ShcD Binds DOCK4, Promotes Ameboid Motility and Metastasis Dissemination, Predicting Poor Prognosis in Melanoma

 ,
,  , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
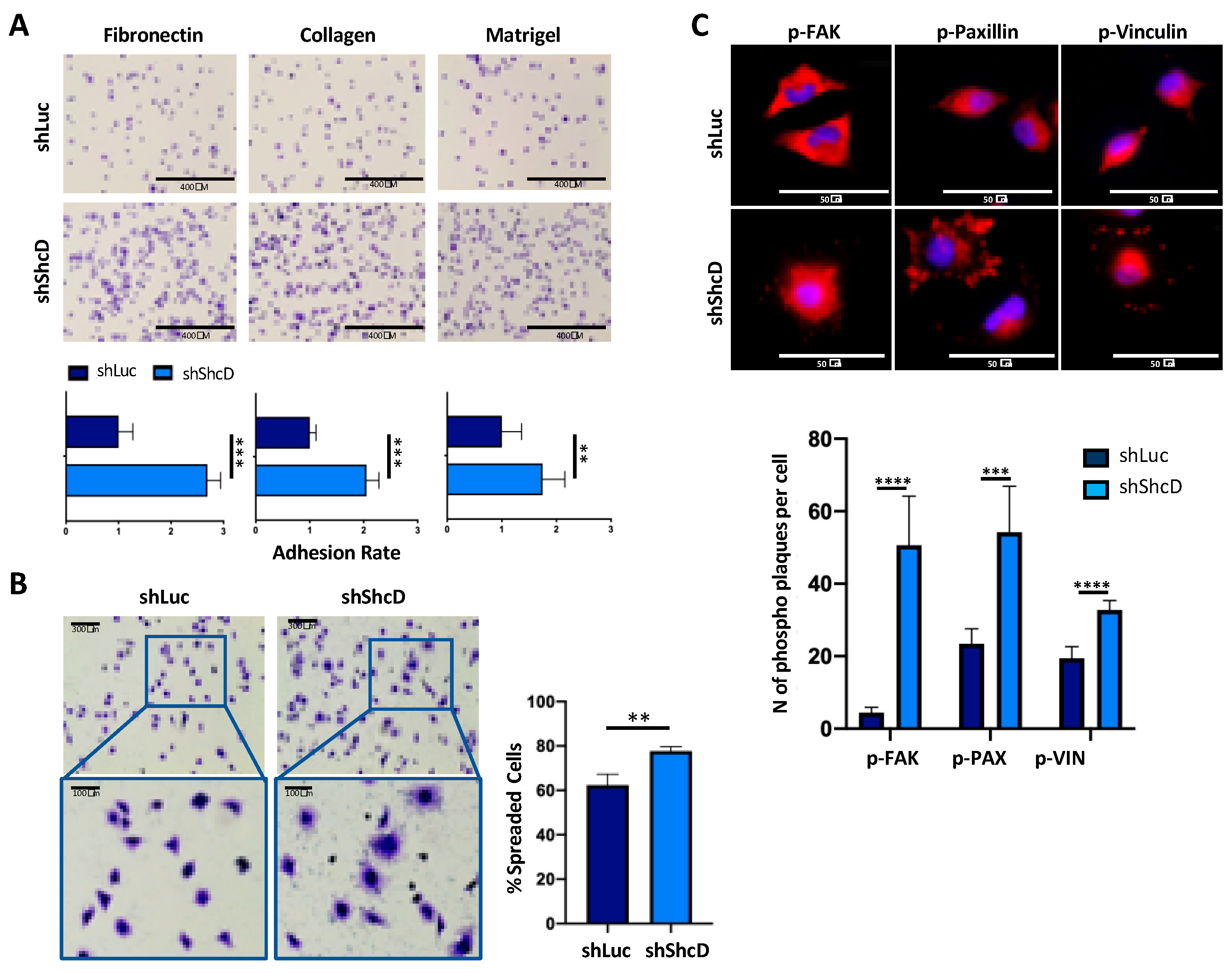
2.1. ShcD Impairs the Adhesive Properties of Melanoma Cells
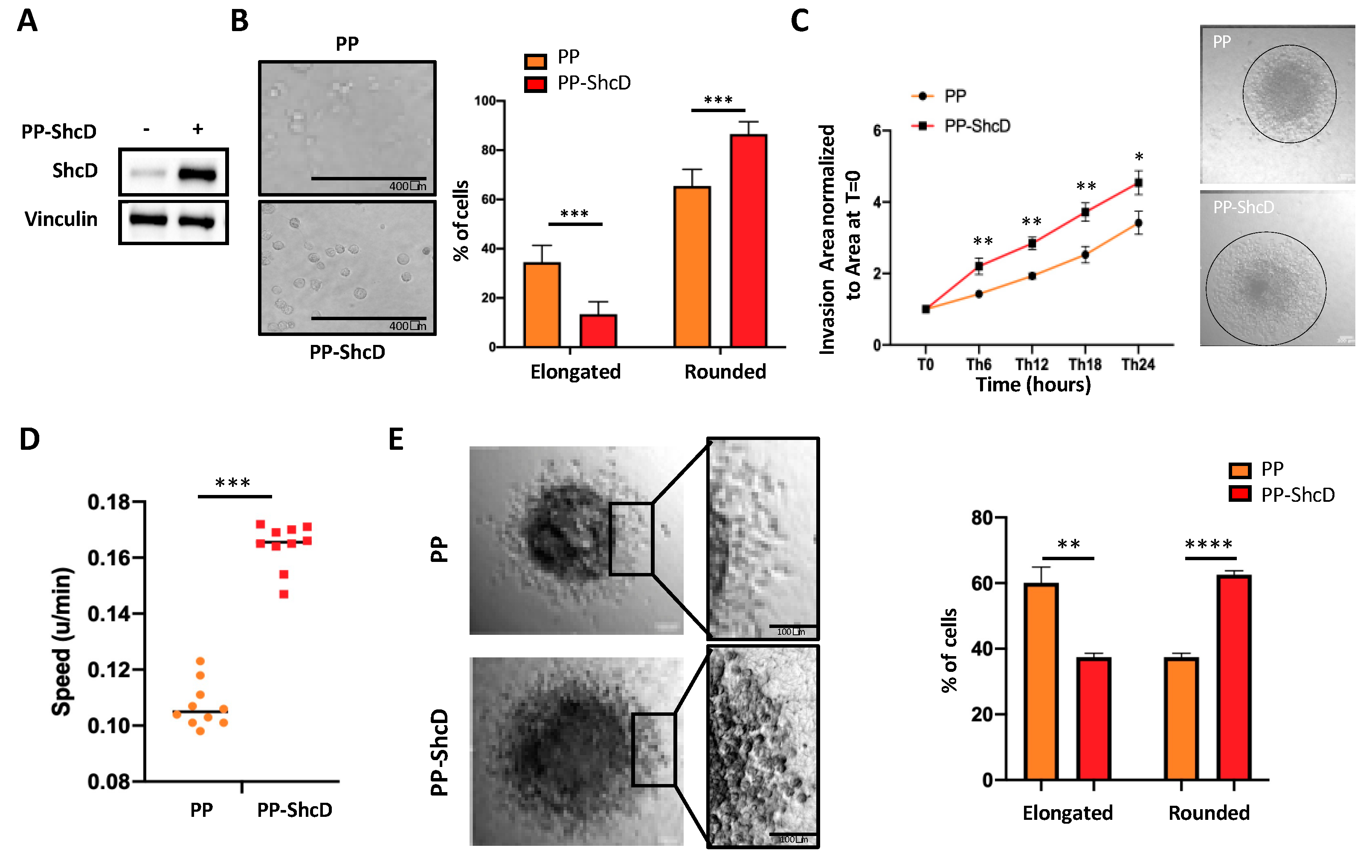
2.2. ShcD Regulates Melanoma Cell Morphology and Sustains Amoeboid Movement of Melanoma Cells in 3D Matrix
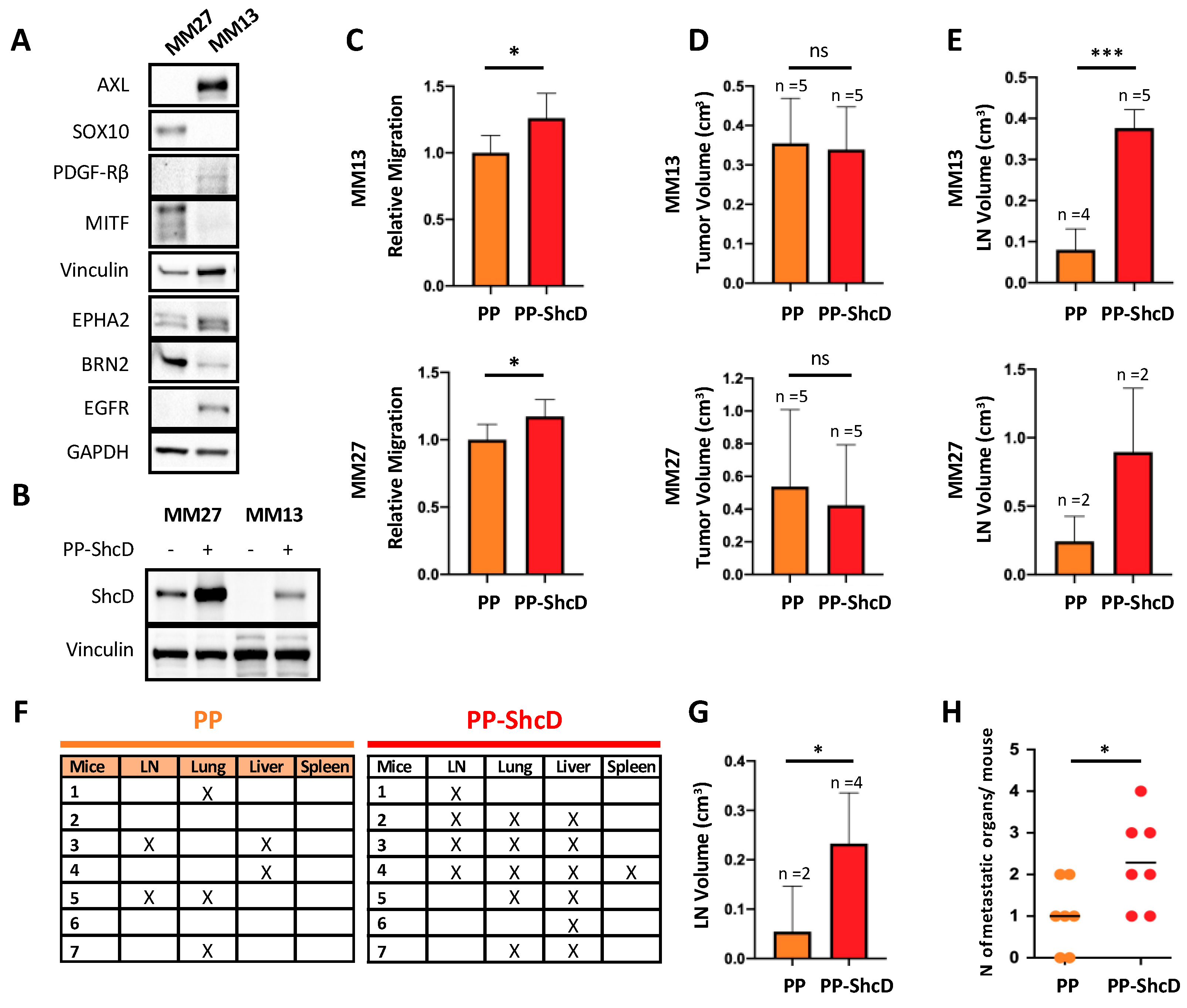
2.3. ShcD Is Crucial for Metastasis Dissemination
2.4. ShcD Expression Correlates with Melanoma Progression and Patient Outcome in Melanoma Patients
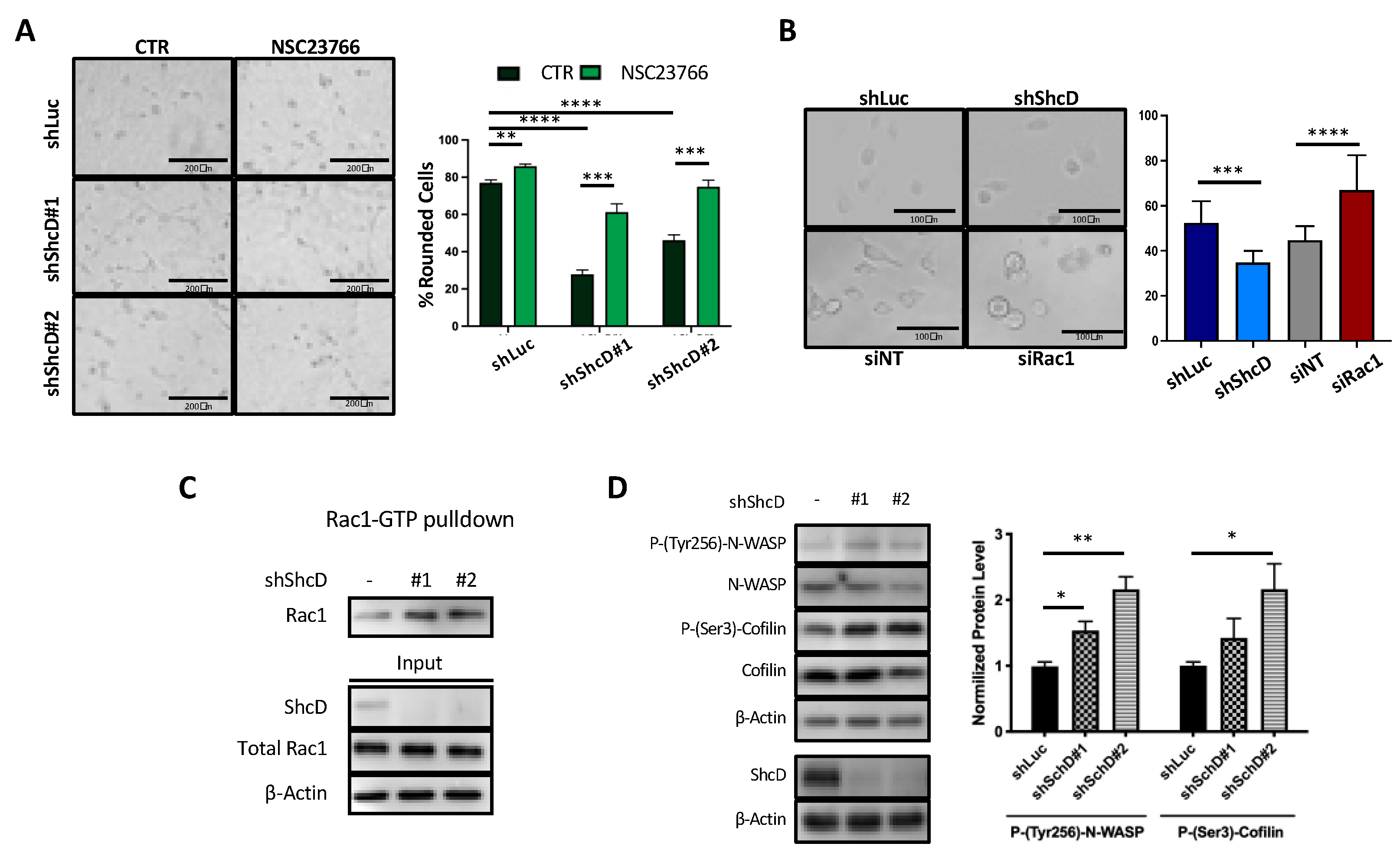
2.5. Rac1 Molecular Pathway Is Altered in ShcD Expressing Melanoma Cells
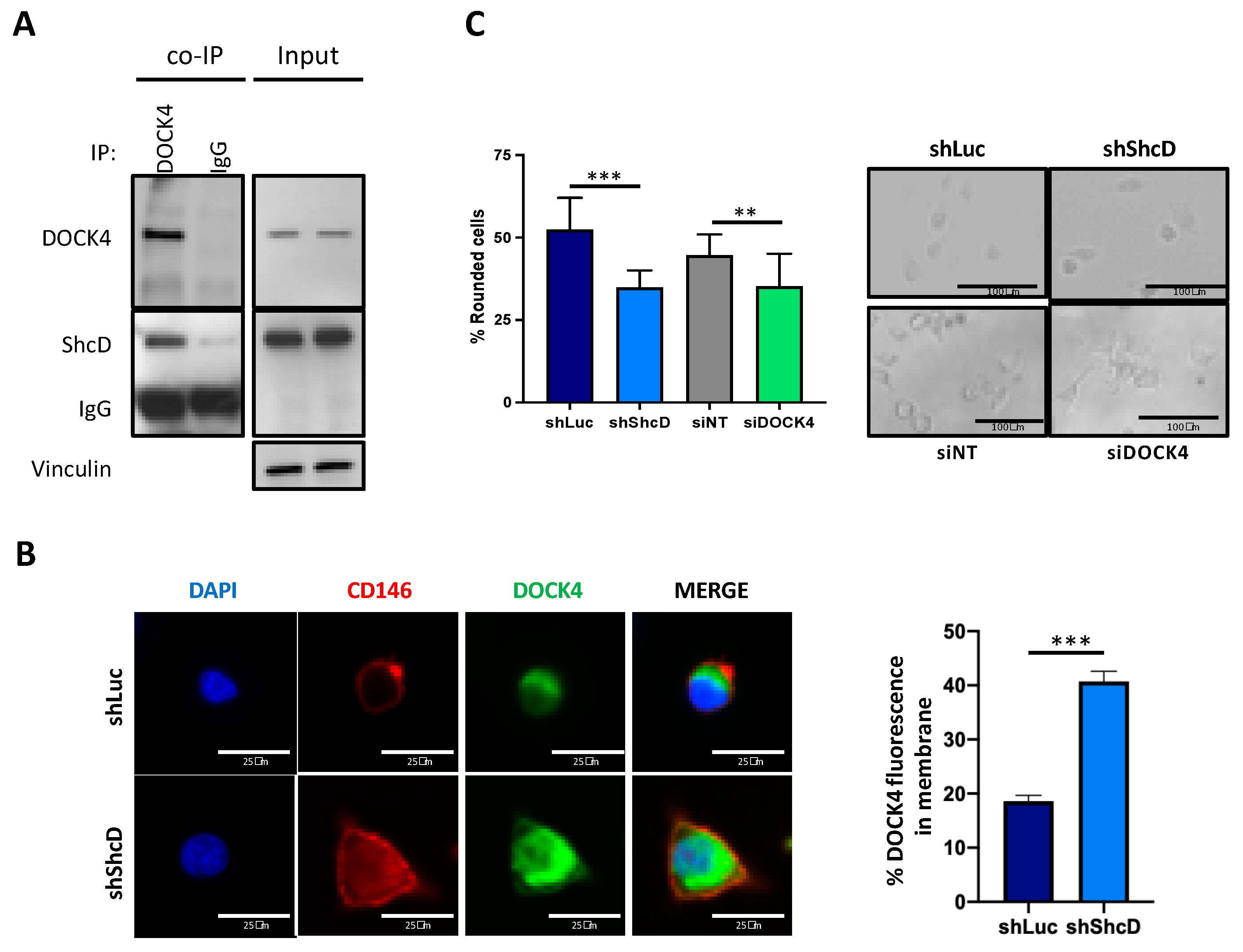
2.6. ShcD Negatively Regulates Rac1 Activation by Preventing Its Binding to DOCK4
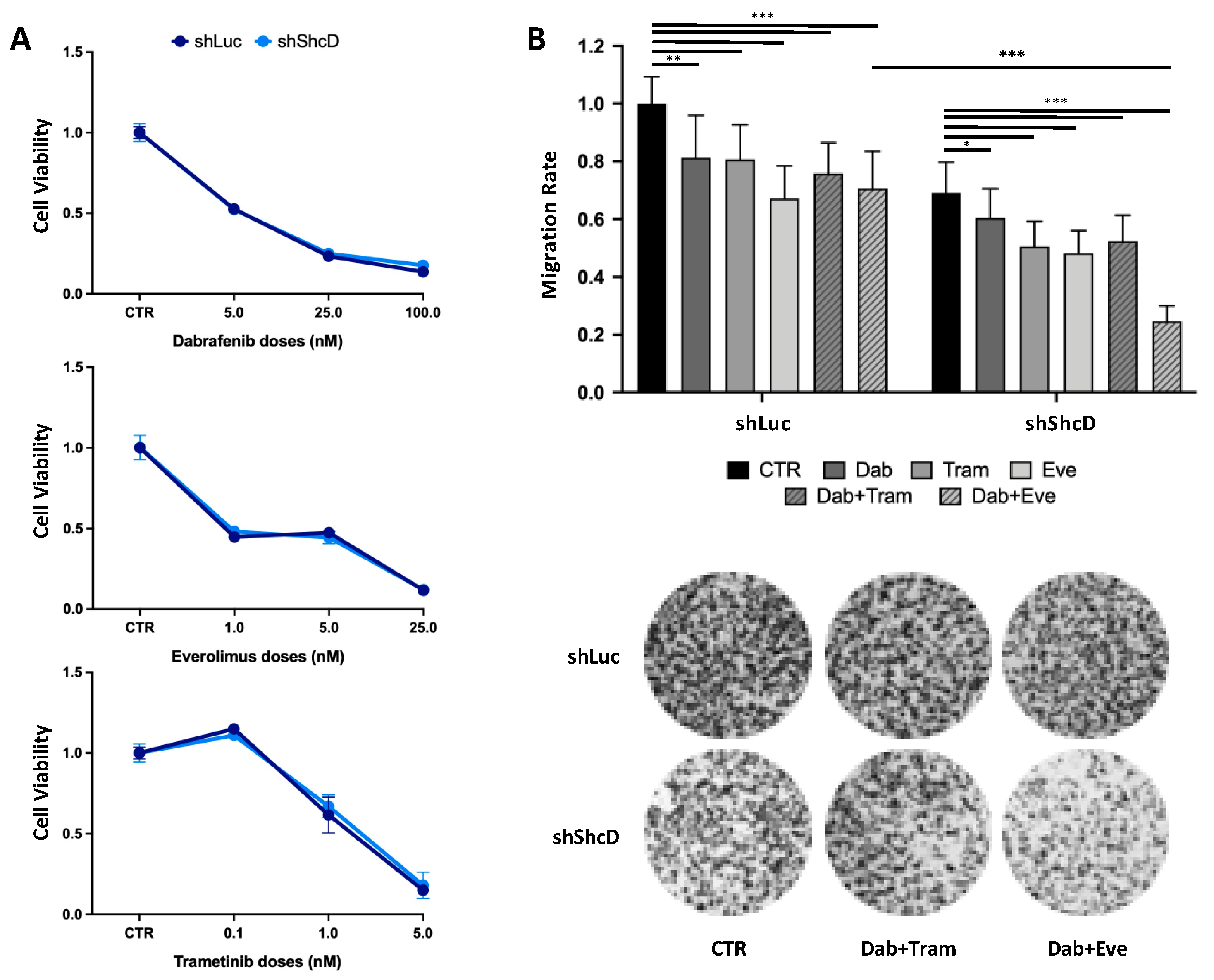
2.7. ShcD Contributes to Resistance of Melanoma Cells to Treatment
3. Discussion
4. Materials and Methods
4.1. Plasmids, Lentiviral Transduction and Cell Culture
4.1.1. Plasmids
4.1.2. Retroviral and Lentiviral Transfections and Infections
4.1.3. siRNA Transfection Protocol
4.1.4. Cell Cultures
4.2. PDX Generation and In Vivo Studies
4.3. Cell Adhesion and Spreading Assays
4.4. Cell Morphology Assessment
4.5. Cell Migration Assay
4.6. Spheroid Invasion Assay and Time-Lapse Analysis
4.7. Immunofluorescence Staining
4.8. Immunoblot and Immunoprecipitation
4.9. ShcD Antibody Generation (Anti-CH2 Clone 47-10-21)
4.10. RAC1 Activation Assay
4.11. Tissue Microarrays (TMA)
4.12. In Situ Hybridization (ISH)
4.13. Drug Treatment
4.14. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Exp. Med. 2010, 207, 11–19. [Google Scholar] [CrossRef]
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011, 21, 736–744. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef]
