Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity



 , ,
, ,  ,
, 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
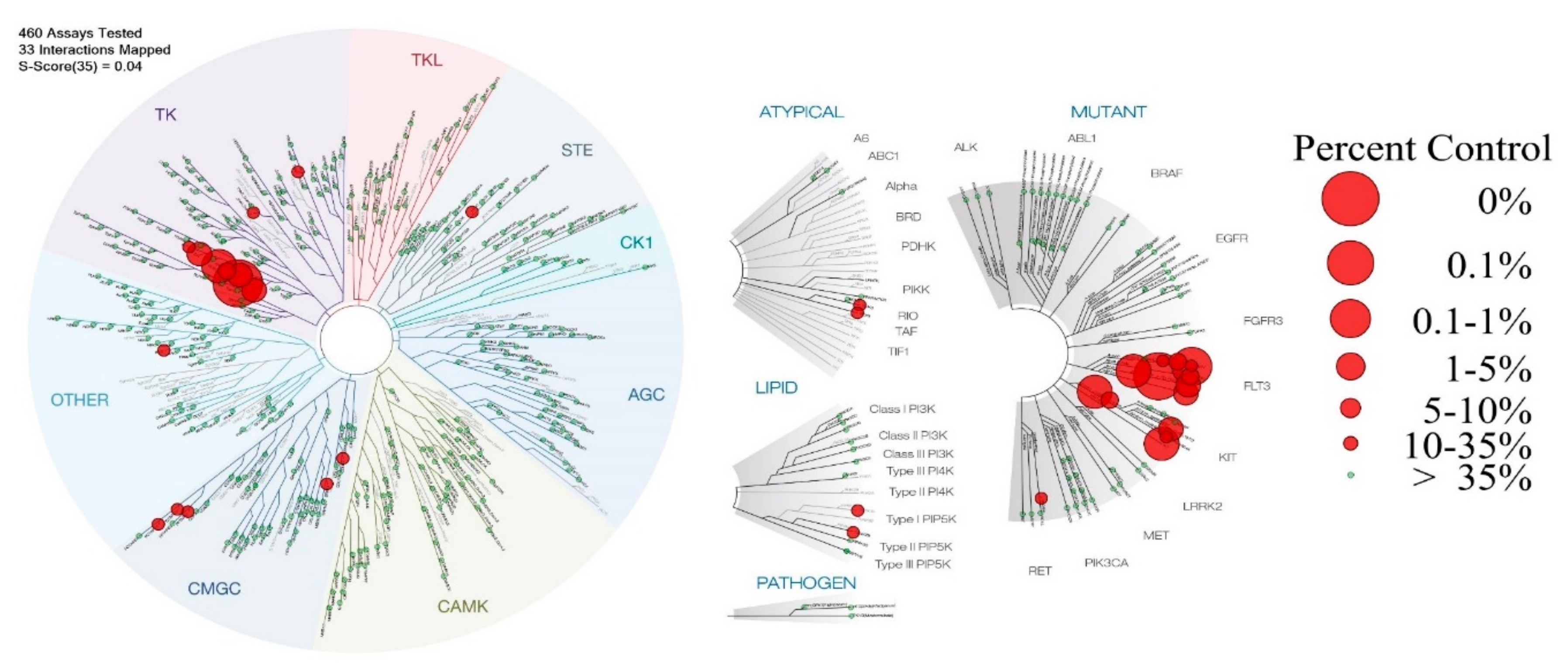
2.1. Kinases’ Binding Affinity for Compound 1
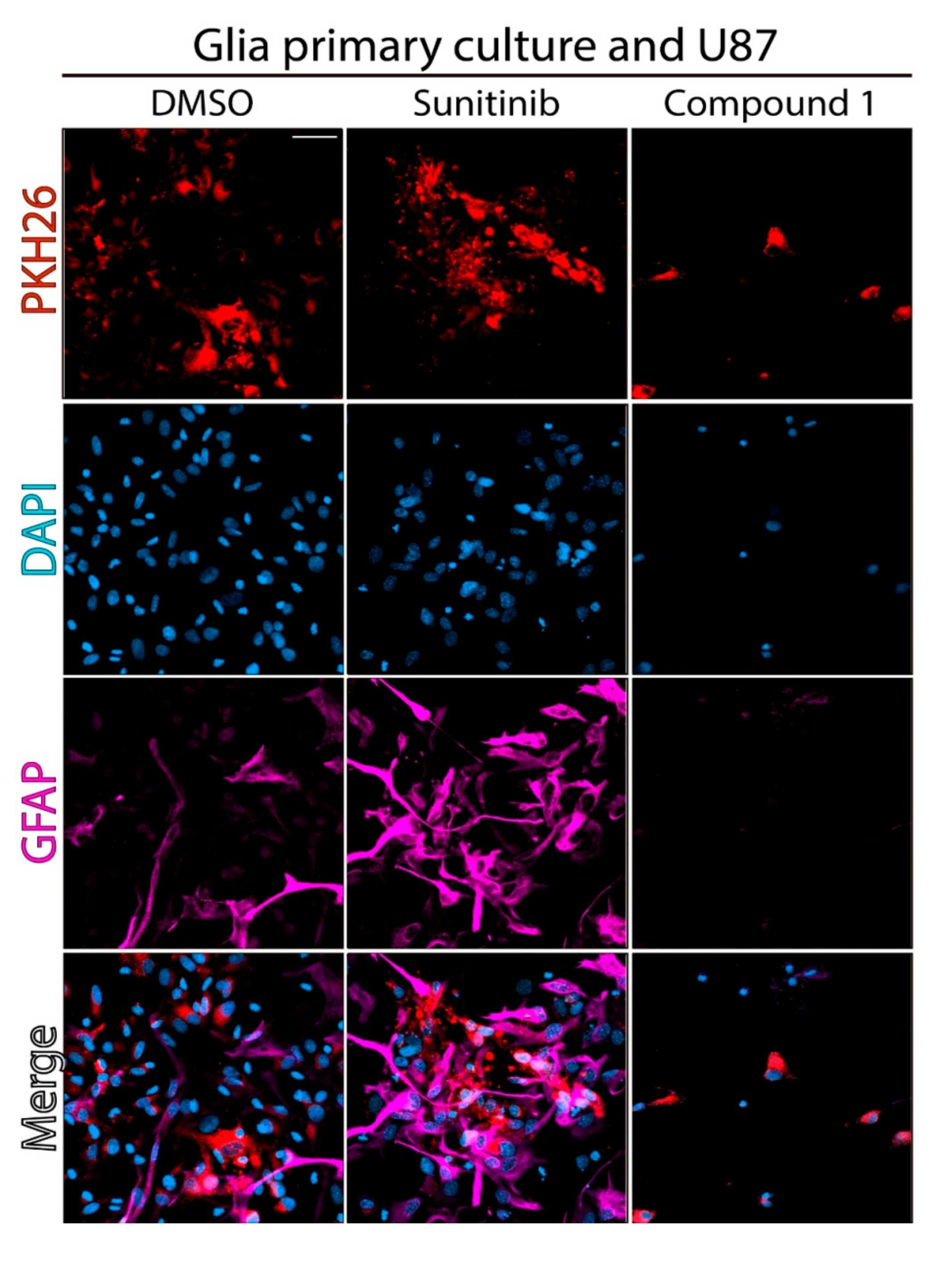
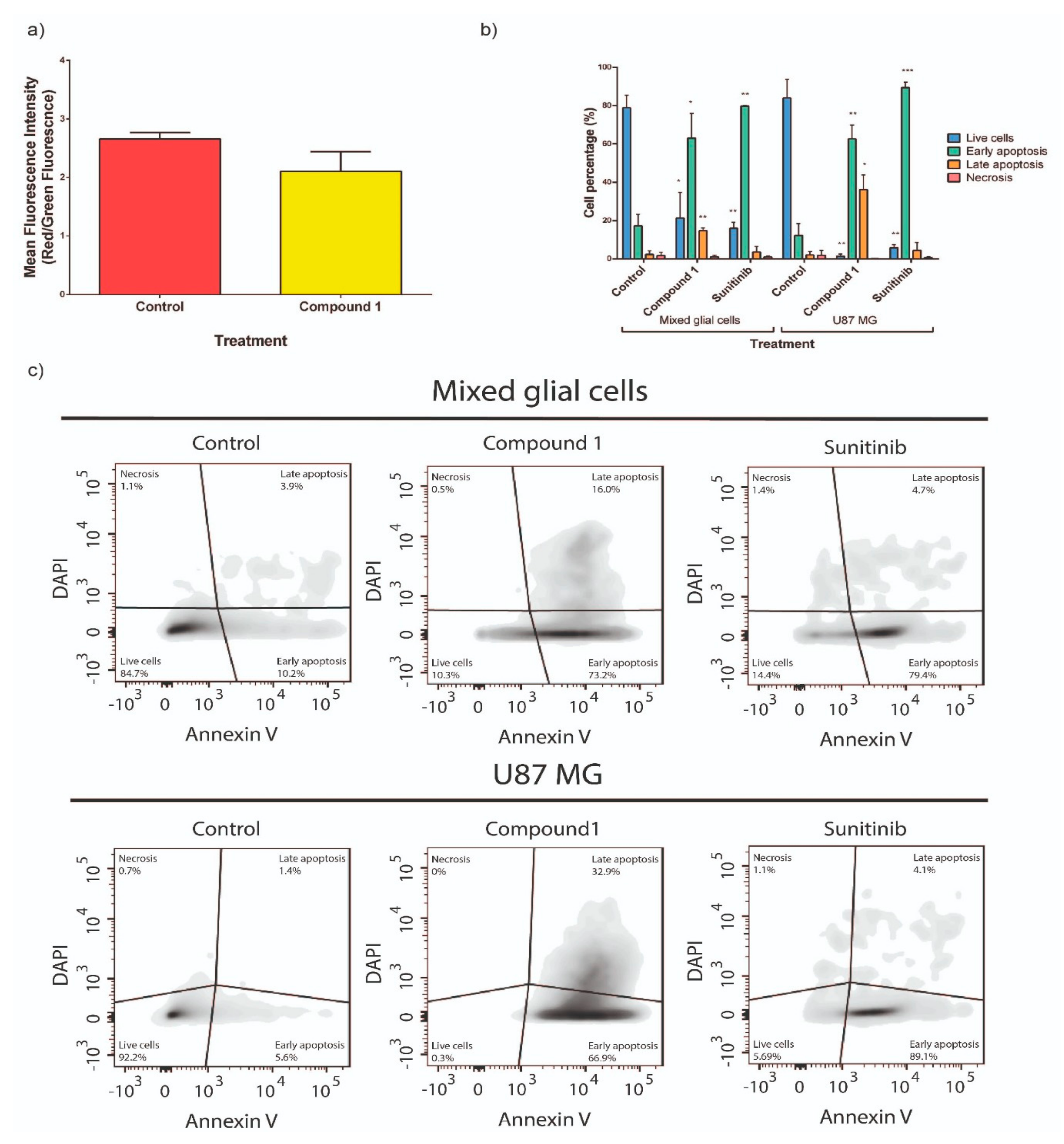
2.2. Effect of Compound 1 on Co-Cultures of Tumor Cells and Normal Astrocytes
2.3. Study of Cellular Death Mechanism Triggered by Compound 1
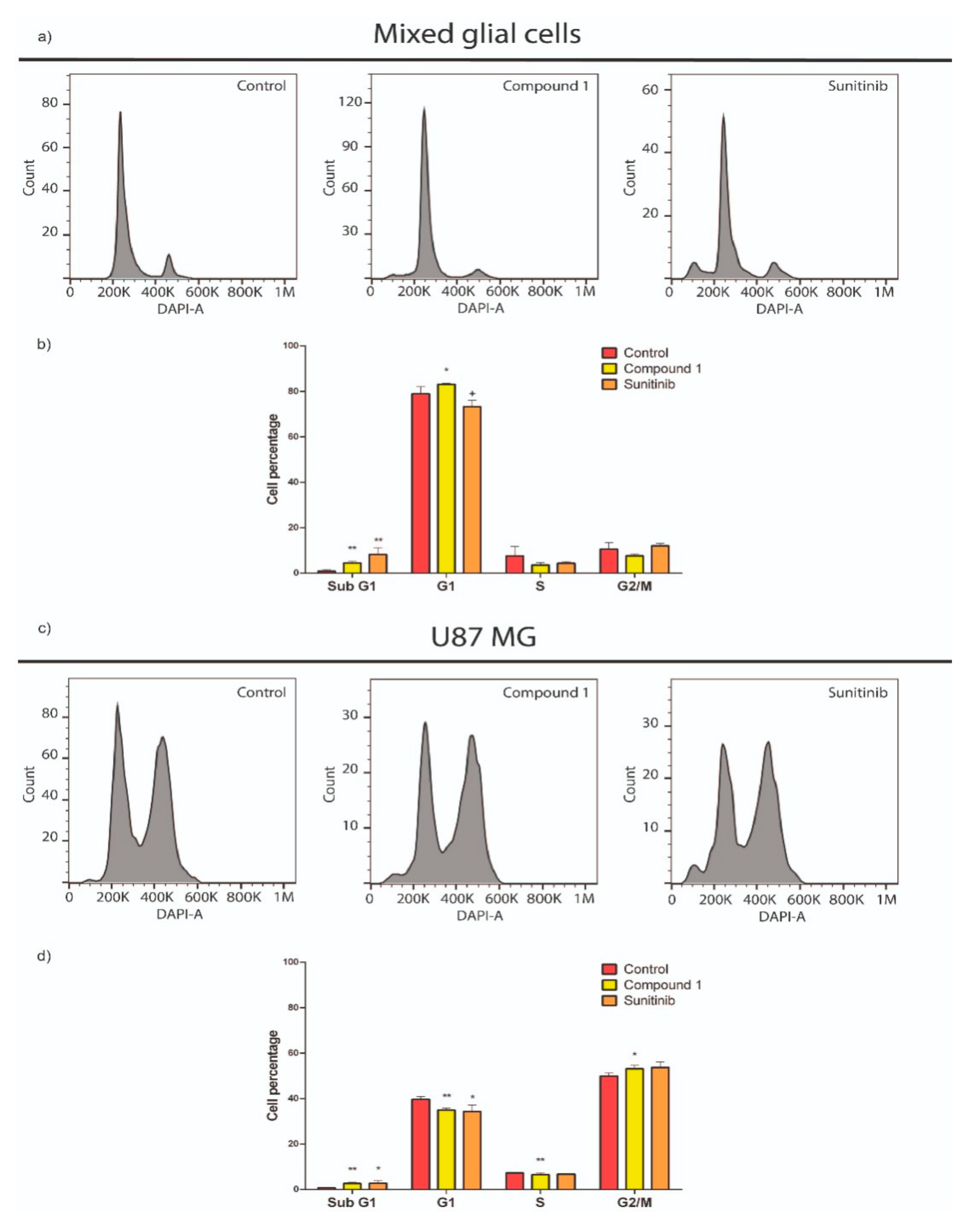
2.4. Compound 1’s Effect on the Cell Cycle
2.5. Drug-Like Properties of Compound 1
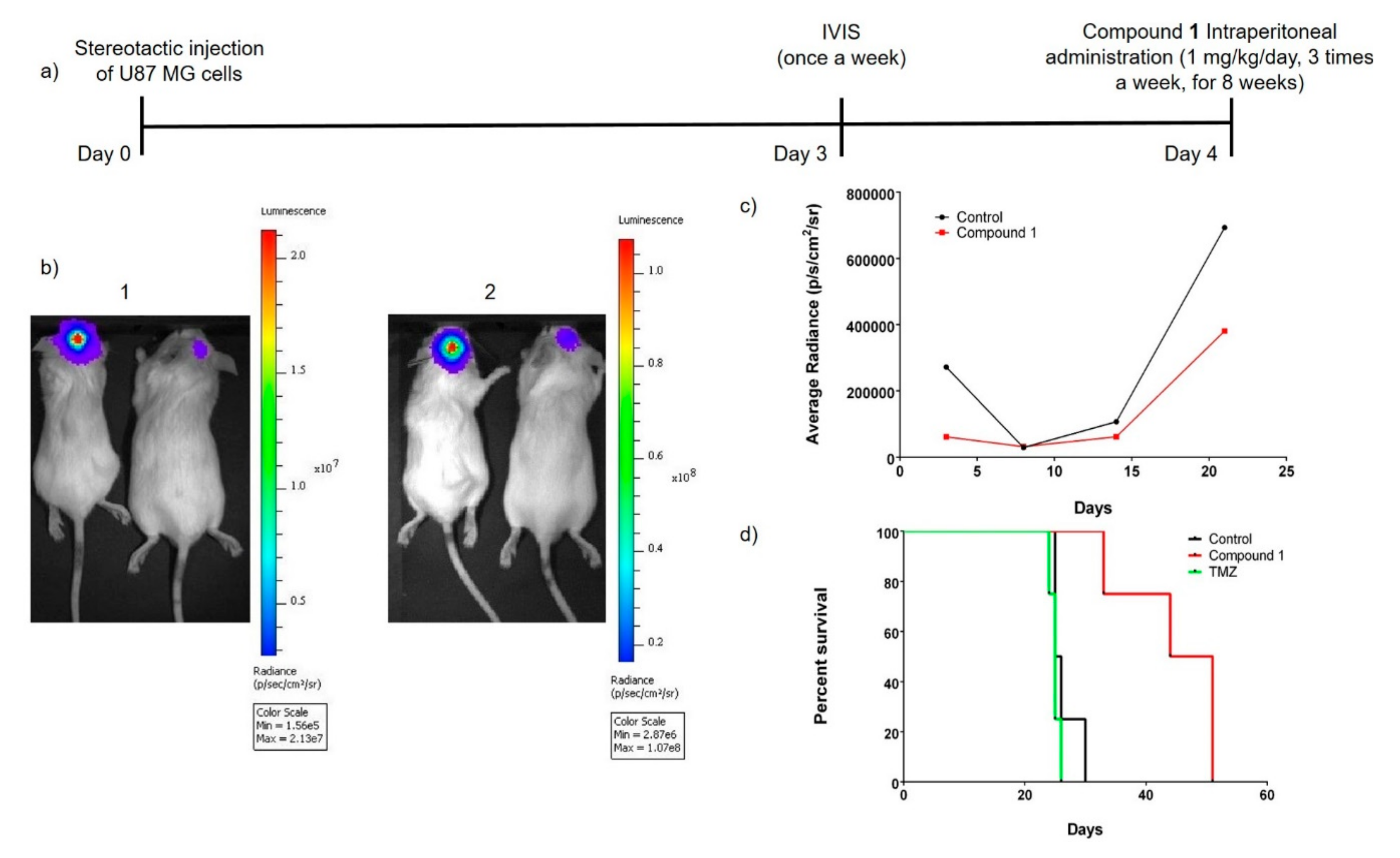
2.6. Study of the In Vivo Anti-Glioblastoma Activity of Compound 1
3. Discussion
4. Materials and Methods
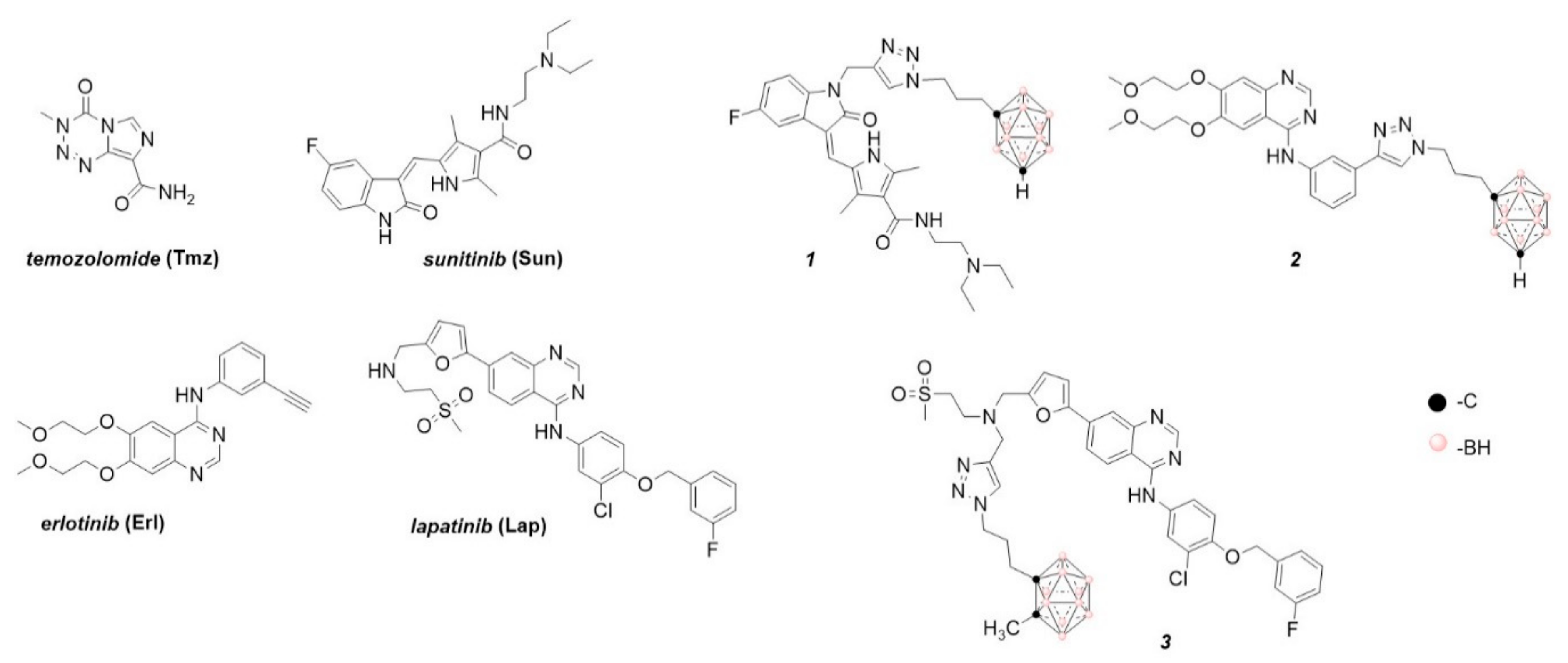
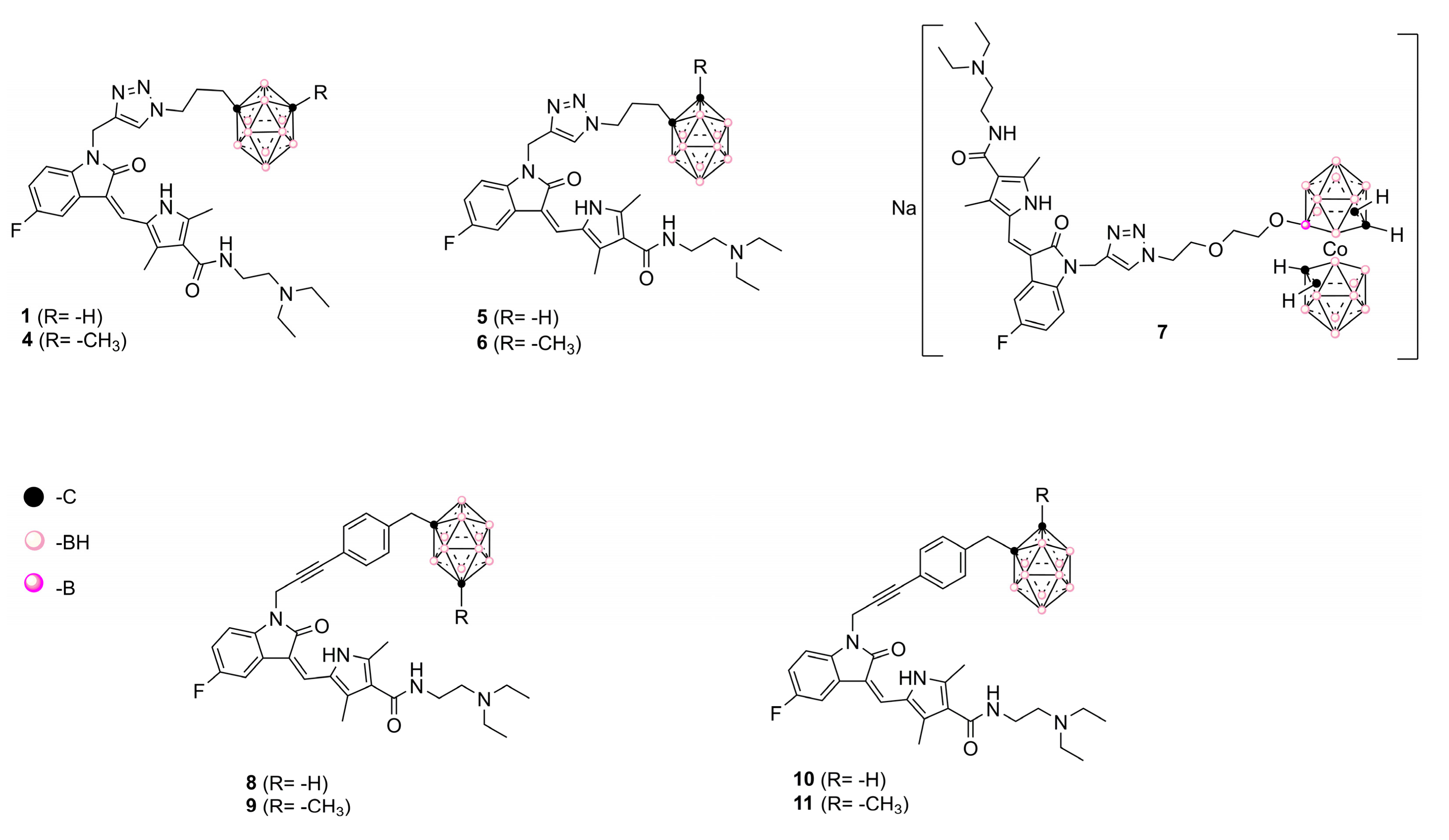
4.1. Chemistry
4.2. Biology
4.2.1. Kinases Assays
4.2.2. PDGFR-β and FLT3 Enzymatic Assays
4.2.3. Confocal Microscopy of Treated Co-Cultures of U87 MG and Astrocytes
4.2.4. Cellular Death Mechanisms
Collapse of the ΔΨm
Phosphatidylserine Exposure Assay
4.2.5. Measurement of Cell Cycle/DNA Content
4.3. Drug-Like Properties
4.3.1. Prediction
4.3.2. Ames Test
4.3.3. BBB In Vitro Passage Assay
BBB Artificial Model
Isolation and Culture of Porcine Brain Micro Vessel Endothelial Cells
Assembling of the Artificial BBB Model
BBB-Passage Protocol
4.4. In Vivo Anti-Glioblastoma Studies
4.4.1. Treatments
4.4.2. Anti-Tumor Evaluation
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fitzmaurice, C.; Dicker, D.; Pain, A.; Hamavid, H.; Moradi-Lakeh, M.; MacIntyre, M.F.; Allen, C.; Hansen, G.M.; Woodbrook, R.; Wolfe, C.; et al. The global burden of cancer 2013. JAMA Oncol. 2015, 1, 505–527. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Curschmann, J.; et al. Radiotherapy plus concomitant and adjuvant Temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III randomized trial comparing the efficacy of Cediranib as monotherapy, and in combination with Lomustine, versus Lomustine alone in patients with recurrent glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef]
