016, 30, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-Z.; Ziffra, J.; Stennett, L.; Bodner, B.; Bonish, B.K.; Chaturvedi, V.; Bennett, F.; Pollock, P.M.; Trent, J.M.; Hendrix, M.J.C.; et al. Proteasome Inhibitors Trigger NOXA-Mediated Apoptosis in Melanoma and Myeloma Cells. Cancer Res. 2005, 65, 6282–6293. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bougie, P.; Wuilleme-Toumi, S.; Menoret, E.; Trichet, V.; Robillard, N.; Philippe, M.; Bataille, R.; Amiot, M. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res. 2007, 67, 5418–5424. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bougie, P.; Menoret, E.; Juin, P.; Dousset, C.; Pellat-Deceunynck, C.; Amiot, M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem. Biophys. Res. Commun. 2011, 413, 460–464. [Google Scholar] [CrossRef]
- Moreau, P.; Chanan-Khan, A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Punnoose, E.A.; Cordero, J.; Munasinghe, W.; et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood 2017, 130, 2392–2400. [Google Scholar] [CrossRef]
- Moreau, P.; Harrison, S.; Cavo, M.; De La Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.T.M.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Updated Analysis of Bellini, a Phase 3 Study of Venetoclax or Placebo in Combination with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 1888. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Costa, L.J.; Stadtmauer, E.A.; Morgan, G.J.; Monohan, G.P.; Kovacsovics, T.; Burwick, N.; Jakubowiak, A.J.; Mobasher, M.; Freise, K.; Ross, J.A.; et al. Phase 2 study of venetoclax plus carfilzomib and dexamethasone in patients with relapsed/refractory multiple myeloma. J. Clin. Oncol. 2018, 36 (Suppl. 15), 8004. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Bueno, O.F.; Maciag, P.C.; Vue, J.; Westrup, S.; Pesko, J.; Ross, J.A.; Salem, A.H.; Gibbs, S.; Moreau, P.; et al. First Analysis from a Phase 1/2 Study of Venetoclax in Combination with Daratumumab and Dexamethasone, +/- Bortezomib, in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2019, 134 (Suppl. 1), 925. [Google Scholar]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Allan, J.; Siddiqi, T.; Advani, R.; Keezer, A.; Gustine, J.; Meid, K.; Dubeau, T.; Lam, J.; Xu, L.; et al. Multicenter prospective phase II study of venetoclax in patients with previously treated Waldenstrom macroglobulinemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, e39–e40. [Google Scholar] [CrossRef]
- Blombery, P. Mechanisms of intrinsic and acquired resistance to venetoclax in B-cell lymphoproliferative disease. Leuk. Lymphoma 2020, 61, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Chiron, D.; Dousset, C.; Brosseau, C.; Touzeau, C.; Maiga, S.; Moreau, P.; Pellat-Deceunynck, C.; Le Gouill, S.; Amiot, M. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget 2015, 6, 8750–8759. [Google Scholar] [CrossRef] [PubMed]
- Bodo, J.; Zhao, X.; Durkin, L.; Souers, A.J.; Phillips, D.C.; Smith, M.R.; Hsi, E.D. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget 2016, 7, 70000–70010. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Chan, Y.C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129. [Google Scholar] [CrossRef]
- Chueh, A.C.; Tse, J.W.T.; Dickinson, M.; Ioannidis, P.; Jenkins, L.; Togel, L.; Tan, B.; Luk, I.; Davalos-Salas, M.; Nightingale, R.; et al. ATF3 Repression of BCL-XL Determines Apoptotic Sensitivity to HDAC Inhibitors across Tumor Types. Clin. Cancer Res. 2017, 23, 5573–5584. [Google Scholar] [CrossRef]
- Gupta, V.A.; Matulis, S.M.; Conage-Pough, J.E.; Nooka, A.K.; Kaufman, J.L.; Lonial, S.; Boise, L.H. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017, 129, 1969–1979. [Google Scholar] [CrossRef]
- Thijssen, R.; Slinger, E.; Weller, K.; Geest, C.R.; Beaumont, T.; van Oers, M.H.; Kater, A.P.; Eldering, E. Resistance to ABT-199 induced by microenvironmental signals in chronic lymphocytic leukemia can be counteracted by CD20 antibodies or kinase inhibitors. Haematologica 2015, 100, e302–e306. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, S.; Ylanko, J.; Shi, Y.; Hariharan, S.; Oakes, C.C.; Brauer, P.M.; Zuniga-Pflucker, J.C.; Leber, B.; Spaner, D.E.; Andrews, D.W. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood 2016, 128, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.S.; Chang, B.Y.; Chang, S.; Tong, T.; Ham, S.; Sherry, B.; Burger, J.A.; Rai, K.R.; Chiorazzi, N. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia 2016, 30, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.; Chang, S.; Pals, S.T.; et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef]
