Shaping the Treatment Paradigm Based on the Current Understanding of the Pathobiology of Multiple Myeloma: An Overview

Abstract
:Simple Summary
Abstract
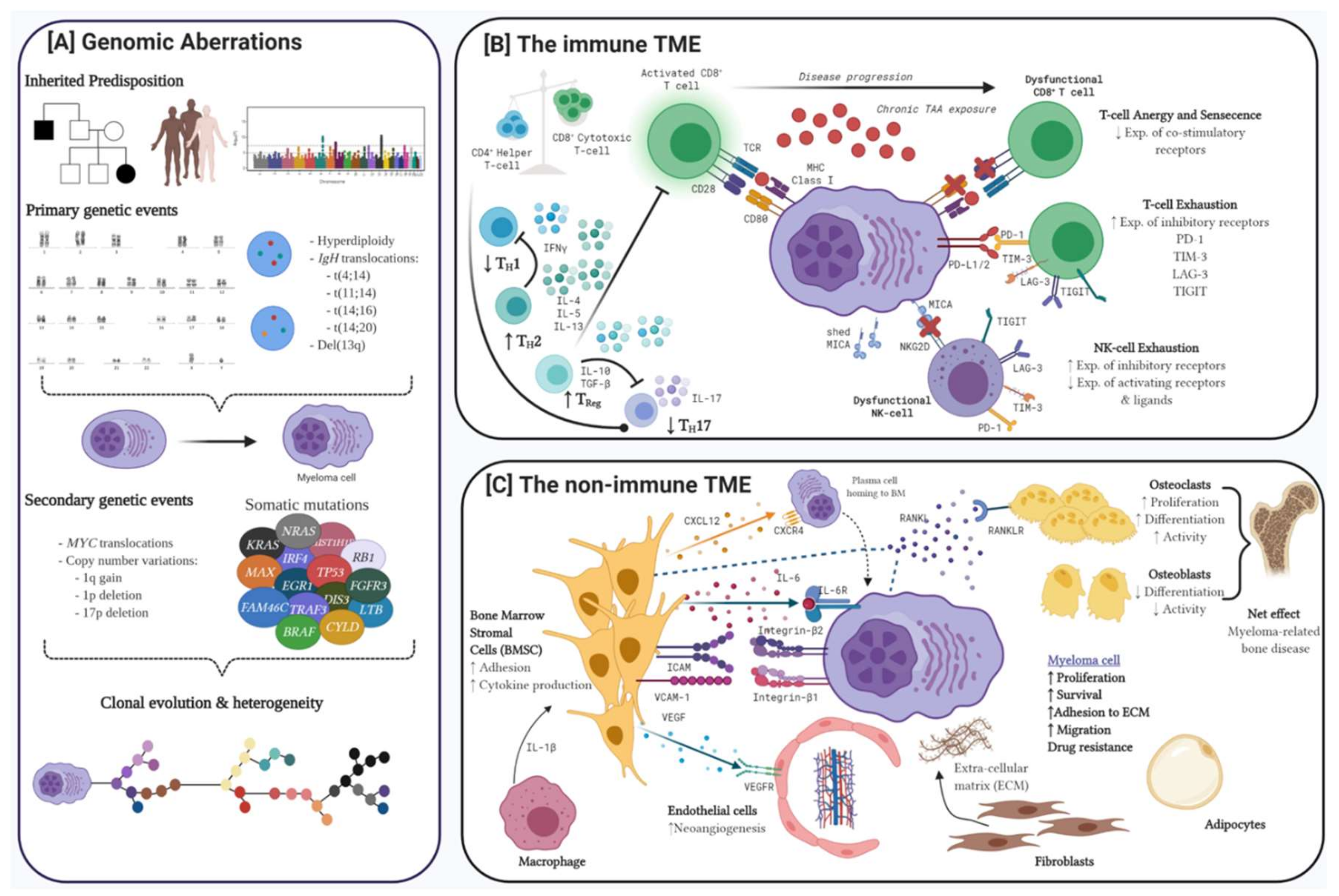
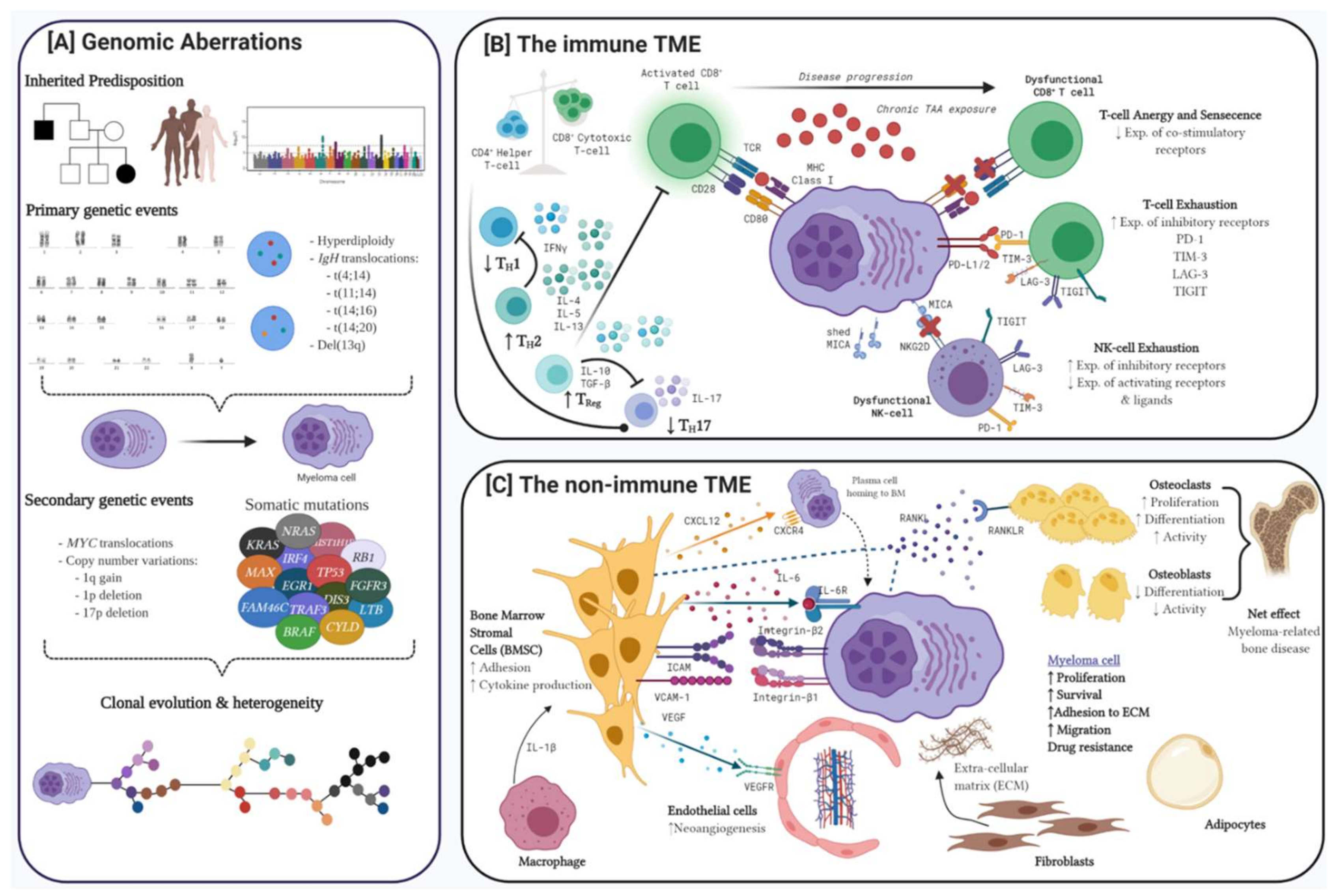
1. Advances on the Pathobiology of Myeloma
1.1. Genomic Aberrations in MM
1.1.1. Hyperdiploidy
1.1.2. Chromosomal Translocations
1.1.3. Copy Number Variations
1.1.4. Somatic Mutations
1.2. The Immune Tumour Microenvironment
1.2.1. T-cell Dysregulation in Myeloma
1.2.2. NK-cell Dysregulation in Myeloma
1.3. The Non-Immune Tumour Microenvironment
2. Advances on the Treatment of Multiple Myeloma
2.1. Treatment of the Newly Diagnosed MM Patient
2.1.1. Induction Therapy
TE-NDMM
TNE-NDMM
2.1.2. Consolidation and/or Maintenance
2.2. Treatment of the Relapsed/Refractory MM Patient
2.2.1. Early Relapse
2.2.2. Later Relapse
Novel Immunotherapies—ADCs, bsAbs, CAR T-cells
Small Molecules—Selinexor, Venetoclax and Iberdomide
3. Future Direction and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Morgan, G.J.; Johnson, D.C.; Weinhold, N.; Goldschmidt, H.; Landgren, O.; Lynch, H.T.; Hemminki, K.; Houlston, R.S. Inherited genetic susceptibility to multiple myeloma. Leukemia 2013, 28, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.S.; Li, N.; Weinhold, N.; Försti, A.; Ali, M.; Van Duin, M.; Thorleifsson, G.; Johnson, D.C.; Chen, B.; Halvarsson, B.-M.; et al. Genome-wide association study identifies multiple susceptibility loci for multiple myeloma. Nat. Commun. 2016, 7, 12050. [Google Scholar] [CrossRef] [PubMed]
- Chesi, M.; Bergsagel, P.L. Advances in the pathogenesis and diagnosis of multiple myeloma. Int. J. Lab. Hematol. 2015, 37, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Smadja, N.V.; Bastard, C.; Brigaudeau, C.; Leroux, D.; Fruchart, C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood 2001, 98, 2229–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyn, C.; Melchor, L.; Murison, A.; Wardell, C.P.; Brioli, A.; Boyle, E.M.; Kaiser, M.F.; Walker, B.A.; Begum, D.B.; Dahir, N.B.; et al. Coexistent hyperdiploidy does not abrogate poor prognosis in myeloma with adverse cytogenetics and may precede IGH translocations. Blood 2015, 125, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Chretien, M.-L.; Corre, J.; Lauwers-Cances, V.; Magrangeas, F.; Cleynen, A.; Yon, E.; Hulin, C.; Leleu, X.; Orsini-Piocelle, F.; Blade, J.-S.; et al. Understanding the role of hyperdiploidy in myeloma prognosis: Which trisomies really matter? Blood 2015, 126, 2713–2719. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, R.; Blood, E.; Rue, M.; Harrington, D.; Oken, M.; Kyle, R.A.; Dewald, G.W.; Van Ness, B.; Van Wier, S.A.; Henderson, K.J.; et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood 2003, 101, 4569–4575. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Sloan, S.; Li, D.; Zhuang, L.; Yi, Q.-L.; Chen, C.I.; Reece, D.; Chun, K.; Stewart, A.K. The t(4;14) is associated with poor prognosis in myeloma patients undergoing autologous stem cell transplant. Br. J. Haematol. 2004, 125, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.G.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Avet-Loiseau, H.; Leleu, X.; Roussel, M.; Moreau, P.; Guerin-Charbonnel, C.; Caillot, D.; Marit, G.; Benboubker, L.; Voillat, L.; Mathiot, C.; et al. Bortezomib Plus Dexamethasone Induction Improves Outcome of Patients With t(4;14) Myeloma but Not Outcome of Patients With del(17p). J. Clin. Oncol. 2010, 28, 4630–4634. [Google Scholar] [CrossRef]
- Mina, R.; Joseph, N.S.; Gay, F.; Kastritis, E.; Petrucci, M.T.; Kaufman, J.L.; Montefusco, V.; Gavriatopoulou, M.; Patriarca, F.; Omedé, P.; et al. Clinical features and survival of multiple myeloma patients harboring t(14;16) in the era of novel agents. Blood Cancer J. 2020, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Attal, M.; Moreau, P.; Charbonnel, C.; Garban, F.; Hulin, C.; Leyvraz, S.; Michallet, M.; Yakoub-Agha, I.; Garderet, L.; et al. Genetic abnormalities and survival in multiple myeloma: The experience of the Intergroupe Francophone du Myélome. Blood 2007, 109, 3489–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Popkova, T.; Benkova, K.; Richterova, P.; Plonkova, H.; Zuchnicka, J.; Broskevicova, L.; et al. Single-agent venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am. J. Hematol. 2018, 94, E35–E37. [Google Scholar] [CrossRef]
- Touzeau, C.; Dousset, C.; Le Gouill, S.; Sampath, D.; Leverson, J.D.; Souers, A.J.; Maïga, S.; Béné, M.C.; Moreau, P.; Pellatdeceunynck, C.; et al. The Bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia 2014, 28, 210–212. [Google Scholar] [CrossRef]
- Touzeau, C.; Maciag, P.; Amiot, M.; Moreau, P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia 2018, 32, 1899–1907. [Google Scholar] [CrossRef]
- Walker, A.B.; Wardell, C.P.; Brioli, A.; Boyle, E.M.; Kaiser, M.F.; Begum, D.B.; Dahir, N.B.; Johnson, D.C.; Ross, F.M.; Davies, F.E.; et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014, 4, e191. [Google Scholar] [CrossRef]
- Franssen, L.E.; Nijhof, I.S.; Couto, S.; Levin, M.-D.; Bos, G.M.; Broijl, A.; Klein, S.K.; Ren, Y.; Wang, M.; Koene, H.R.; et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematology 2018, 103, e368–e371. [Google Scholar] [CrossRef]
- Saxe, D.; Seo, E.-J.; Bergeron, M.B.; Han, J.-Y. Recent advances in cytogenetic characterization of multiple myeloma. Int. J. Lab. Hematol. 2019, 41, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Xu, Y.; Shi, L.; Shizhen, Z.; Deng, S.; Xie, Z.; Sui, W.; Zhan, F.; Qiu, L. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica 2013, 99, 353–359. [Google Scholar] [CrossRef]