- Lombardi, G.; Pambuku, A.; Bellu, L.; Farina, M.; Della Puppa, A.; Denaro, L.; Zagonel, V. Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit. Rev. Oncol./Hematol. 2017, 111, 94–102. [Google Scholar] [CrossRef]
- Goodwin, C.R.; Rath, P.; Oyinlade, O.; Lopez, H.; Mughal, S.; Xia, S.; Li, Y.; Kaur, H.; Zhou, X.; Ahmed, A.K.; et al. Crizotinib and Erlotinib inhibits growth of c-Met + /EGFRvIII + primary human glioblastoma xenografts. Clin. Neurol. Neurosurg. 2018, 171, 26–33. [Google Scholar] [CrossRef]
- Papaetis, G.S.; Syrigos, K.N. Sunitinib: A multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs 2009, 23, 377–389. [Google Scholar] [CrossRef]
- Giannopoulou, E.; Dimitropoulos, K.; Argyriou, A.A.; Koutras, A.K.; Dimitrakopoulos, F.; Kalofonos, H.P. An in vitro study, evaluating the effect of sunitinib and/or lapatinib on two glioma cell lines. Investig. New Drugs 2010, 28, 554–560. [Google Scholar] [CrossRef] [PubMed]
- de Bouard, S.; Herlin, P.; Christensen, J.G.; Lemoisson, E.; Gauduchon, P.; Raymond, E.; Guillamo, J.-S. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro-Oncol. 2007, 9, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Balaña, C.; Gil, M.J.; Perez, P.; Reynes, G.; Gallego, O.; Ribalta, T.; Capellades, J.; Gonzalez, S.; Verger, E. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: Results of a phase II study. Target Oncol. 2014, 9, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Nowosielski, M.; Haybaeck, J.; Embacher, S.; Stockhammer, F.; Gotwald, T.; Holzner, B.; Capper, D.; Preusser, M.; Marosi, C.; et al. A single-arm phase II Austrian/German multicenter trial on continuous daily sunitinib in primary glioblastoma at first recurrence (SURGE 01-07). Neuro-Oncol. 2014, 16, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Cerecetto, H.; Couto, M. Medicinal chemistry of boron-bearing compounds for BNCT-glioma treatment: Current challenges and perspectives. In Glioma: Contemporary Diagnostic and Therapeutic Approaches; Omerhodzic, I., Arnautovic, K., Eds.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Barth, R.F.; Vicente, M.H.; Harling, O.K.; Kiger, W.S.; Riley, K.J.; Binns, P.J.; Wagner, F.M.; Suzuki, M.; Aihara, T.; Kato, I.; et al. Current status of boron neutron capture therapy of high grade gliomas and recurrent head and neck cancer. Radiat. Oncol. 2012, 7, 1–21. [Google Scholar] [CrossRef]