- Roberts, A.W.; Huang, D.C.S.; Czabotar, P.E.; Westerman, D.A.; Seymour, J.F.; Anderson, M.A.; Conway, T.; Thijssen, R.; McBean, M.; Chen, X.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernández-Sánchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef] [PubMed]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ren, Y.; Lawlor, M.; Shah, B.D.; Park, P.M.C.; Lwin, T.; Wang, X.; Liu, K.; Wang, M.; Gao, J.; et al. BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models. Cancer Cell 2019, 35, 752–766.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kanagal-Shamanna, R.; Navsaria, L.; Ok, C.Y.; Zhang, S.; Nomie, K.; Han, G.; Hao, D.; Hill, H.A.; Jiang, C.; et al. Efficacy of venetoclax in high risk relapsed mantle cell lymphoma (MCL)—Outcomes and mutation profile from venetoclax resistant MCL patients. Am. J. Hematol. 2020, 95, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Birkinshaw, R.W.; Nguyen, T.; Gong, J.N.; Thompson, E.R.; Xu, Z.; Westerman, D.A.; Czabotar, P.E.; Dickinson, M.; Huang, D.C.S.; et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br. J. Haematol. 2019, 186, e188–e191. [Google Scholar] [CrossRef]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef]
- Grabow, S.; Delbridge, A.R.; Aubrey, B.J.; Vandenberg, C.J.; Strasser, A. Loss of a Single Mcl-1 Allele Inhibits MYC-Driven Lymphomagenesis by Sensitizing Pro-B Cells to Apoptosis. Cell Rep. 2016, 14, 2337–2347. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Shi, C.X.; Sereduk, C.; Yin, H.; Mousses, S.; Stewart, A.K. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012, 72, 757–768. [Google Scholar] [CrossRef]
- Van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, A.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Vikstrom, I.; Carotta, S.; Luthje, K.; Peperzak, V.; Jost, P.J.; Glaser, S.; Busslinger, M.; Bouillet, P.; Strasser, A.; Nutt, S.L.; et al. Mcl-1 Is Essential for Germinal Center Formation and B Cell Memory. Science 2010, 330, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Peperzak, V.; Vikstrom, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Dzhagalov, I.; Dunkle, A.; He, Y.W. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J. Immunol. 2008, 181, 521–528. [Google Scholar] [CrossRef]
- Huntington, N.D.; Puthalakath, H.; Gunn, P.; Naik, E.; Michalak, E.M.; Smyth, M.J.; Tabarias, H.; Degli-Esposti, M.A.; Dewson, G.; Willis, S.N.; et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat. Immunol. 2007, 8, 856–863. [Google Scholar] [CrossRef]
- Omari, S.; Waters, M.; Naranian, T.; Kim, K.; Perumalsamy, A.L.; Chi, M.; Greenblatt, E.; Moley, K.H.; Opferman, J.T.; Jurisicova, A. Mcl-1 is a key regulator of the ovarian reserve. Cell Death Dis. 2015, 6, e1755. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Brennan, M.S.; Chang, C.; Tai, L.; Lessene, G.; Strasser, A.; Dewson, G.; Kelly, G.L.; Herold, M.J. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 2018, 132, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Hird, A.W.; Tron, A.E. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol. Ther. 2019, 198, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A.; Myers, T.G.; Scudiero, D.; Kitada, S.; Reed, J.C.; Fornace, A.J., Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000, 60, 6101–6110. [Google Scholar]