- Schmidt, T.M.; Barwick, B.G.; Joseph, N.; Heffner, L.T.; Hofmeister, C.C.; Bernal, L.; Dhodapkar, M.V.; Gupta, V.A.; Jaye, D.L.; Wu, J.; et al. Gain of Chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.D.; Ross, F.M.; Walker, B.A.; Wardell, C.P.; Tapper, W.J.; Chiecchio, L.; Dagrada, G.; Konn, Z.J.; Gregory, W.M.; Jackson, G.H.; et al. Mapping of Chromosome 1p Deletions in Myeloma Identifies FAM46C at 1p12 and CDKN2C at 1p32.3 as Being Genes in Regions Associated with Adverse Survival. Clin. Cancer Res. 2011, 17, 7776–7784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, M.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Hayman, S.R.; Hwa, Y.L.; et al. Prognostic implications of abnormalities of chromosome 13 and the presence of multiple cytogenetic high-risk abnormalities in newly diagnosed multiple myeloma. Blood Cancer J. 2017, 7, e600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Leone, P.E.; Chiecchio, L.; Dickens, N.J.; Jenner, M.W.; Boyd, K.; Johnson, D.C.; Gonzalez, D.; Dagrada, G.P.; Protheroe, R.K.M.; et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood 2010, 116, e56–e65. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Painuly, U.; Rajkumar, S.V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; Dingli, D.; et al. Natural history of multiple myeloma with de novo del(17p). Blood Cancer J. 2019, 9, 32. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Tian, E.; Qu, P.; Mathur, P.; Schinke, C.; Van Rhee, F.; Zangari, M.; Rasche, L.; Weinhold, N.; Alapat, D.; et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica 2017, 102, e364–e367. [Google Scholar] [CrossRef] [Green Version]
- Perrot, A.; Lauwers-Cances, V.; Tournay, E.; Hulin, C.; Chretien, M.-L.; Royer, B.; Dib, M.; Decaux, O.; Jaccard, A.; Belhadj, K.; et al. Development and Validation of a Cytogenetic Prognostic Index Predicting Survival in Multiple Myeloma. J. Clin. Oncol. 2019, 37, 1657–1665. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef]
- Egan, J.B.; Shi, C.-X.; Tembe, W.; Christoforides, A.; Kurdoglu, A.; Sinari, S.; Middha, S.; Asmann, Y.; Schmidt, J.; Braggio, E.; et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012, 120, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal evolution in myeloma: The impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019, 104, 1440–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, G.H.; Davies, F.; Pawlyn, C.; Cairns, D.A.; Striha, A.; Collett, C.; Hockaday, A.; Jones, J.R.; Kishore, B.; Garg, M.; et al. Lenalidomide maintenance versus observation for patients with newly diagnosed multiple myeloma (Myeloma XI): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, N.U.; Sperling, A.S.; Bolli, N.; Wedge, D.C.; Van Loo, P.; Tai, Y.-T.; Shammas, M.A.; Fulciniti, M.; Samur, M.K.; Richardson, P.G.; et al. Differential and limited expression of mutant alleles in multiple myeloma. Blood 2014, 124, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Dosani, T.; Carlsten, M.; Maric, I.; Landgren, O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J. 2015, 5, e306. [Google Scholar] [CrossRef] [Green Version]
- Ogawara, H.; Handa, H.; Yamazaki, T.; Toda, T.; Yoshida, K.; Nishimoto, N.; Al-Ma’Quol, W.H.S.; Kaneko, Y.; Matsushima, T.; Tsukamoto, N.; et al. High Th1/Th2 ratio in patients with multiple myeloma. Leuk. Res. 2005, 29, 135–140. [Google Scholar] [CrossRef]
- Raitakari, M.; Brown, R.D.; Sze, D.; Yuen, E.; Barrow, L.; Nelson, M.; Pope, B.; Esdale, W.; Gibson, J.; Joshua, D.E. T-cell expansions in patients with multiple myeloma have a phenotype of cytotoxic T cells. Br. J. Haematol. 2000, 110, 203–209. [Google Scholar] [CrossRef]
- Almeida, M.M.-A.; Maria, J.M.; Guillermo, M.-N.; Josefina, G.; Jose, H.; Gema, M.; Jesus, F.S.M.; Alberto, O. Characterization of bone marrow T cells in monoclonal gammopathy of undetermined significance, multiple myeloma, and plasma cell leukemia demonstrates increased infiltration by cytotoxic/Th1 T cells demonstrating a squed TCR-Vβ repertoire. Cancer 2006, 106, 1296–1305. [Google Scholar] [CrossRef]
- Dhodapkar, M.V.; Krasovsky, J.; Osman, K.; Geller, M.D. Vigorous Premalignancy-specific Effector T Cell Response in the Bone Marrow of Patients with Monoclonal Gammopathy. J. Exp. Med. 2003, 198, 1753–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Magalhães, R.J.P.; Vidriales, M.B.; Paiva, B.; Fernández, C.; García-Sanz, R.; Mateos, M.-V.; Gutiérrez, N.C.; Lecrevisse, Q.; Blanco, J.F.; Hernández, J.; et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 2012, 98, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; Mateos, M.V.; Sanchez-Abarca, L.I.; Puig, N.; Vidriales, M.-B.; López-Corral, L.; Sánchez, L.A.C.; Hernandez, M.T.; Bargay, J.; De Arriba, F.; et al. Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: A longitudinal analysis. Blood 2016, 127, 1151–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Chen, S.; Huang, J.; Chen, Y.; Yang, L.; Wang, C.; Zhong, J.; Lu, Y.; Wang, L.; Zhu, K.; et al. Increased exhausted CD8+T cells with programmed death-1, T-cell immunoglobulin and mucin-domain-containing-3 phenotype in patients with multiple myeloma. Asia-Pacific J. Clin. Oncol. 2018, 14, e266–e274. [Google Scholar] [CrossRef]
- Woyach, J.A.; Pennell, M.; Huang, Y.; Benson, D.M.; Efebera, Y.A.; Chaudhry, M.; Hughes, T.; Woyach, J.A.; Byrd, J.C.; Zhang, S.; et al. T Cell Transcriptional Profiling and Immunophenotyping Uncover LAG3 as a Potential Significant Target of Immune Modulation in Multiple Myeloma. Biol. Blood Marrow Transplant. 2020, 26, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Khan, R.; Joshi, S.; Kumar, L.; Sharma, A. Dysregulation in T helper 1/T helper 2 cytokine ratios in patients with multiple myeloma. Leuk. Lymphoma 2010, 51, 920–927. [Google Scholar] [CrossRef]
- Noonan, K.; Marchionni, L.; Anderson, J.; Pardoll, D.; Roodman, G.D.; Borrello, I. A novel role of IL-17–producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood 2010, 116, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Beyer, M.; Kochanek, M.; Giese, T.; Endl, E.; Weihrauch, M.R.; Knolle, P.A.; Classen, S.; Schultze, J.L. In vivo peripheral expansion of naive CD4+CD25highFoxP3+ regulatory T cells in patients with multiple myeloma. Blood 2006, 107, 3940–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2013, 55, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, K.; Kaminska, W.; Hus, I.; Dmoszynska, A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: Detailed characterisation of immune status in multiple myeloma. Br. J. Cancer 2012, 106, 546–552. [Google Scholar] [CrossRef]