- Orgaz, J.L.; Sanz-Moreno, V. Emerging molecular targets in melanoma invasion and metastasis. Pigment. Cell Melanoma Res. 2013, 26, 39–57. [Google Scholar] [CrossRef]
- Sahai, E. Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 2005, 15, 87–96. [Google Scholar] [CrossRef]
- Paluch, E.K.; Raz, E. The role and regulation of blebs in cell migration. Curr. Opin. Cell Biol. 2013, 25, 582–590. [Google Scholar] [CrossRef]
- Ruprecht, V.; Wieser, S.; Callan-Jones, A.; Smutny, M.; Morita, H.; Sako, K.; Barone, V.; Ritsch-Marte, M.; Sixt, M.; Voituriez, R.; et al. Cortical Contractility Triggers a Stochastic Switch to Fast Amoeboid Cell Motility. Cell 2015, 160, 673–685. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuzé, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and Low Adhesion Induce Fast Amoeboid Migration of Slow Mesenchymal Cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Virós, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 Signaling Cooperate to Control Actomyosin Contractility in Tumor Cells and Stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Tozluoğlu, M.; Tournier, A.L.; Jenkins, R.P.; Hooper, S.; Bates, P.A.; Sahai, E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol. 2013, 15, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Marshall, C.J. Rho-GTPase signaling drives melanoma cell plasticity. Cell Cycle 2009, 8, 1484–1487. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Sanz-Moreno, V.; Self, A.; Godi, A.; Marshall, C.J. DOCK10-Mediated Cdc42 Activation Is Necessary for Amoeboid Invasion of Melanoma Cells. Curr. Biol. 2008, 18, 1456–1465. [Google Scholar] [CrossRef]