- Capoulat, M.E.; Sauzet, N.; Valda, A.A.; Gagetti, L.; Guillaudin, O.; Lebreton, L.; Maire, D.; Mastinu, P.; Praena, J.; Riffard, Q.; et al. Neutron spectrometry of the 9Be(d (1.45 MeV), n)10B reaction for accelerator-based BNCT. Nucl. Instrum. Methods Phys. Res. B Beam Interact. Mater. At. 2019, 445, 57–62. [Google Scholar] [CrossRef]
- Kumada, H.; Naito, F.; Hasegawa, K.; Kobayashi, H.; Kurihara, T.; Takada, K.; Onishi, T.; Sakurai, K.; Matsumura, A.; Sakae, T. Development of LINAC-based neutron source for boron neutron capture therapy in University of Tsukuba. Plasma Fusion Res. 2018, 13, 2406006. [Google Scholar] [CrossRef]
- Gordillo, N.; Montseny, E.; Sobrevilla, P. State of the art survey on MRI brain tumor segmentation. Magn. Reson. Imaging 2013, 31, 1426–1438. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.R.; Stocchetti, N.; Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008, 7, 728–741. [Google Scholar] [CrossRef]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.; Mastandrea, I.; Cabrera, M.; Cabral, P.; Teixidor, F.; Cerecetto, H.; Vinas, C. Small-molecule kinase-inhibitors-loaded boron cluster as hybrid agents for glioma-cell-targeting therapy. Chem. Eur. J. 2017, 23, 9233–9238. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.; García, M.F.; Alamón, C.; Cabrera, M.; Cabral, P.; Merlino, A.; Teixidor, F.; Cerecetto, H.; Vinas, C. Discovery of potent EGFR inhibitors through the incorporation of a 3D-aromatic-boron-rich-cluster into the 4-anilinoquinazoline scaffold: Potential drugs for glioma treatment. Chem. Eur. J. 2018, 24, 3122–3126. [Google Scholar] [CrossRef]
- Couto, M.; Alamón, C.; Sánchez, C.; Dávila, B.; Fernández, M.; Lecot, N.; Cabral, P.; Teixidor, F.; Viñas, C.; Cerecetto, H. Carboranylanilinoquinazoline EGFR-inhibitors: Toward “lead-to-candidate” stage in the drug-development pipeline. Future Med. Chem. 2019, 11, 2273–2285. [Google Scholar] [CrossRef]