- Yang, J.; Chu, Y.; Yang, X.; Gao, D.; Zhu, L.; Yang, X.; Wan, L.; Li, M. Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.M.; Devarakonda, S.; Bumma, N.; Chaudhry, M.; Benson, D.M. Potential of NK cells in multiple Myeloma therapy. Expert Rev. Hematol. 2019, 12, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Vanneman, M.; Munshi, N.C.; Tai, Y.-T.; Prabhala, R.H.; Ritz, J.; Neuberg, D.; Anderson, K.C.; Carrasco, D.R.; Dranoff, G. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc. Natl. Acad. Sci. USA 2008, 105, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Costelloa, R.; Boehrer, A.; Sanchez, C.; Mercier, D.; Baier, C.; Le Treut, T.; Sébahoun, G. Differential expression of natural killer cell activating receptors in blood versus bone marrow in patients with monoclonal gammopathy. Immunology 2013, 139, 338–341. [Google Scholar] [CrossRef]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti–PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058. [Google Scholar] [CrossRef] [Green Version]
- Ullah, T.R. The role of CXCR4 in multiple myeloma: Cells’ journey from bone marrow to beyond. J. Bone Oncol. 2019, 17, 100253. [Google Scholar] [CrossRef]
- Liang, X.; Song, E. The role of bone marrow stromal cells in blood diseases and clinical significance as a crucial part of the hematopoietic microenvironment. Ann. Blood 2020, 5, 2. [Google Scholar] [CrossRef]
- Markovina, S.; Callander, N.; O’Connor, S.L.; Xu, G.; Shi, Y.; Leith, C.; Kim, K.; Trivedi, P.; Kim, J.; Hematti, P.; et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol. Cancer 2010, 9, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, G.; Hazlehurst, L.; Lynch, C.C. Dissecting the multiple myeloma-bone microenvironment reveals new therapeutic opportunities. J. Mol. Med. 2016, 94, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment approaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [Green Version]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.P.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J., Jr.; Anderson, L.D.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.C.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Landgren, O.; Devlin, S.; Boulad, M.; Mailankody, S. Role of MRD status in relation to clinical outcomes in newly diagnosed multiple myeloma patients: A meta-analysis. Bone Marrow Transplant. 2016, 51, 1565–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of Minimal Residual Disease with Superior Survival Outcomes in Patients With Multiple Myeloma. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Corre, J.; Lauwers-Cances, V.; Chretien, M.-L.; Robillard, E.N.; Leleu, X.; Hulin, C.; Gentil, E.C.; Arnulf, B.; Belhadj, K.; et al. Evaluation of Minimal Residual Disease (MRD) By Next Generation Sequencing (NGS) Is Highly Predictive of Progression Free Survival in the IFM/DFCI 2009 Trial. Blood 2015, 126, 191. [Google Scholar] [CrossRef]
- Paiva, B.; Cedena, M.-T.; Puig, N.; Arana, P.; Vidriales, M.B.; Cordon, L.; Flores-Montero, J.; Gutierrez, N.C.; Martín-Ramos, M.-L.; Martinez-Lopez, J.; et al. Minimal residual disease monitoring and immune profiling in multiple myeloma in elderly patients. Blood 2016, 127, 3165–3174. [Google Scholar] [CrossRef]
- Cavo, M.; Terpos, E.; Nanni, C.; Moreau, P.; Lentzsch, S.; Zweegman, S.; Hillengass, J.; Engelhardt, M.; Usmani, S.Z.; Vesole, D.H.; et al. Role of 18F-FDG PET/CT in the diagnosis and management of multiple myeloma and other plasma cell disorders: A consensus statement by the International Myeloma Working Group. Lancet Oncol. 2017, 18, e206–e217. [Google Scholar] [CrossRef]
- Pugh, T.J. Circulating Tumour DNA for Detecting Minimal Residual Disease in Multiple Myeloma. Semin. Hematol. 2018, 55, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Thoren, K.L. Mass spectrometry methods for detecting monoclonal immunoglobulins in multiple myeloma minimal residual disease. Semin. Hematol. 2018, 55, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Cavo, M.; Gay, F.; Beksac, M.; Pantani, L.; Petrucci, M.T.; Dimopoulos, M.A.; Dozza, L.; Van Der Holt, B.; Zweegman, S.; Oliva, S.; et al. Autologous haematopoietic stem-cell transplantation versus bortezomib–melphalan–prednisone, with or without bortezomib–lenalidomide–dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): A multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020, 7, e456–e468. [Google Scholar] [CrossRef]
- Quach, H.; Joshua, D.; Ho, J.; Szer, J.; Spencer, A.; Harrison, S.J.; Mollee, P.; Roberts, A.W.; Horvath, N.; Talulikar, D.; et al. Treatment of patients with multiple myeloma who are eligible for stem cell transplantation: Position statement of the Myeloma Foundation of Australia Medical and Scientific Advisory Group. Intern. Med. J. 2015, 45, 94–105. [Google Scholar] [CrossRef]
- Mina, R.; Bringhen, S.; Wildes, T.M.; Zweegman, S.; Rosko, A.E. Approach to the Older Adult with Multiple Myeloma. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Rosiñol, L.; Oriol, A.; Rios, R.; Sureda, A.; Blanchard, M.J.; Hernández, M.T.; Martínez-Martínez, R.; Moraleda, J.M.; Jarque, I.; Bargay, J.; et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood 2019, 134, 1337–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Jacobus, S.J.; Cohen, A.D.; Weiss, M.; Callander, N.S.; Singh, A.A.; Parker, T.L.; Menter, A.R.; Yang, X.; Parsons, B.M.; et al. Carfilzomib, lenalidomide, and dexamethasone (KRd) versus bortezomib, lenalidomide, and dexamethasone (VRd) for initial therapy of newly diagnosed multiple myeloma (NDMM): Results of ENDURANCE (E1A11) phase III trial. J. Clin. Oncol. 2020, 38, LBA3. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Costa, L.J.; Chhabra, S.; Godby, K.N.; Medvedova, E.; Cornell, R.F.; Hall, A.C.; Silbermann, R.W.; Innis-Shelton, R.; Dhakal, B.; DeIdiaquez, D.; et al. Daratumumab, Carfilzomib, Lenalidomide and Dexamethasone (Dara-KRd) Induction, Autologous Transplantation and Post-Transplant, Response-Adapted, Measurable Residual Disease (MRD)-Based Dara-Krd Consolidation in Patients with Newly Diagnosed Multiple Myeloma (NDMM). Blood 2019, 134, 860. [Google Scholar] [CrossRef]