- Westbrook, J.A.; Wood, S.L.; Cairns, D.A.; McMahon, K.; Gahlaut, R.; Thygesen, H.; Shires, M.; Roberts, S.; Marshall, H.; Oliva, M.R.; et al. Identification and validation of DOCK4 as a potential biomarker for risk of bone metastasis development in patients with early breast cancer. J. Pathol. 2019, 247, 381–391. [Google Scholar] [CrossRef]
- Kobayashi, M.; Harada, K.; Negishi, M.; Katoh, H. Dock4 forms a complex with SH3YL1 and regulates cancer cell migration. Cell. Signal. 2014, 26, 1082–1088. [Google Scholar] [CrossRef]
- Hiramoto, K.; Negishi, M.; Katoh, H. Dock4 is regulated by RhoG and promotes Rac-dependent cell migration. Exp. Cell Res. 2006, 312, 4205–4216. [Google Scholar] [CrossRef]
- Turco, M.Y.; Furia, L.; Dietze, A.; Diaz, L.F.; Ronzoni, S.; Sciullo, A.; Simeone, A.; Constam, D.B.; Faretta, M.; Lanfrancone, L. Cellular Heterogeneity During Embryonic Stem Cell Differentiation to Epiblast Stem Cells Is Revealed by the ShcD/RaLP Adaptor Protein. STEM CELLS 2012, 30, 2423–2436. [Google Scholar] [CrossRef][Green Version]
- Fagiani, E.; Giardina, G.; Luzi, L.; Cesaroni, M.; Quarto, M.; Capra, M.; Germano, G.; Bono, M.; Capillo, M.; Pelicci, P.; et al. RaLP, a New Member of the Src Homology and Collagen Family, Regulates Cell Migration and Tumor Growth of Metastatic Melanomas. Cancer Res. 2007, 67, 3064–3073. [Google Scholar] [CrossRef]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Investig. Dermatol. 2020, 1–9. [Google Scholar] [CrossRef]
- Geiger, B.; Yamada, K.M. Molecular Architecture and Function of Matrix Adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2016, 11, 5–27. [Google Scholar] [CrossRef]
- Georgouli, M.; Herraiz, C.; Crosas-Molist, E.; Fanshawe, B.; Maiques, O.; Perdrix, A.; Pandya, P.; Rodriguez-Hernandez, I.; Ilieva, K.M.; Cantelli, G.; et al. Regional Activation of Myosin II in Cancer Cells Drives Tumor Progression via a Secretory Cross-Talk with the Immune Microenvironment. Cell 2019, 176, 757–774.e23. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Mooter, L.L.V.D.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Tomasevic, N.; Jia, Z.; Russell, A.J.; Fujii, T.; Hartman, J.J.; Clancy, S.; Wang, M.; Béraud, C.; Wood, K.W.; Sakowicz, R. Differential Regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistrty 2007, 46, 3494–3502. [Google Scholar] [CrossRef]
- Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Margheri, F.; Laurenzana, A.; Fibbi, G.; Pimpinelli, N.; Calorini, L. Everolimus selectively targets vemurafenib resistant BRAFV600E melanoma cells adapted to low pH. Cancer Lett. 2017, 408, 43–54. [Google Scholar] [CrossRef]
- Kulesa, P.M.; Kasemeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like pnenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.M.; Morrison, J.A.; Kulesa, P.M. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment. Cell Melanoma Res. 2012, 25, 573–583. [Google Scholar] [CrossRef]
- Hawley, S.P.; Wills, M.K.B.; Rabalski, A.J.; Bendall, A.J.; Jones, N. Expression patterns of ShcD and Shc family adaptor proteins during mouse embryonic development. Dev. Dyn. 2010, 240, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Champeval, D.; Rambow, F.; LaRue, L. Transcriptomic Analysis of Mouse Embryonic Skin Cells Reveals Previously Unreported Genes Expressed in Melanoblasts. J. Investig. Dermatol. 2012, 132, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Arozarena, I.; Bischof, H.; Gilby, D.; Belloni, B.; Dummer, R.; Wellbrock, C. In melanoma, beta-catenin is a suppressor of invasion through a cell-type specific mechanism. Oncogene 2011, 30, 4531–4543. [Google Scholar] [CrossRef] [PubMed]
- Strub, T.; Kobi, D.; Koludrovic, D.; Davidson, I. A POU3F2-MITF-SHC4 Axis in Phenotype Switching of Melanoma Cells. In Research on Melanoma—A Glimpse into Current Directions and Future Trends; IntechOpen: London, UK, 2011. [Google Scholar]