- Couto, M.; Alamón, C.; García, M.F.; Kovacs, M.; Trias, E.; Nievas, S.; Pozzi, E.C.; Curotto, P.; Thorp, S.I.; Dagrosa, M.A.; et al. Closo-carboranyl- and metallacarboranyl (1,2,3)triazolyl-decorated Lapatinib-scaffold for cancer therapy combining tyrosine kinase inhibition and boron neutron capture therapy. Cells 2020, 9, 1408. [Google Scholar] [CrossRef]
- Couto, M.; Alamón, C.; Nievas, S.; Perona, M.; Dagrosa, M.A.; Teixidor, F.; Cabral, P.; Vinas, C.; Cerecetto, H. Bimodal therapeutic agents against glioblastoma, one of the most lethal forms of cancer. Chem. Eur. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.D.; Vajkoczy, P.; Shawver, L.K.; Thurnher, A.; Liang, C.; Mohammadi, M.; Schlessinger, J.; Ullrich, A.; Hubbard, S.R.; Blake, R.A.; et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000, 60, 4152–4160. [Google Scholar] [PubMed]
- Sun, L.; Liang, C.; Shirazian, S.; Zhou, Y.; Miller, T.; Cui, J.; Fukuda, J.Y.; Chu, J.-Y.; Nematalla, A.; Wang, X.; et al. Discovery of 5-[5-Fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2003, 46, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Drevs, J.; Medinger, M.; Schmidt-Gersbach, C.; Weber, R.; Unger, C. Receptor tyrosine kinases: The main targets for new anticancer therapy. Curr. Drug Targets. 2003, 4, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis, and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Hückel’s rule of aromaticity categorizes aromatic closo boron hydride clusters. Chem. Eur. J. 2016, 22, 7437–7443. [Google Scholar] [CrossRef]
- Poater, J.; Viñas, C.; Bennour, I.; Escayola, S.; Solà, M.; Teixidor, F. Too persistent to give up: Aromaticity in boron clusters survives radical structural changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. [Google Scholar] [CrossRef]
- Fox, M.A.; Hughes, A.K. Cage C–H⋯X interactions in solid-state structures of icosahedral carboranes. Coord. Chem. Rev. 2004, 248, 457–476. [Google Scholar] [CrossRef]