- Weisel, K.; Asemissen, A.M.; Besemer, B.; Haenel, M.; Blau, I.W.; Goerner, M.; Ko, Y.-D.; Dürig, J.; Staib, P.; Mann, C.; et al. Depth of response to isatuximab, carfilzomib, lenalidomide, and dexamethasone (Isa-KRd) in front-line treatment of high-risk multiple myeloma: Interim analysis of the GMMG-CONCEPT trial. J. Clin. Oncol. 2020, 38, 8508. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Ailawadhi, S.; Sexton, R.; Hoering, A.; Lipe, B.; Hita, S.; Durie, B.G.; Zonder, J.A.; Dhodapkar, M.V.; Callander, N.S.; et al. Primary analysis of the randomized phase II trial of bortezomib, lenalidomide, dexamthasone with/without elotuzumab for newly diagnosed, high-risk multiple myeloma (SWOG-1211). J. Clin. Oncol. 2020, 38, 8507. [Google Scholar] [CrossRef]
- Facon, T.; Dimopoulos, M.A.; Dispenzieri, A.; Catalano, J.V.; Belch, A.; Cavo, M.; Pinto, A.; Weisel, K.; Ludwig, H.; Bahlis, N.J.; et al. Final analysis of survival outcomes in the phase 3 FIRST trial of up-front treatment for multiple myeloma. Blood 2018, 131, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Durie, B.G.M.; Hoering, A.; Sexton, R.; Abidi, M.H.; Epstein, J.; Rajkumar, S.V.; Dispenzieri, A.; Kahanic, S.P.; Thakuri, M.C.; Reu, F.J.; et al. Longer term follow-up of the randomized phase III trial SWOG S0777: Bortezomib, lenalidomide and dexamethasone vs. lenalidomide and dexamethasone in patients (Pts) with previously untreated multiple myeloma without an intent for immediate autologous stem cell transplant (ASCT). Blood Cancer J. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Gay, F.; Cerrato, C.; Scalabrini, D.R.; Galli, M.; Belotti, A.; Zamagni, E.; Ledda, A.; Grasso, M.; Angelucci, E.; Liberati, A.M.; et al. Carfilzomib-Lenalidomide-Dexamethasone (KRd) Induction-Autologous Transplant (ASCT)-Krd Consolidation Vs KRd 12 Cycles Vs Carfilzomib-Cyclophosphamide-Dexamethasone (KCd) Induction-ASCT-KCd Consolidation: Analysis of the Randomized Forte Trial in Newly Diagnosed Multiple Myeloma (NDMM). Blood 2018, 132, 121. [Google Scholar] [CrossRef]
- Gay, F.; Cerrato, C.; Petrucci, M.T.; Zambello, R.; Gamberi, B.; Ballanti, S.; Omedè, P.; Palmieri, S.; Troia, R.; Spada, S.; et al. Efficacy of carfilzomib lenalidomide dexamethasone (KRd) with or without transplantation in newly diagnosed myeloma according to risk status: Results from the FORTE trial. J. Clin. Oncol. 2019, 37, 8002. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Mai, K.E.; Salwender, H.; Bertsch, U.; Miah, K.; Kunz, C.; Fenk, R.; Blau, I.; Scheid, C.; Martin, H.; et al. Bortezomib, lenalidomide and dexamethasone with or without elotuzomab as induction therapy for newly-diagnosed, transplant-eligible multiple myeloma. In Proceedings of the EHA, Frankfurt, Germany, February 2020. [Google Scholar]
- Palumbo, A.; Bringhen, S.; Mateos, M.-V.; LaRocca, A.; Facon, T.; Kumar, S.; Offidani, M.; McCarthy, P.; Evangelista, A.; Lonial, S.; et al. Geriatric assessment predicts survival and toxicities in elderly myeloma patients: An International Myeloma Working Group report. Blood 2015, 125, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Dimopoulos, M.A.; Meuleman, N.; Belch, A.; Mohty, M.; Chen, W.-M.; Kim, K.; Zamagni, E.; Rodriguez-Otero, P.; Renwick, W.; et al. A simplified frailty scale predicts outcomes in transplant-ineligible patients with newly diagnosed multiple myeloma treated in the FIRST (MM-020) trial. Leukemia 2020, 34, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanan-Khan, A.; Giralt, S. Importance of Achieving a Complete Response in Multiple Myeloma, and the Impact of Novel Agents. J. Clin. Oncol. 2010, 28, 2612–2624. [Google Scholar] [CrossRef]
- Bringhen, S.; Mateos, M.V.; Zweegman, S.; LaRocca, A.; Falcone, A.P.; Oriol, A.; Rossi, D.; Cavalli, S.; Wijermans, P.; Mina, R.; et al. Age and organ damage correlate with poor survival in myeloma patients: Meta-analysis of 1435 individual patient data from 4 randomized trials. Haematologica 2013, 98, 980–987. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, E.K.; Laubach, J.P.; Yee, A.J.; Chen, T.; Huff, C.A.; Basile, F.G.; Wade, P.M.; Paba-Prada, C.E.; Ghobrial, I.M.; Schlossman, R.L.; et al. A phase 2 study of modified lenalidomide, bortezomib and dexamethasone in transplant-ineligible multiple myeloma. Br. J. Haematol. 2018, 182, 222–230. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Richardson, P.G.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; et al. Bortezomib Plus Melphalan and Prednisone Compared with Melphalan and Prednisone in Previously Untreated Multiple Myeloma: Updated Follow-Up and Impact of Subsequent Therapy in the Phase III VISTA Trial. J. Clin. Oncol. 2010, 28, 2259–2266. [Google Scholar] [CrossRef] [Green Version]
- Reeder, C.B.; Reece, D.E.; Kukreti, V.; Chen, C.; Trudel, S.; Hentz, J.; Noble, B.; Pirooz, N.A.; Spong, J.E.; Piza, J.G.; et al. Cyclophosphamide, bortezomib and dexamethasone induction for newly diagnosed multiple myeloma: High response rates in a phase II clinical trial. Leukemia 2009, 23, 1337–1341. [Google Scholar] [CrossRef]
- Reeder, C.; Reece, D.E.; Kukreti, V.; Chen, C.I.; Trudel, S.; Laumann, K.; Hentz, J.; Pirooz, N.A.; Piza, J.G.; Tiedemann, R.; et al. Once- versus twice-weekly bortezomib induction therapy with CyBorD in newly diagnosed multiple myeloma. Blood 2010, 115, 3416–3417. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. New Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef]
- Cavo, M.; Tacchetti, P.; Patriarca, F.; Petrucci, M.T.; Pantani, L.; Galli, M.; Di Raimondo, F.; Crippa, C.; Zamagni, E.; Palumbo, A.; et al. Bortezomib with thalidomide plus dexamethasone compared with thalidomide plus dexamethasone as induction therapy before, and consolidation therapy after, double autologous stem-cell transplantation in newly diagnosed multiple myeloma: A randomised phase 3 study. Lancet 2010, 376, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.; Lauwers-Cances, V.; Robillard, N.; Hulin, C.; Leleu, X.; Benboubker, L.; Marit, G.; Moreau, P.; Pegourie, B.; Caillot, D.; et al. Front-line transplantation program with lenalidomide, bortezomib, and dexamethasone combination as induction and consolidation followed by lenalidomide maintenance in patients with multiple myeloma: A phase II study by the Intergroupe Francophone du Myélome. J. Clin. Oncol. 2014, 32, 2712–2717. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Pasquini, M.C.; Blackwell, B.; Hari, P.; Bashey, A.; Devine, S.; Efebera, Y.; Ganguly, S.; Gasparetto, C.; Geller, N.; et al. Autologous Transplantation, Consolidation, and Maintenance Therapy in Multiple Myeloma: Results of the BMT CTN 0702 Trial. J. Clin. Oncol. 2019, 37, 589–597. [Google Scholar] [CrossRef]