- Weichenthal, M.; Senel, G.; Both, M.; Senkpiehl, I.; Lekic, D.; Kähler, K.C.; Ugurel, S.; Leiter, U.M.; Mohr, P.; Hauschild, A.; et al. Evaluation of the Melanoma Tumor Burden Score (MTBS) in a real-world setting. J. Clin. Oncol. 2017, 35, 9565. [Google Scholar] [CrossRef]
- Ahmed, S.B.; Prigent, S.A. Insights into the Shc Family of Adaptor Proteins. J. Mol. Signal. 2017, 12, 2. [Google Scholar] [CrossRef]
- Katoh, H.; Negishi, M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nat. Cell Biol. 2003, 424, 461–464. [Google Scholar] [CrossRef]
- Hiramoto-Yamaki, N.; Takeuchi, S.; Ueda, S.; Harada, K.; Fujimoto, S.; Negishi, M.; Katoh, H. Ephexin4 and EphA2 mediate cell migration through a RhoG-dependent mechanism. J. Cell Biol. 2010, 190, 461–477. [Google Scholar] [CrossRef]
- Saci, A.; Cantley, L.C.; Carpenter, C.L. Rac1 Regulates the Activity of mTORC1 and mTORC2 and Controls Cellular Size. Mol. Cell 2011, 42, 50–61. [Google Scholar] [CrossRef]
- Kinsella, T.; Mencarelli, A.; Valtieri, M.; Riganelli, D.; Grignani, F.; Lanfrancone, L.; Peschle, C.; Nolan, G.P.; Pelicci, P.G. High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res. 1998, 58, 14–19. [Google Scholar]
- Confalonieri, S.; Quarto, M.; Goisis, G.; Nuciforo, P.; Donzelli, M.; Jodice, G.; Pelosi, G.; Viale, G.; Pece, S.; Di Fiore, P.P. Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 2009, 28, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Capra, M.; Nuciforo, P.G.; Confalonieri, S.; Quarto, M.; Bianchi, M.; Nebuloni, M.; Boldorini, R.; Pallotti, F.; Viale, G.; Gishizky, M.L.; et al. Frequent Alterations in the Expression of Serine/Threonine Kinases in Human Cancers. Cancer Res. 2006, 66, 8147–8154. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ShcD in Primary Tissue | ShcD in Metastatic Tissue | |||||||
