Comprehensive Gene Mutation Profiling of Circulating Tumor DNA in Ovarian Cancer: Its Pathological and Prognostic Impact

 ,
,
Abstract
:Simple Summary
Abstract
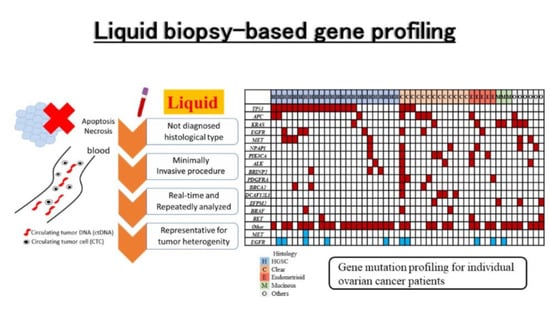
1. Introduction
2. Results
2.1. Flow Chart of Patients’ Enrollment and Clinical Characteristics
2.2. Association of cfDNA Levels with Clinical Stage and Histological Subtype
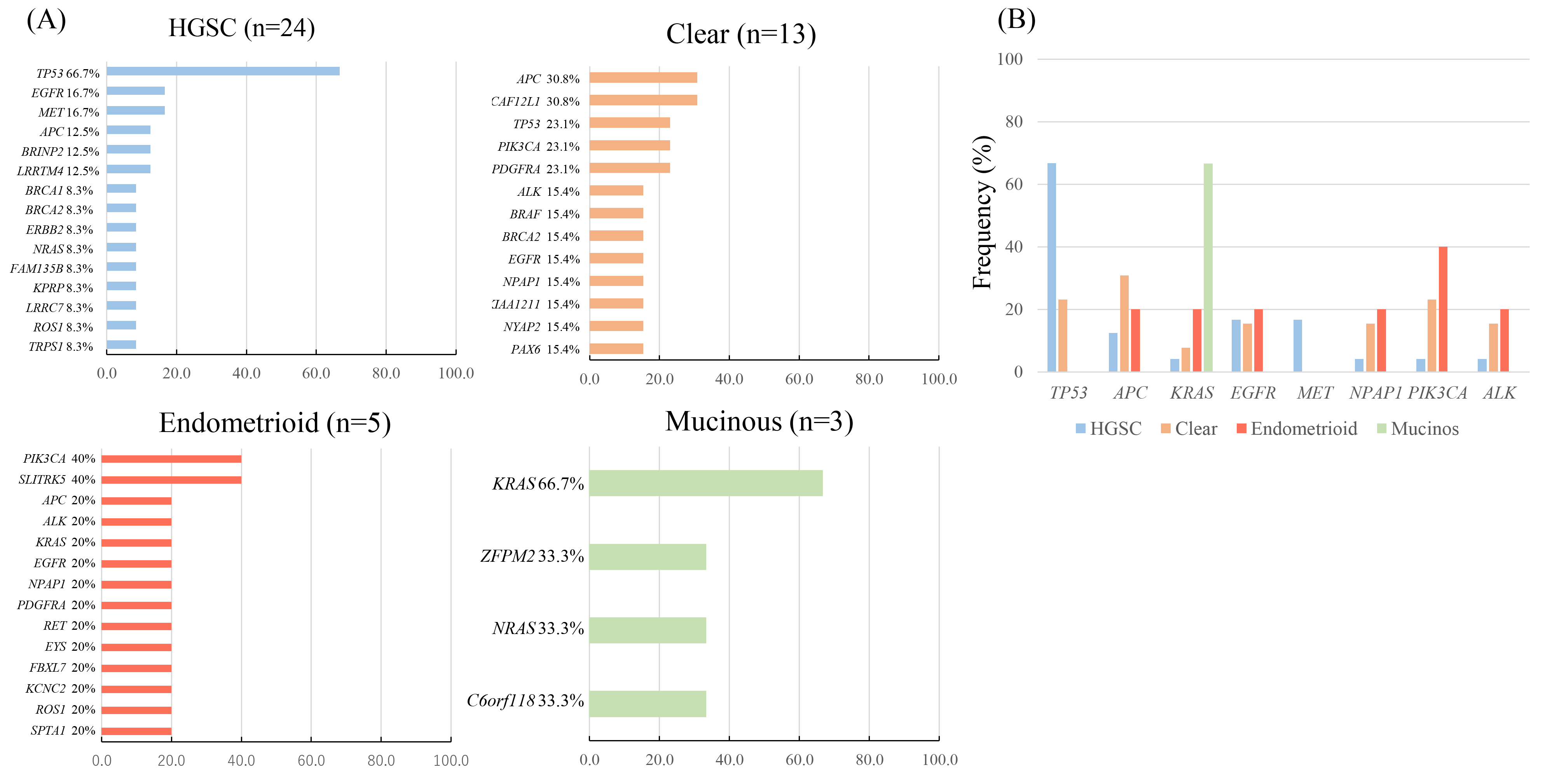
2.3. Profiling of Genetic Somatic Mutation in Ovarian Cancer Using CAPP-Seq
2.4. The Concordance of TP53 Mutations between ctDNA and Tissue DNA
2.5. Association of Blood Tumor Mutation Burden (bTMB) with Disease Stage and Histological Subtype
2.6. Impact of cfDNA Concentration and Pathogenic Mutations on Patient Survival
2.7. Monitoring of Changes in ctDNA-Based Genetic Profiles During Treatment Course
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Circulating Tumor DNA Extraction
4.3. Circulating Tumor DNA Sequencing
4.4. Tumor DNA Extraction
4.5. Tumor DNA Sequencing
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Bois, A.; Pfisterer, J. Future options for first-line therapy of advanced ovarian cancer. Int. J. Gynecol. Cancer 2005, 15 (Suppl. 1), 42–50. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. The dualistic model of ovarian carcinogenesis. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Blair, B.G.; Bardelli, A.; Park, B.H. Somatic alterations as the basis for resistance to targeted therapies. J. Pathol. 2013, 232, 244–254. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- McDermott, U.; Downing, J.R.; Stratton, M.R. Genomics and the continuum of cancer care. N. Engl. J. Med. 2011, 364, 340–350. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravergna, G.; et al. P2.08 Emergence of kras mutations and acquired resistance to anti egfr therapy in colorectal cancer. Ann. Oncol. 2012, 23, v24. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nat. Cell Biol. 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Oliner, K.K.; Parker, A.A.; Siena, S.; Van Cutsem, E.; Huang, J.J.; Humblet, Y.; Van Laethem, J.-L.; André, T.; Wiezorek, J.J.; et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 1902–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Wang, J.; Xu, Y.; Ding, X.; Li, M.; Jiang, F.; Xu, L.; Yin, R. Circulating tumor DNA is effective for the detection of EGFR mutation in non-small cell lung cancer: A meta-analysis. Cancer Epidemiol. Biomark. Prev. 2014, 24, 206–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.-H.; Barrett, J.C.; Jänne, P.A. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- The cancer genome atlas research network integrated genomic analyses of ovarian carcinoma. Nat. Cell Biol. 2011, 474, 609–615. [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [Green Version]