- Mai, E.K.; Benner, A.; Bertsch, U.; Brossart, P.; Hänel, A.; Kunzmann, V.; Naumann, R.; Neben, K.; Egerer, G.; Ho, A.D.; et al. Singleversustandem high-dose melphalan followed by autologous blood stem cell transplantation in multiple myeloma: Long-term results from the phase III GMMG-HD2 trial. Br. J. Haematol. 2016, 173, 731–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Pasquini, M.C.; Blackwell, B.; Knust, K.; Bashey, A.; Devine, S.M.; Efebera, Y.A.; Ganguly, S.; Gasparetto, C.; Geller, N.; et al. Comparison of Autologous Hematopoietic Cell Transplant (autoHCT), Bortezomib, Lenalidomide (Len) and Dexamethasone (RVD) Consolidation with Len Maintenance (ACM), Tandem Autohct with Len Maintenance (TAM) and Autohct with Len Maintenance (AM) for up-Front Treatment of Patients with Multiple Myeloma (MM): Primary Results from the Randomized Phase III Trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702-StaMINA Trial). Blood 2016, 128. [Google Scholar] [CrossRef]
- Cavo, M.; Petrucci, M.T.; Di Raimondo, F.; Zamagni, E.; Gamberi, B.; Crippa, C.; Marzocchi, G.; Grasso, M.; Ballanti, S.; Vincelli, D.I.; et al. Upfront Single Versus Double Autologous Stem Cell Transplantation for Newly Diagnosed Multiple Myeloma: An Intergroup, Multicenter, Phase III Study of the European Myeloma Network (EMN02/HO95 MM Trial). Blood 2016, 128, 991. [Google Scholar] [CrossRef]
- Mccarthy, P.L.; Holstein, S.A.; Petrucci, M.T.; Richardson, P.G.; Hulin, C.; Tosi, P.; Bringhen, S.; Musto, P.; Anderson, K.C.; Caillot, D.; et al. Lenalidomide Maintenance After Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: A Meta-Analysis. J. Clin. Oncol. 2017, 35, 3279–3289. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Gay, F.; Schjesvold, F.; Beksac, M.; Hajek, R.; Weisel, K.C.; Goldschmidt, H.; Maisnar, V.; Moreau, P.; Min, C.K.; et al. Oral ixazomib maintenance following autologous stem cell transplantation (TOURMALINE-MM3): A double-blind, randomised, placebo-controlled phase 3 trial. Lancet 2019, 393, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Spicka, I.; Quach, H.; Oriol, A.; Hajek, R.; Garg, M.; Beksac, M.; Bringhen, S.; Katodritou, E.; Chng, W.J.; et al. Ixazomib vs placebo maintenance for newly diagnosed multiple myeloma (NDMM) patients not undergoing autologous stem cell transplant (ASCT): The phase III TOURMALINE-MM4 trial. J. Clin. Oncol. 2020, 38, 8527. [Google Scholar] [CrossRef]
- Moreau, P.; Siegel, D.S.; Goldschmidt, H.; Niesvizky, R.; Bringhen, S.; Orlowski, R.Z.; Blaedel, J.; Yang, Z.; Dimopoulos, M.A. Subgroup Analysis of Patients with Biochemical or Symptomatic Relapse at the Time of Enrollment in the Endeavor Study. Blood 2018, 132, 3243. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Moreau, P.; Mateos, M.-V.; Berenson, J.R.; Weisel, K.; Lazzaro, A.; Song, K.; Dimopoulos, M.A.; Huang, M.; Zahlten-Kumeli, A.; Stewart, A.K. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): Interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018, 19, 953–964. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. New Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Sonneveld, P.; Hungria, V.; Nooka, A.K.; Estell, J.A.; Barreto, W.; Corradini, P.; Min, C.-K.; Medvedova, E.; Weisel, K.; et al. Daratumumab, Bortezomib, and Dexamethasone Versus Bortezomib and Dexamethasone in Patients With Previously Treated Multiple Myeloma: Three-year Follow-up of CASTOR. Clin. Lymphoma Myeloma Leuk. 2020, 20, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.-A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Spicka, I.; Risse, M.-L.; Asset, G.; et al. Isatuximab plus carfilzomib and dexamethasone vs carfilzomib and dexamethasone in relapsed/refractory multiple myeloma (IKEMA): Intermin analysis of a phase 3, radnomised, open-label study. In Proceedings of the EHA, Online conference. December 2020. [Google Scholar]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. The Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; Leblanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.-V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.A.; San-Miguel, J.; Belch, A.; White, D.; Benboubker, L.; Cook, G.; Leiba, M.; Morton, J.; Ho, P.J.; Kim, K.; et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of POLLUX. Haematologica 2018, 103, 2088–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenson, J.R.; Cartmell, A.; Bessudo, A.; Lyons, R.M.; Harb, W.A.; Tzachanis, D.; Coleman, M.; Boccia, R.V.; Rifkin, R.M.; Patel, P.; et al. Updated results from CHAMPION-1, a phase I/II study investigating weekly carfilzomib with dexamethasone for patients (Pts) with relapsed or refractory multiple myeloma (RRMM). J. Clin. Oncol. 2015, 33, 8527. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Niesvizky, R.; Weisel, K.; Siegel, D.S.; Hajek, R.; Mateos, M.-V.; Cavo, M.; Huang, M.; Zahlten-Kumeli, A.; Moreau, P. Once- versus twice-weekly carfilzomib in relapsed and refractory multiple myeloma by select patient characteristics: Phase 3 A.R.R.O.W. study subgroup analysis. Blood Cancer J. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Stewart, K.A.; Dimopoulos, M.; Siegel, D.; Facon, T.; Berenson, J.; Raje, N.; Berdeja, J.G.; Orlowski, R.Z.; Yang, H.; et al. Once-weekly (70 mg/m2) vs twice-weekly (56 mg/m2) dosing of carfilzomib in patients with relapsed or refractory multiple myeloma: A post hoc analysis of the ENDEAVOR, A.R.R.O.W. and CHAMPION-1 trials. Cancer Med. 2020, 9, 2989–2996. [Google Scholar] [CrossRef] [Green Version]