- Donati, A.; Ristori, S.; Bonechi, C.; Panza, L.; Martini, G.; Rossi, C. Evidences of strong C−H⋯·O bond in an o-carboranyl β-lactoside in solution. J. Am. Chem. Soc. 2002, 124, 8778. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, H.; Yan, H.; Zou, W.; Cremer, D. B–H⋯π interaction: A new type of nonclassical hydrogen bonding. J. Am. Chem. Soc. 2016, 138, 4334–4337. [Google Scholar] [CrossRef]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer. 2010, 10, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Abbeele, A.D.V.D.; Druker, B.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Woulfe, K.C.; Durand, J.-B.; Vagnozzi, R.; Kramer, D.; Chu, T.F.; Beahm, C.; Chen, M.H.; Force, T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin. Transl. Sci. 2009, 2, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hasinoff, B.B.; Patel, D.; O’Hara, K.A. Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor Sunitinib. Mol. Pharmacol. 2008, 74, 1722–1728. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Latuske, E.-M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jucker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef]
- Nagao, H.; Ijiri, K.; Hirotsu, M.; Ishidou, Y.; Yamamoto, T.; Nagano, S.; Takizawa, T.; Nakashima, K.; Komiya, S.; Setoguchi, T. Role of GLI2 in the growth of human osteosarcoma. J. Pathol. 2011, 224, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Pan, L.; Che, X.; Cui, D.; Li, C. Gli1 inhibition induces cell-cycle arrest and enhanced apoptosis in brain glioma cell lines. J. Neuro-oncol. 2010, 98, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.B.; Prêle, C.M.; Baltic, S.; Arthur, P.G.; Creaney, J.; Watkins, D.N.; Thompson, P.J.; Mutsaers, S.E. Mitochondria-derived reactive oxygen species drive GANT61-induced mesothelioma cell apoptosis. Oncotarget 2015, 6, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhou, L.-F.; Yang, B.; Zhao, H.-J.; Wang, Y.-Q.; Li, X.-Y.; Ye, Y.-P.; Chen, F.-Y. The loss of a sugar chain at C(3) position enhances Stemucronatoside K-induced apoptosis, cell cycle arrest and hedgehog pathway inhibition in HT-29 cells. Chem. Biodivers. 