|---|---|---|---|---|---|---|---|---|
| No. of Patients, n (%) | Negative | Positive | p-Values | No. of Patients, n (%) | Negative | Positive | p-Values | |
| No. of patients, n (%) | 183 (100%) | 120 (66%) | 63 (34%) | 94 (100%) | 40 (43%) | 54 (57%) | <001 | |
| At diagnosis | Antecedent CM | |||||||
| Sex | 0.46 | |||||||
| Female | 97 (54%) | 67 (56%) | 30 (50%) | 29 (31%) | 13 (32%) | 16 (30%) | 0.76 | |
| Men | 83 (46%) | 53 (44%) | 30 (50%) | 65 (69%) | 27 (68%) | 38 (70%) | ||
| Age at diagnosis | 0.21 | |||||||
| Median | 56 | 54 | 59 | 48 | 50 | 45 | 0.02 | |
| Interquartile range | (44, 67) | (43, 65) | (46, 69) | (32, 59) | (41, 62) | (24, 55) | ||
| Breslow thickness mm | <0001 | |||||||
| Median | 1.0 | 0.5 | 2.4 | 2.8 | 1.9 | 3.1 | 0.14 | |
| Interquartile range | (0.5, 3.1) | (0.3, 1.6) | (1.2, 4.7) | (1.5, 5.0) | (0.9,4.8) | (1.8, 5.4) | ||
| Breslow thickness mm | <0001 | 0.12 | ||||||
| 0.01–1.0 | 91 (51) | 80 (67) | 11 (18) | (<0001 †) | 9 (25) | 7 (67) | 2 (4) | (0.02) |
| 1.01–2.0 | 26 (14) | 14 (12) | 12 (20) | 20 (21) | 8 (17) | 12 (22) | ||
| 2.01–4.0 | 31 (17) | 15 (13) | 16 (27) | 16 (17) | 3 (20) | 13 (24) | ||
| >4.0 | 31 (17) | 10 (8) | 21 (35) | 25 (27) | 10 (8) | 15 (28) | ||
| Unknown | 1 (1) | 1 (1) | 0 (0) | 24 (1) | 12 (30) | 12 (22) | ||
| Site of primary | 0.75 | 0.23 | ||||||
| Extremity | 83 (45%) | 51 (43%) | 30 (50%) | (0.28 ‡) | 30 (32%) | 12 (30%) | 18 (33%) | (0.73 ‡) |
| Trunk | 78 (43%) | 54 (45%) | 23 (38%) | 38 (41%) | 16 (40%) | 22 (41%) | ||
| Head and neck | 13 (7%) | 9 (8%) | 4 (7%) | 6 (6%) | 1 (3%) | 5 (9%) | ||
| Other | 7 (4%) | 4 (3%) | 3 (5%) | 5 (5%) | 4 (10%) | 1 (2%) | ||
| Occults | 2 (1%) | 2 (1%) | 0 (0%) | 15 (16%) | 7 (18%) | 8 (15%) | ||
| Ulceration | <0001 | 0.04 | ||||||