- Weisel, K.; Spencer, A.; Lentzsch, S.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.-D.; Bosi, A.; et al. Daratumumab, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma: Subgroup analysis of CASTOR based on cytogenetic risk. J. Hematol. Oncol. 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Weisel, K.; Moreau, P.; Anderson, L.D.; White, D.; San-Miguel, J.; Sonneveld, P.; Engelhardt, M.; Jenner, M.; Corso, A.; et al. Pomalidomide, bortezomib, and dexamethasone for multiple myeloma previously treated with lenalidomide (OPTIMISMM): Outcomes by prior treatment at first relapse. Leukemia 2020. [CrossRef]
- Dimopoulos, M.A.; Stewart, A.K.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.; Mihaylov, G.G.; Goranova-Marinova, V.; et al. Carfilzomib, lenalidomide, and dexamethasone in patients with relapsed multiple myeloma categorised by age: Secondary analysis from the phase 3 ASPIRE study. Br. J. Haematol. 2017, 177, 404–413. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Usmani, S.Z.; San-Miguel, J.; Bahlis, N.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; et al. Four-Year Follow-up of the Phase 3 Pollux Study of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) Alone in Relapsed or Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 1866. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; Mcgehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; Van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Lee, L.; Bounds, D.; Paterson, J.; Herledan, G.; Sully, K.; Seestaller-Wehr, L.M.; Fieles, W.E.; Tunstead, J.; Mccahon, L.; Germaschewski, F.M.; et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br. J. Haematol. 2016, 174, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Nooka, A.K.; Stockerl-Goldstein, K.; Quach, H.; Forbes, A.; Mateos, M.-V.; Khot, A.; Tan, A.; Abonour, R.; Chopra, B.; Rogers, R.; et al. DREAMM-6: Safety and tolerability of belantamab mafodotin in combination with bortezomib/dexamethasone in relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8502. [Google Scholar] [CrossRef]
- Hipp, S.; Tai, Y.-T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef]
- Cho, S.-F.; Lin, L.; Xing, L.; Wen, K.; Yu, T.; Hsieh, P.A.; Li, Y.; Munshi, N.C.; Wahl, J.; Matthes, K.; et al. AMG 701 Potently Induces Anti-Multiple Myeloma (MM) Functions of T Cells and IMiDs Further Enhance Its Efficacy to Prevent MM Relapse In Vivo. Blood 2019, 134, 135. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kronke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermudez, A.; De la Rubia, J.; Mateos, M.V.; Ocio, E.M.; Rodriguez-Otero, P.; San Miguel, J.; Li, S.; Sarmiento, R.; et al. Intermin results from the first phase I clinical study of the B-cell maturation antigen (BCMA) 2+1 T cell engager (TCE) CC-93269 in patients (pts) with relapsed/refractory multiple myeloma (RRMM). In Proceedings of the European Haematology Association, Frankfurt, Germany, 21–24 June 2001; p. 295025. [Google Scholar]
- Mateos, M.V.; Usmani, S.Z.; Nahi, H.; Krishnan, A.Y.; San-Miguel, J.; Oriol, A.; Rosinol, L.; Chari, A.; Adams, H., III; Girgis, S.; et al. A phase 1 study of Teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, for the treatment of relapsed and/or refractory multiple myeloma (RRMM). In Proceedings of the European Haematology Association, Frankfurt, Germany, 21–24 June 2001; p. 295026. [Google Scholar]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; DiCapua Siegel, D.S.; Bensinger, W.; Cota, M.; et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): Update of the phase 1/2 EVOLVE study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.J.; Shah, N.; Jagannath, S.; Berdej, J.G.; Lonial, S.; Raje, N.S.; DiCapua Siegel, D.S.; Lin, Y.; Oriol, A.; et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J. Clin. Oncol. 2020, 38, 8503. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Singh, I.; Zudaire, E.; Yeh, T.-M.; Allred, A.J.; Olyslager, Y.; Banerjee, A.; Goldberg, J.D.; et al. Update of CARTITUDE-1: A phase Ib/II study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed CAR-T-cell therapy, in relapsed/refractory multiple myeloma. J. Clin. Oncol. 2020, 38, 8505. [Google Scholar] [CrossRef]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)–a novel class of anti-cancer agents. J. Hematol Oncol 2014, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, U.H.; Senapedis, W.; Baloglu, E.; Unger, T.J.; Chari, A.; Vogl, D.; Cornell, R.F. Clinical Implications of Targeting XPO1-mediated Nuclear Export in Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Senapedis, W.T.; Baloglu, E.; Landesman, Y. Clinical translation of nuclear export inhibitors in cancer. Semin. Cancer Biol. 2014, 27, 74–86. [Google Scholar] [CrossRef]
- Schmidt, J.; Braggio, E.; Kortuem, K.M.; Egan, J.B.; Zhu, Y.X.; Xin, C.S.; Tiedemann, R.E.; Palmer, S.E.; Garbitt, V.M.; Mccauley, D.; et al. Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT. Leukemia 2013, 27, 2357–2365. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Sutherland, H.; White, D.; Sebag, M.; Lentzsch, S.; Kotb, R.; Venner, C.P.; Gasparetto, C.; Del Col, A.; Neri, P.; et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood 2018, 132, 2546–2554. [Google Scholar] [CrossRef] [Green Version]
- Vogl, D.T.; Dingli, D.; Cornell, R.F.; Huff, C.A.; Jagannath, S.; Bhutani, D.; Zonder, J.; Baz, R.; Nooka, A.; Richter, J.; et al. Selective Inhibition of Nuclear Export with Oral Selinexor for Treatment of Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 859–866. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral selinexor–dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Podar, K.; Shah, J.; Chari, A.; Richardson, P.G.; Jagannath, S. Selinexor for the treatment of multiple myeloma. Expert Opin. Pharmacother. 2020, 21, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Delimpasi, S.; Simonova, M.; Spicka, I.; Pour, L.; Kryachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; et al. Weekly selinexor, bortezomib, and dexamethasone (SVd) versus twice weekly bortezomib and dexamethasone (Vd) in patients with multiple myeloma (MM) after one to three prior therapies: Initial results of the phase III BOSTON study. J. Clin. Oncol. 2020, 38, 8501. [Google Scholar] [CrossRef]