2016, 13, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Organisation for Economic Co-Operation and Development (OECD). OECD Guidelines for Testing of Chemicals: Acute Oral Toxicity—Acute Toxic Class Method. OECD 423, 2001. Available online: https://ntp.niehs.nih.gov/iccvam/suppdocs/feddocs/oecd/oecd_gl423.pdf (accessed on 1 January 2020).
- Malina, K.C.K.; Cooper, I.; Teichberg, V.I. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Res. 2009, 1284, 12–21. [Google Scholar] [CrossRef]
- Aparicio-Blanco, J.; Romero, I.A.; Male, D.K.; Slowing, K.; García-García, L.; Torres-Suárez, A. Cannabidiol enhances the passage of lipid nanocapsules across the blood-brain barrier both in vitro and in vivo. Mol. Pharm. 2019, 16, 1999–2010. [Google Scholar] [CrossRef]
- Le Joncour, V.; Karaman, S.; Laakkonen, P.M. Predicting in vivo payloads delivery using a blood-brain tumor-barrier in a dish. J. Vis. Exp. 2019, 146, e59384. [Google Scholar] [CrossRef]
- Bhupathiraju, N.V.S.D.K.; Hu, X.; Zhou, Z.; Fronczek, F.R.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Vicente, M.G.H. Synthesis and in vitro evaluation of BBB permeability, tumor cell uptake, and cytotoxicity of a series of carboranylporphyrin conjugates. J. Med. Chem. 2014, 57, 6718–6728. [Google Scholar] [CrossRef]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Carbon-Mangels, M.; Hutter, M.C. Selecting relevant descriptors for classification by Bayesian estimates: A comparison with decision trees and support vector machines approaches for disparate data sets. Mol. Inform. 2011, 30, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.L.M.; Glen, R.C.; Mitchell, J.B.O. Development and comparison of hERG blocker classifiers: Assessment on different datasets yields markedly different results. Mol. Inform. 2011, 30, 443–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Wang, J.; Chen, L.; Zhang, L.; Yu, H.; Hou, T. ADMET evaluation in drug discovery. 12. Development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharm. 2012, 9, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Filimonov, D.; Zakharov, A.; Xie, W.; Huang, Y.; Zhu, F.; Shen, T.; Yalo, J.; Poroikov, V.V. Computer-aided prediction of rodent carcinogenicity by PASS and CISOC-PSCT. QSAR Comb. Sci. 2009, 28, 806–810. [Google Scholar] [CrossRef]