- Lessene, G.; Czabotar, P.E.; Sleebs, B.E.; Zobel, K.; Lowes, K.N.; Adams, J.M.; Baell, J.B.; Colman, P.M.; Deshayes, K.; Fairbrother, W.J.; et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.-F.; Hasvold, L.; Wang, L.; Wang, X.; Petros, A.M.; Park, C.H.; Boghaert, E.R.; Catron, N.D.; Chen, J.; Colman, P.M.; et al. Discovery of a Potent and Selective BCL-XL Inhibitor with in Vivo Activity. ACS Med. Chem. Lett. 2014, 5, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, ra40–ra279. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mondello, P.; Erazo, T.; Tannan, N.B.; Asgari, Z.; de Stanchina, E.; Nanjangud, G.; Seshan, V.E.; Wang, S.; Wendel, H.G.; et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc. Natl. Acad. Sci. USA 2018, 115, 12034–12039. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Younes, A.; Heller, D.; Manova-Todorova, K.; de Stanchina, E.; Seshan, V.; Hagen, C.; Asgari, Z.; Ferreira, M.D.S.; Manzari, M.T.; Bala, N.B. Dual Inhibition of MCL1 and BCL2 in Lymphoma Using Tumor Targeted Nanoparticles. Blood 2019, 134 (Suppl. 1), 305. [Google Scholar]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| ClinicalTrials.Gov Identifier | Other Study ID Numbers | Intervention | Disease | Study Phase(s) | Publications |
|---|---|---|---|---|---|
| VEN monotherapy | |||||
| NCT01328626 | M12-175 | VEN | R/R CLL | 1 | [46] |
| NCT01889186 | M13-982 | VEN | R/R CLL with del(17p) | 2 | [49,71] |
| NCT02141282 | M14-032 | VEN | R/R CLL after BCR inhibitor therapy | 2 | [50,51] |
| VEN combination therapy | |||||
| NCT01682616 | M13-365 | VEN + R | R/R CLL/SLL | 1b | [53] |
| NCT02005471 | GO28667, MURANO | VEN + R | R/R CLL | 3 | [54,72] |
| NCT02242942 | BO25323, CLL14 | VEN + G | CLL | 3 | [57] |
| NCT02756897 | 2015-0860 | VEN + IBR | R/R CLL | 2 | [65] |
| NCT02427451 | OSU-14266, NCI-2015-00252 | VEN + IBR + G | R/R CLL | 1b/2 | [67] |
| NCT02401503 | CLL2-BAG, 2014-000580-40 | Bendamustine + VEN + G | CLL | 2 | [70] |
| Clinicaltrials.Gov Identifier | Other Study ID Numbers | Intervention | Disease | Study Phase(s) | Publications |
|---|---|---|---|---|---|
| VEN monotherapy | |||||
| NCT01328626 | M12-175 | VEN | R/R NHL (including MCL, FL, DLBCL, RT-DLBCL, WM, and MZL) | 1 | [74] |
| VEN combination therapy | |||||
| NCT02471391 | 14/148, AIM | VEN + IBR | MCL | 2 | [75] |
| NCT03112174 | PCYC-1143-CA, SYMPATICO | VEN + IBR | MCL | 3 | N/A |
| NCT02055820 | GO27878, 2013-003749-40, CAVALLI | VEN + R-/G-CHOP | NHL (including FL and DLBCL) | 1b/2 | [76] |
| Clinicaltrials.Gov Identifier | Other Study ID Numbers | Intervention | Disease | Study Phase(s) | Publications |
|---|---|---|---|---|---|
| VEN monotherapy | |||||
| NCT01794520 | M13-367, 2012-000589-38 | VEN | R/R MM | 1/2 | [80] |
| VEN combination therapy | |||||
| NCT01794507 | M12-901, 2011-004626-10 | VEN + bortezomib + dexamethasone | R/R MM | 1 | [85] |
| NCT02755597 | M14-031, 2015-004411-20 | VEN + bortezomib + dexamethasone | R/R MM | 3 | [86] |
| NCT02899052 | M15-538 | VEN + carfilzomib + dexamethasone | R/R MM | 2 | [89] |
| NCT03399539 | MC168C, NCI-2017-02456 | VEN + ixazomib citrate + dexamethasone | R/R MM | 1/2 | N/A |
| NCT03314181 | M15-654, 2017-002099-26 | VEN + daratumumab + dexamethasone +/- bortezomib | R/R MM | 2 | [91] |
| NCT03312530 | BO39813, 2017-000830-68 | Cobimetinib + VEN +/- atezolizumab | R/R MM | 1b/2 | N/A |
| Clinicaltrials.Gov Identifier | Other Study ID Numbers | Intervention | Route of Administration | Disease | Study Phase(s) |
|---|---|---|---|---|---|
| NCT03218683 | D6910C00001 | AZD-5991 | IV | R/R hematologic malignancies | 1 |
| NCT02992483 | CMIK665 × 2101, 2016-003624-22 | MIK665 | IV | R/R MM, R/R NHL | 1 |
| NCT02675452 | 20150161, 2015-004777-32 | AMG-176 | IV | R/R MM | 1 |
| NCT03465540 | 20170173 | AMG-397 | PO | R/R MM, R/R NHL | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, V.S.; Xu, Z.-F.; Huang, D.C.S.; Thijssen, R. BH3 Mimetics for the Treatment of B-Cell Malignancies—Insights and Lessons from the Clinic. Cancers 2020, 12, 3353. https://doi.org/10.3390/cancers12113353
Lin VS, Xu Z-F, Huang DCS, Thijssen R. BH3 Mimetics for the Treatment of B-Cell Malignancies—Insights and Lessons from the Clinic. Cancers. 2020; 12(11):3353. https://doi.org/10.3390/cancers12113353
Chicago/Turabian StyleLin, Victor S., Zhuo-Fan Xu, David C. S. Huang, and Rachel Thijssen. 2020. "BH3 Mimetics for the Treatment of B-Cell Malignancies—Insights and Lessons from the Clinic" Cancers 12, no. 11: 3353. https://doi.org/10.3390/cancers12113353
APA StyleLin, V. S., Xu, Z.-F., Huang, D. C. S., & Thijssen, R. (2020). BH3 Mimetics for the Treatment of B-Cell Malignancies—Insights and Lessons from the Clinic. Cancers, 12(11), 3353. https://doi.org/10.3390/cancers12113353