- Kumar, S.; Kaufman, J.L.; Gasparetto, C.; Mikhael, J.; Vij, R.; Pegourie, B.; Benboubker, L.; Facon, T.; Amiot, M.; Moreau, P.; et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood 2017, 130, 2401–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Harrison, S.; Cavo, M.; De La Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.T.M.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Updated Analysis of Bellini, a Phase 3 Study of Venetoclax or Placebo in Combination with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2019, 134, 1888. [Google Scholar] [CrossRef]
- Harrison, S.; Cavo, M.; De La Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.T.M.; Salwender, H.; Suzuki, K.; Kim, I.; Moreau, P.; et al. T(11;14) and High BCL2 Expression Are Predictive Biomarkers of Response to Venetoclax in Combination with Bortezomib and Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma: Biomarker Analyses from the Phase 3 Bellini Study. Blood 2019, 134, 142. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Zhang, W.; Man, H.-W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; Lebrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (CC-220) with improved degradation of ikaros and aiolos. J. Med. Chem. 2018, 61, 535–542. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Lu, L.; Amatangelo, M.; Chiu, H.; Gandhi, A.K.; Pourdehnad, M.; Klippel, A.; Thakurta, A. CC-220 Is a potent cereblon modulating agent that displays anti-proliferative, pro-apoptotic and immunomodulatory activity on sensitive and resistant multiple myeloma cell lines. Blood 2016, 128, 1591. [Google Scholar] [CrossRef]
- Amatangelo, M.; Bjorklund, C.C.; Kang, J.; Polonskaia, A.; Viswanatha, S.; Thakurta, A. Iberdomide (CC-220) Has Synergistic Anti-Tumor and Immunostimulatory Activity Against Multiple Myeloma in Combination with Both Bortezomib and Dexamethasone, or in Combination with Daratumumab in Vitro. Blood 2018, 132, 1935. [Google Scholar] [CrossRef]
- Lonial, S.; Van de Donk, N.W.C.J.; Popat, R.; Zonder, J.A.; Minnema, M.C.; Larsen, J.; Nguyen, T.V.; Chen, M.S.; Bensmaine, A.; Cota, M.; et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2019, 37, 8006. [Google Scholar] [CrossRef]
- Richardson, P.G.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martinez, J.; Mateos, M.-V.; Rodríguez Otero, P.; Lonial, S.; Popat, R.; Oriol, A.; et al. First-in-human phase I study of the novel CELMoD agent CC-92480 combined with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8500. [Google Scholar] [CrossRef]

{kind=link}
| Aberration Category | Genomic Aberration | Involved Oncogenes and TSG | Frequency, % | Prognosis | |
|---|---|---|---|---|---|
| Primary genetic events | Hyperdiploidy | Chromosomes 3, 5, 7, 9, 11, 15, 19 and 21 trisomy | Overexpression of MYC, NFκB and MAPK pathways | 50 | Favourable |
| Chromosomal translocations | t(4;14) | IGH-MMSET/FGF3 | 10–15 | Adverse | |
| t(11;14) | IGH-CCND1 | 15–20 | Neutral | ||
| t(14;16) | IGH-MAF | 5 | Adverse | ||
| t(14;20) | IGH-MAFB | 1 | Adverse | ||
| Secondary genetic events | Chromosomal translocations | MYC translocations | Multiple partners incl. IGH, IGL, IGK, FAM46C, CCND1, XBP1 | 15–20 | Neutral or Adverse |
| Copy number variations | 1q gain | CKS1B ANP32E BCL-9 | 35–40 | Adverse | |
| 1p deletion | FAM46C FAF1 CDKN2C | 30 | Adverse | ||
| 13q deletion * | RB1 | 45–50 | Neutral or Adverse | ||
| 17p deletion | TP53 | 10 | Adverse | ||
| Clinical Study | Response Rate, % | Med PFS, Months | Overall Survival | |||||
|---|---|---|---|---|---|---|---|---|
| Study Population | Regimen | ORR | CR | ≥VGPR | MRDneg | |||
| Newly diagnosed multiple myeloma, transplant eligible (TE) | ||||||||
| IFM 2009 [78] | Phase III n = 700 Med age = 59y | VRd (x3) > VRd (x5) > R maint. vs. VRd (x3) > ASCT + VRd (x2) > R maint. | 97 vs. 98 | 48 vs. 59 p = 0.03 | 77 vs. 88 p = 0.001 | 65 vs. 79 p < 0.001 | 36 vs. 50 HR 0.65 p < 0.001 | 4yr OS 82 vs. 81% HR =1.16 p = 0.87 |
| PETHEMA/GEM2012 [82] | Phase III n = 458 Med age = 58y | VRd (x6) > ASCT > VRd (x2) | 81 | 44 | 75 | 45.2 (@10−6) 65.9 (@10−4) | NR | NR |
| EMN02/HO95 [79] | Phase III n = 1197 Med age = 58y | VCD induction 1st randomisation to ASCT vs. VMP intensification [results reported here] 2nd randomisation to VRd vs. observation | 95 vs. 95 | 44 vs. 40 | 84 vs. 77 | 56.7 vs. 41.9 HR = 0.73 p = 0.0001 | 5yr OS 75.1 vs. 71.6% HR = 0.90 p = 0.35 | |
| ENDURANCE [83] | NDMM * No immediate intention for ASCT n = 1087 Med age = 65y * del(17p), t(14;16) & t(14;20) excluded | VRd vs. KRd Followed by second randomisation to R maintenance until PD vs. 2 yrs. | 84 vs. 87 p = 0.13 | 15 vs. 18 p = 0.261 | 65 vs. 74 p = 0.002 | 34.4 vs. 34.6 HR 1.04 p = 0.70 | 3yr OS 84 vs. 83% HR 0.98 p = 0.923 | |
| FORTE [84,85] | Phase III n = 474 Med age = 57y | (A) KCd > ASCT > KCd (n = 159) (B) KRd > ASCT > KRd (n = 158) (C) KRd (x12) (n = 157) | 60 vs. 61 | 89 vs. 87 | 58 vs. 54 | Data immature | Data immature | |
| Comparisons are between cohort (B) KRd > ASCT > KRd and cohort (C) KRd (x12) | ||||||||
| CASSIOPEIA [84] | Phase III n = 1085 Med age = 59y | VTd vs. Dara-VTd > ASCT > VTd or Dara-VTd consolidation > 2nd randomisation to Dara maintenance vs. observation until PD | Response assessment at D100 post ASCT | |||||
| 89.9 vs. 92.6 p = 0.11 | 26 vs. 39 p < 0.0001 | 78 vs. 83 p = 0.024 | 20 vs. 34 p < 0.0001 | 18m PFS 85 vs. 93% HR 0.43 p < 0.0001 | Data immature | |||
| GRIFFIN [69] | Phase III n = 207 Med age = 60y | VRd (x4) vs. Dara-VRd > ASCT > x2 consolidation > 2nd randomisation to R (x26) vs. Dara-R maintenance | Response assessment at end of consolidation | |||||
| 92 vs. 98 p = 0.016 | 32 vs. 42.4 p = 0.068 | 73 vs. 91 p = 0.0014 | 16.5 vs. 47.1 | NR at med f/u 22.1m | Data immature | |||
| MASTER [85] | Phase II n = 81 Med age = 61y | Dara-KRd (x4) > ASCT > Dara-KRd (MRD-directed x4-9) > R maintenance | Response assessment at end of consolidation | |||||
| 100 | 95 | 100 | 82% (@10−5) | Data immature | Data immature | |||
| GMMG-CONCEPT [86] | * Both TE & TNE Phase II High risk pts n = 153, interim report on n = 50 | Isa-KRd TE: Isa-KRD (x6) > ASCT; n = 46 TNE: Isa-KRd (x8); n = 4 | 100 | 46 | 90 | 31 (20/33 pts) | Data immature | Data immature |