- Przybyl, J.; Chabon, J.J.; Spans, L.; Ganjoo, K.; Vennam, S.; Newman, A.M.; Forgo, E.; Varma, S.; Zhu, S.; Debiec-Rychter, M.; et al. Combination approach for detecting different types of alterations in circulating tumor DNA in leiomyosarcoma. Clin. Cancer Res. 2018, 24, 2688–2699. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arend, R.; Londoño, A.I.; Montgomery, A.M.; Smith, H.J.; Dobbin, Z.C.; Katre, A.A.; Martinez, A.; Yang, E.S.; Alvarez, R.D.; Huh, W.K.; et al. Molecular response to neoadjuvant chemotherapy in high-grade serous ovarian carcinoma. Mol. Cancer Res. 2018, 16, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwahashi, N.; Sakai, K.; Noguchi, T.; Yahata, T.; Toujima, S.; Nishio, K.; Ino, K. A comprehensive gene mutation analysis of liquid biopsy samples from patients with metastatic colorectal cancer to the ovary: A case report. Oncol. Lett. 2018, 16, 6431–6436. [Google Scholar] [CrossRef]
- Iwahashi, N.; Sakai, K.; Noguchi, T.; Yahata, T.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Liquid biopsy-based comprehensive gene mutation profiling for gynecological cancer using cancer personalized profiling by deep sequencing. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Sakai, K.; Iwahashi, N.; Matsuda, K.; Matsukawa, H.; Yahata, T.; Toujima, S.; Nishio, K.; Ino, K. Changes in the gene mutation profiles of circulating tumor DNA detected using CAPP‑Seq in neoadjuvant chemotherapy‑treated advanced ovarian cancer. Oncol. Lett. 2020, 19, 2713–2720. [Google Scholar] [CrossRef]
- Mardis, E.; Na, R.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; et al. Faculty Opinions recommendation of Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Faculty Opinions—Post-Publication Peer Review of the Biomedical Literature. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-C.; Wang, D.; Jin, L.; Yao, H.-W.; Zhang, J.-H.; Wang, J.; Zhao, X.-M.; Shen, C.-Y.; Chen, W.; Wang, X.-L.; et al. Circulating tumor DNA detectable in early- and late-stage colorectal cancer patients. Biosci. Rep. 2018, 38, 20180322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.A.; Hachiya, T.; Iwaya, T.; Kume, K.; Matsuo, T.; Kawasaki, K.; Abiko, Y.; Akasaka, R.; Matsumoto, T.; Otsuka, K.; et al. Individualized mutation detection in circulating tumor DNA for monitoring colorectal tumor burden using a cancer-associated gene sequencing panel. PLoS ONE 2016, 11, e0146275. [Google Scholar] [CrossRef] [PubMed]