- Zhu, H.; Martin, T.M.; Ye, L.; Sedykh, A.; Young, D.M.; Tropsha, A. Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol. 2009, 22, 1913–1921. [Google Scholar] [CrossRef]
- Wang, J.; Krudy, G.; Hou, T.; Zhang, W.; Holland, G.; Xu, X. Development of reliable aqueous solubility models and their application in druglike analysis. J. Chem. Inf. Model. 2007, 47, 1395–1404. [Google Scholar] [CrossRef]
- Vainshtein, A.; Veenman, L.; Shterenberg, A.; Singh, S.; Masarwa, A.; Dutta, B.; Island, B.; Tsoglin, E.; Levin, E.; Leschiner, S.; et al. Quinazoline-based tricyclic compounds that regulate programmed cell death, induce neuronal differentiation, and are curative in animal models for excitotoxicity and hereditary brain disease. Cell Death Discov. 2015, 1, 15027. [Google Scholar] [CrossRef]
- Sosunov, A.; Wu, X.; McGovern, R.; Mikell, C.; McKhann, G.M.; Goldman, J.E. Abnormal mitosis in reactive astrocytes. Acta Neuropathol. Commun. 2020, 8, 47. [Google Scholar] [CrossRef]
- Zeno, S.; Zaaroor, M.; Leschiner, S.; Veenman, L.; Gavish, M. CoCl2 induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry 2009, 48, 4652–4661. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50,U87 MG(μM) 1,2 | IC50,C6(μM) 1,2 | IC50,HT-29 (μM) 1,2 | References |
|---|---|---|---|---|
| 1 | 8.0 ± 0.3 | 6.9 ± 0.5 | <6.25 (0.45 ± 0.04%) | [22] |
| Sun | 32 ± 4 | 36 ± 10 | 6.25 ± 0.04 | [18,20] |
| 2 | 70 ± 5 | 30 ± 5 | 25.0 ± 5.0 | [18,20] |
| Erl | 63 ± 5 | >100 | >100 3 | [18,20] |
| 3 | 10.0 ± 0.2 | 11.8 ± 0.4 | >100 (73 ± 6) | [21] |
| Lap | 54 ± 14 | >100 (89 ± 5) | 6.25 ± 0.05 | [21] |
| DiscoveRx Gene Symbol | Entrez Gene Symbol | Percent of Remaining Enzymatic Activity 2 |
|---|---|---|
| PDGFR-α | PDGFRA | 2.6 |
| PDGFR-β | PDGFRB | 0.05 |
| FLT1 3 | FLT1 | 14 |
| FLT3 | FLT3 | 2.1 |
| FLT3(D835V) | FLT3 | 0.1 |
| FLT3(ITD) | FLT3 | 2.3 |
| FLT3(ITD,D835V) | FLT3 | 0.55 |
| FLT3(ITD,F691L) | FLT3 | 0 |