| SWOG 1211 [87] | Phase II High risk pts n = 103 Med age = 62.4y | VRd (x8) vs. Elo-VRd(x8) > attenuated VRd maintenance | 88 vs. 83 | 6 vs. 2.1 | 34 vs. 31 HR 0.97 p = 0.449 | NR vs. 68 HR 1.28 p = 0.239 | ||
| Newly diagnosed multiple myeloma, transplant ineligible (TNE) | ||||||||
| FIRST [88] | Phase III n = 1623 | Rd continuous vs. Rd (18 months) vs. MPT | Response assessment for Rd cohort only | |||||
| 81 | 22 | 48 | 26 | 59.1 | ||||
| SWOG S0777 [89] | Both TE and TNE with no immediate intention for ASCT n = 460 Age ≥ 65 = 43% | Rd (x6) vs. VRd (x8) > R maintenance | 78.8 vs 90.2 | 12.1 vs. 24.2 | 53.2 vs. 74.9 | 29 vs. 41 HR 0.74 p = 0.003 | 69 vs. NR HR 0.71 p = 0.0114 | |
| ALCYONE [68] | Phase III n = 706 Med age = 71y | VMP (x9) vs. Dara-VMP f/b Dara maintenance in Dara-VMP cohort | 73.9 vs. 90.9 p < 0.0001 | 25 vs. 46 p < 0.0001 | 50 vs. 73 p < 0.0001 | 7 vs. 28 p < 0.0001 | 18.1 vs. NR p < 0.001 | 36m OS estimate 67.9 vs. 78% HR 0.60 p = 0.0003 |
| MAIA [67] | Phase III n = 737 Med age = 73y | Rd vs. Dara-Rd | 81.3 vs. 92.9 p < 0.001 | 24.9 vs. 47.6 p < 0.01 | 53.1 vs. 79.3 p < 0.01 | 7.3 vs. 24.2 p < 0.001 | 31.9 vs. NR HR 0.56 p < 0.001 | Data immature |
| Median Progression Free Survival (mPFS), Months | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Study Population | Regimen | Med f/u (m) | Overall | Rnaïve | Rexp. | Rref. | PIexp. | PIref. | |
| Proteasome Inhibitor-based | |||||||||
| ENDEAVOR [112,113] | Phase III, n = 929 Med # PL = 2 (1–2) Prior R = 38% Prior V = 54% | Kd vs. Vd | 11.9 | 18.7 vs. 9.4 HR 0.53 p < 0.0001 | 12.9 vs. 7.3 HR 0.67 | 8.6 vs. 6.6 HR 0.80 | 15.6 vs. 8.1 HR 0.56 | ||
| A.R.R.O.W. [114] | Phase III, n = 478 Med # PL = 2–3 Prior R = 84% (74.5% ref.) Prior V = 98.9% (42% ref.) | Kd 70QW vs. Kd 27BIW | 12.6 | 11.2 vs. 7.6 HR 0.69 p = 0.0029 | HR 0.72 | HR 0.76 | HR 0.73 | ||
| CASTOR [115,116] | Phase III, n = 498 Med # PL = 2 (1–10) Prior IMiD = 75.7% Prior PI = 68.5% | Dara-Vd vs. Vd | 40 | 16.7 vs. 7.1 HR 0.31 p < 0.0001 | 16.7 (Dara-Vd) | 9.5 (Dara-Vd) | 7.8 (Dara-Vd) | ||
| CANDOR [117] | Phase III, n = 466 Med # PL = 2 (1–2) Prior R = 42.2% (33% ref.) Prior V = 90.3% (29% ref.) | Dara-Kd vs. Kd | 17 | NR vs. 15.8 HR 0.63 p = 0.0027 | HR 0.71 | HR 0.53 | HR 0.47 | ||
| IKEMA [118] | Phase III, n = 302 1–3 prior lines Prior R = 78% (33% ref.) Prior PI = 90% (33% ref.) | Isa-Kd vs. Kd | 20.7 | NR vs. 19.2 HR 0.53 p = 0.007 | |||||
| Pomalidomide-based | |||||||||
| Study population | Regimen | Med f/u (m) | Overall | Rnaïve | Rexp. | Rref. | PIexp. | PIref. | |
| OPTIMISMM [119] | Phase III, n = 559 1–3 prior lines Prior R = 100% (70% ref.) Prior V = 72% (10% ref.) | VPd vs. Vd | 15.9 | 11.2 vs. 7.1 HR 0.61 p < 0.0001 | 11.2 | 9.5 | |||
| ELOQUENT-3 [120] | Phase III, n = 117 Med # PL = 3 (range 2–8) Prior R = 99% (87% ref.) Prior V = 100% (80% ref.) | Elo-Pd vs. Pd | 9.1 | 10.3 vs. 4.7 HR 0.54 p = 0.008 | 10.2 | ||||
| ICARIA [121] | Phase III, n = 307 Med # PL = 3 (range 2–4) Prior R = 100% (93% ref.) Prior V = 100% (76% ref.) | Isa-Pd vs. Pd | 11.6 | 11.5 vs. 6.5 HR 0.60 p = 0.001 | |||||
| Lenalidomide-based | |||||||||
| ASPIRE [122] | Phase III, n = 792 Med # PL = 2 (1–3) Prior R = 19.8% Prior V = 65.8% | KRd vs. Rd | 26.3 vs. 17.6 HR 0.69 p = 0.0001 | 28.7 (KRd) n = 317 | 19.4 (KRd) n = 79 | 9.3 (KRd) n = 57 | |||
| TOURMALINE-MM1 [123] | Phase III, n= 722 1–3 prior lines Prior R = 12% Prior V =69% | Ixa-Rd vs. Rd | 14.7 | 20.6 vs. 14.7 HR 0.74 p = 0.01 | NR vs. 17.5 HR 0.74 | 18.4 vs. 13.6 HR 0.74 | |||
| ELOQUENT-2 [124] | Phase Ib-II, n= 646 Med # PL = 2 (1–4) Prior R = 6% Prior V = 70% | Elo-Rd vs. Rd | 24.5 | 19.4 vs. 14.9 HR 0.70 p < 0.001 | |||||
| POLLUX [125] | Phase III, n = 569 Med # PL = 1 (1–11) Prior R = 55.1% (3.7% ref.) Prior V = 86.1% (18.1% ref.) | Dara-Rd vs. Rd | 45.8 vs. 17.5 HR 0.43 p < 0.0001 | 38.6 vs. 18.6 HR 0.34 | |||||
| EVOLVE [145] | KarMMa [146] | CARTITUDE-1 [147] | |
|---|---|---|---|
| Product | Orvacabtagene autoleucel [orva-cel] | Idecabtagene vicleucel [ide-cel] | JNJ-4528 |
| Target Antigen | BCMA | BCMA | BCMA |
| n | 62 | 128 | 29 |
| Median age (range) | 61 (33–77) | 61 (33–78) | 60 (50–75) |
| High risk cytogenetics *, % | 41 | 35 | 27 |
| Median prior lines of therapy, (range) | 6 (3–18) | 6 (3–16) | 5 (3–18) |
| Triple-refractory, % | 94 | 84 | 86 |
| Penta-refractory, % | 48 | 26 | 31 |
| Bridging chemotherapy required, % | 63 | 88 | 79 |
| Median follow-up time, months | 6.9 | 13.3 | 9.0 |
| ORR, % | 92 | 73 | 100 |
| sCR/CR, % | 36 | 33 | 86 |
| ≥VGPR, % | 68 | 53 | 97 |
| MRDneg, % | 84 | 94 | 81 |
| Median duration of response, months | NR | 10.7 [19.0 months in CR pts] | NR |
| Median PFS, months | NR mPFS (lowest dose cohort) = 9.3 m | 8.8 (5.6–11.6) [20.2 months in CR pts] | NR 9 month PFS = 86% |
| Median OS, months | NR | 19.4 (18.2-NR) | NR |
| Any grade CRS, % | 89 | 84 | 93 |
| ≥ Grade 3 CRS, % | 3 | 6 | 7 |
| Median time to CRS, days (range) | 2 (1–4) | 1 (1–12) | 7 (2–12) |
| Median duration of CRS, days (range) | 4 (1–10) | 5 (1–63) | 4 (2–64) |
| Any grade ICANS, % | 13 | 17 | 10 |
| ≥ Grade 3 ICANS, % | 3 | 3 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ninkovic, S.; Quach, H. Shaping the Treatment Paradigm Based on the Current Understanding of the Pathobiology of Multiple Myeloma: An Overview. Cancers 2020, 12, 3488. https://doi.org/10.3390/cancers12113488
Ninkovic S, Quach H. Shaping the Treatment Paradigm Based on the Current Understanding of the Pathobiology of Multiple Myeloma: An Overview. Cancers. 2020; 12(11):3488. https://doi.org/10.3390/cancers12113488
Chicago/Turabian StyleNinkovic, Slavisa, and Hang Quach. 2020. "Shaping the Treatment Paradigm Based on the Current Understanding of the Pathobiology of Multiple Myeloma: An Overview" Cancers 12, no. 11: 3488. https://doi.org/10.3390/cancers12113488
APA StyleNinkovic, S., & Quach, H. (2020). Shaping the Treatment Paradigm Based on the Current Understanding of the Pathobiology of Multiple Myeloma: An Overview. Cancers, 12(11), 3488. https://doi.org/10.3390/cancers12113488