- Lapin, M.; Oltedal, S.; Tjensvoll, K.; Buhl, T.; Smaaland, R.; Garresori, H.; Javle, M.; Glenjen, N.I.; Abelseth, B.K.; Gilje, B.; et al. Fragment size and level of cell-free DNA provide prognostic information in patients with advanced pancreatic cancer. J. Transl. Med. 2018, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tissot, C.; Toffart, A.-C.; Villar, S.; Souquet, P.-J.; Merle, P.; Moro-Sibilot, D.; Pérol, M.; Zavadil, J.; Brambilla, C.; Olivier, M.; et al. Circulating free DNA concentration is an independent prognostic biomarker in lung cancer. Eur. Respir. J. 2015, 46, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.; Lee, R.; Garner, G.; Dhomen, N.; Lorigan, P.; et al. Plasma total cell-free DNA (cfDNA) is a surrogate biomarker for tumour burden and a prognostic biomarker for survival in metastatic melanoma patients. Eur. J. Cancer 2018, 88, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical utility of plasma cell-free DNA in adult patients with newly diagnosed glioblastoma: A pilot prospective study. Clin. Cancer Res. 2020, 26, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Hayano, T.; Yokota, Y.; Hosomichi, K.; Nakaoka, H.; Yoshihara, K.; Adachi, S.; Kashima, K.; Tsuda, H.; Moriya, T.; Tanaka, K.; et al. Molecular characterization of an intact p53 pathway subtype in high-grade serous ovarian cancer. PLoS ONE 2014, 9, e114491. [Google Scholar] [CrossRef]
- Ueno, Y.; Enomoto, T.; Otsuki, Y.; Sugita, N.; Nakashima, R.; Yoshino, K.; Kuragaki, C.; Ueda, Y.; Aki, T.; Ikegami, H.; et al. Prognostic significance of p53 mutation in suboptimally resected advanced ovarian carcinoma treated with the combination chemotherapy of paclitaxel and carboplatin. Cancer Lett. 2006, 241, 289–300. [Google Scholar] [CrossRef]
- Kuo, K.-T.; Mao, T.-L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.-L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [Green Version]
- Cybulska, P.; Paula, A.D.C.; Tseng, J.; Leitao, M.M., Jr.; Bashashati, A.; Huntsman, D.G.; Nazeran, T.M.; Aghajanian, C.; Abu-Rustum, N.R.; Delair, D.F.; et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol. Oncol. 2019, 154, 516–523. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husseinzadeh, N.; Husseinzadeh, H.D. mTOR inhibitors and their clinical application in cervical, endometrial and ovarian cancers: A critical review. Gynecol. Oncol. 2014, 133, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.L.; Russell, K.; Millis, S.; Gatalica, Z.; Bender, R.; Voss, A. Molecular profiling of clear cell ovarian cancers: Identifying potential treatment targets for clinical trials. Int. J. Gynecol. Cancer 2016, 26, 648–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal, E.; Beer, D.G.; Ramnath, N. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J. Thorac. Oncol. 2015, 10, e9–e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.R.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Lassus, H.; Sihto, H.; Leminen, A.; Joensuu, H.; Isola, J.; Nupponen, N.N.; Butzow, R. Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J. Mol. Med. 2006, 84, 671–681. [Google Scholar] [CrossRef]
- Nemtsova, M.V.; Kalinkin, A.I.; Kuznetsova, E.B.; Bure, I.V.; Alekseeva, E.A.; Bykov, I.I.; Khorobrykh, T.V.; Mikhaylenko, D.S.; Tanas, A.S.; Kutsev, S.I.; et al. Clinical relevance of somatic mutations in main driver genes detected in gastric cancer patients by next-generation DNA sequencing. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bejar, R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica 2014, 99, 956–964. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef]
- Narahara, M.; Higasa, K.; Nakamura, S.; Tabara, Y.; Kawaguchi, T.; Ishii, M.; Matsubara, K.; Matsuda, F.; Yamada, R. Large-scale east-asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 2014, 9, e100924. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Median) | 60 (28–82) |
|---|---|
| Stage | No. (%) |
| I | 15 (29) |
| II | 5 (10) |
| III | 23 (45) |
| IV | 8 (16) |
| Histology | No. (%) |
| High-grade serous carcinoma (HGSC) | 24 (47) |
| Clear cell carcinoma | 13 (25) |
| Endometrioid carcinoma | 5 (10) |
| Mucinous carcinoma | 3 (6) |
| Others* | 6 (12) |
| Treatment | No. (%) |
| Primary surgery | 35 (69) |
| Neoadjuvant chemotherapy | 16 (31) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguchi, T.; Iwahashi, N.; Sakai, K.; Matsuda, K.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Comprehensive Gene Mutation Profiling of Circulating Tumor DNA in Ovarian Cancer: Its Pathological and Prognostic Impact. Cancers 2020, 12, 3382. https://doi.org/10.3390/cancers12113382
Noguchi T, Iwahashi N, Sakai K, Matsuda K, Matsukawa H, Toujima S, Nishio K, Ino K. Comprehensive Gene Mutation Profiling of Circulating Tumor DNA in Ovarian Cancer: Its Pathological and Prognostic Impact. Cancers. 2020; 12(11):3382. https://doi.org/10.3390/cancers12113382
Chicago/Turabian StyleNoguchi, Tomoko, Naoyuki Iwahashi, Kazuko Sakai, Kaho Matsuda, Hitomi Matsukawa, Saori Toujima, Kazuto Nishio, and Kazuhiko Ino. 2020. "Comprehensive Gene Mutation Profiling of Circulating Tumor DNA in Ovarian Cancer: Its Pathological and Prognostic Impact" Cancers 12, no. 11: 3382. https://doi.org/10.3390/cancers12113382