| Yes | 46 (25%) | 14 (12%) | 32 (51%) | (<0001) | 32 (34%) | 9 (23%) | 23 (43%) | (0.04) |
| No | 131 (72%) | 102 (85%) | 29 (46%) | 23 (24%) | 13 (32%) | 10 (18%) | ||
| Unknown | 6 (3%) | 4 (3%) | 2 (3%) | 39 (42%) | 18 (45%) | 21 (39%) | ||
| Mitotic rate | <0001 | 0.27 | ||||||
| Mitoses <1 mm2 | 64 (35%) | 60 (50%) | 4 (6%) | (<0001) | 32 (34%) | 16 (40%) | 16 (30%) | (0.06) |
| Mitoses ≥1 mm2 | 114 (62%) | 55 (46%) | 59 (94%) | 48 (51%) | 18 (45%) | 30 (55%) | ||
| Unknown | 5 (3%) | 5 (4%) | 0 (0%) | 14 (15%) | 6 (15%) | 8 (15%) | ||
| Histo-pathologic subtype | 0.001 | 0.83 | ||||||
| NM | 53 (29%) | 24 (20%) | 29 (46%) | (0.0004¥) | 12 (13%) | 4 (10%) | 8 (15%) | (0.63¥) |
| SSM | 125 (68%) | 93 (77%) | 32 (51%) | 37 (39%) | 16 (40%) | 21 (39%) | ||
| Other | 4 (2%) | 2 (2%) | 2 (3%) | 5 (5%) | 2 (5%) | 3 (5%) | ||
| Unknown | 1 (1%) | 1 (1%) | 0 (0%) | 40 (43%) | 18 (45%) | 22 (41%) | ||
| AJCC stage | 0.0004 | 0.33 | ||||||
| I | 105 (58%) | 83 (69%) | 22 (35%) | (0.01) | 7 (7%) | 4 (10%) | 3 (5%) | (0.02§) |
| II | 33 (18%) | 16 (13%) | 17 (27%) | 19 (20%) | 10 (25%) | 9 (17%) | ||
| III | 31 (17%) | 16 (13%) | 15 (24%) | 46 (49%) | 18 (45%) | 28 (52%) | ||
| IV | 6 (3%) | 2 (2%) | 4 (6%) | 3 (3%) | 0 (0%) | 3 (5%) | ||
| Unknown | 8 (4%) | 3 (3%) | 5 (8%) | 19 (20%) | 8 (20%) | 11 (21%) | ||
| Lymph-nodes status | 0.02 | 0.33 | ||||||
| Positive | 35 (19%) | 17 (14%) | 18 (28%) | (0.03) | 46 (49%) | 17 (43%) | 18 (54%) | (0.02) |
| Negative | 136 (74%) | 94 (78%) | 42 (67%) | 26 (28%) | 15 (37%) | 11 (20%) | ||
| Unknown | 12 (7%) | 9 (8%) | 3 (5%) | 22 (23%) | 8 (20%) | 14 (26%) | ||
| ShcD evaluation | ||||||||
| First metastasis | 0.01 | |||||||
| Regional metastasis | 76 (81%) | 28 (70%) | 48 (89%) | (0.94) | ||||
| Distant metastasis | 17 (18%) | 12 (30%) | 5 (9%) | |||||
| Unknown | 1 (1%) | 0 (0%) | 1 (2%) | |||||