| KIT | KIT | 0.25 |
| KIT(V559D) | KIT | 0.45 |
| KIT(V559D,T670I) | KIT | 1.4 |
| Cpd | HIA 1 | Subcellular Localization | CYP-Subs 2 | hERG 3 | Carc 4 | LD50 5 | LogS 6 | Viol, Rule 5 7 |
|---|---|---|---|---|---|---|---|---|
 | (+) | Mit 8 | 3A4 | w | (−) | 2.69 | −3.48 | 1 9 |
 | (+) | Mit | 3A4 | w | (−) | 2.66 | −3.24 | 0 |
 | (+) | Mit | - | w | (−) | 2.53 | −1.63 | 0 |

| Ames Test | BBB 1 | |||
|---|---|---|---|---|
| TA98 | TA100 | |||
| Dose (μg/plate) | Number of revertants | before BBB | [1] = 15 ± 5 μM | |
| Positive control 2 | 295 ± 11 | 704 ± 6 | after BBB | [1] = 48 ± 4 μM |
| 0.45 | 12 ± 1 | 67 ± 3 | ||
| 0.15 | 9 ± 1 | 56 ± 3 | ||
| 0.05 | 7.5 ± 1.5 | 46.5 ± 1.5 | ||
| 0.016 | 6 ± 1 | 37 ± 6 | ||
| 0.005 | 8 ± 1 | 29.5 ± 0.5 | ||
| 0 3 | 12 ± 2 | 95.6 ± 5.7 | ||
| Mutagenic | (−) | (−) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alamón, C.; Dávila, B.; García, M.F.; Sánchez, C.; Kovacs, M.; Trias, E.; Barbeito, L.; Gabay, M.; Zeineh, N.; Gavish, M.; et al. Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity. Cancers 2020, 12, 3423. https://doi.org/10.3390/cancers12113423
Alamón C, Dávila B, García MF, Sánchez C, Kovacs M, Trias E, Barbeito L, Gabay M, Zeineh N, Gavish M, et al. Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity. Cancers. 2020; 12(11):3423. https://doi.org/10.3390/cancers12113423
Chicago/Turabian StyleAlamón, Catalina, Belén Dávila, María Fernanda García, Carina Sánchez, Mariángeles Kovacs, Emiliano Trias, Luis Barbeito, Martín Gabay, Nidal Zeineh, Moshe Gavish, and et al. 2020. "Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity" Cancers 12, no. 11: 3423. https://doi.org/10.3390/cancers12113423
APA StyleAlamón, C., Dávila, B., García, M. F., Sánchez, C., Kovacs, M., Trias, E., Barbeito, L., Gabay, M., Zeineh, N., Gavish, M., Teixidor, F., Viñas, C., Couto, M., & Cerecetto, H. (2020). Sunitinib-Containing Carborane Pharmacophore with the Ability to Inhibit Tyrosine Kinases Receptors FLT3, KIT and PDGFR-β, Exhibits Powerful In Vivo Anti-Glioblastoma Activity. Cancers, 12(11), 3423. https://doi.org/10.3390/cancers12113423