| Events during follow-up | ||||||||
| First recurrence * | 0.04 | |||||||
| Yes | 41 (23%) | 22 (19%) | 19 (32%) | (0.08) | ||||
| No | 136 (77%) | 96 (81%) | 40 (68%) | |||||
| Deaths | 0.05 | 0.32 | ||||||
| Yes | 64 (35%) | 36 (30%) | 28 (44%) | (0.39) | 54 (57%) | 11 (69%) | 43 (55%) | (0.40) |
| No | 119 (65%) | 84 (70%) | 35 (56%) | 40 (43%) | 5 (31%) | 35 (45%) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aladowicz, E.; Granieri, L.; Marocchi, F.; Punzi, S.; Giardina, G.; Ferrucci, P.F.; Mazzarol, G.; Capra, M.; Viale, G.; Confalonieri, S.; et al. ShcD Binds DOCK4, Promotes Ameboid Motility and Metastasis Dissemination, Predicting Poor Prognosis in Melanoma. Cancers 2020, 12, 3366. https://doi.org/10.3390/cancers12113366
Aladowicz E, Granieri L, Marocchi F, Punzi S, Giardina G, Ferrucci PF, Mazzarol G, Capra M, Viale G, Confalonieri S, et al. ShcD Binds DOCK4, Promotes Ameboid Motility and Metastasis Dissemination, Predicting Poor Prognosis in Melanoma. Cancers. 2020; 12(11):3366. https://doi.org/10.3390/cancers12113366
Chicago/Turabian StyleAladowicz, Ewa, Letizia Granieri, Federica Marocchi, Simona Punzi, Giuseppina Giardina, Pier Francesco Ferrucci, Giovanni Mazzarol, Maria Capra, Giuseppe Viale, Stefano Confalonieri, and et al. 2020. "ShcD Binds DOCK4, Promotes Ameboid Motility and Metastasis Dissemination, Predicting Poor Prognosis in Melanoma" Cancers 12, no. 11: 3366. https://doi.org/10.3390/cancers12113366
APA StyleAladowicz, E., Granieri, L., Marocchi, F., Punzi, S., Giardina, G., Ferrucci, P. F., Mazzarol, G., Capra, M., Viale, G., Confalonieri, S., Gandini, S., Lotti, F., & Lanfrancone, L. (2020). ShcD Binds DOCK4, Promotes Ameboid Motility and Metastasis Dissemination, Predicting Poor Prognosis in Melanoma. Cancers, 12(11), 3366. https://doi.org/10.3390/cancers12113366