Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Lines, Co-Culture Experiments, and Pharmacologic Reagents
2.3. Cell Proliferation Assay
2.4. Apoptosis and Cell Viability Assays
2.5. Western Blot Assays
2.6. Reverse Protein Phase Array (RPPA) Analysis
2.7. Statistical Analyses
3. Results
3.1. LY3009120 Induces Anti-Leukemia Effects in AML Cells Harboring NRAS or FLT3 Mutations
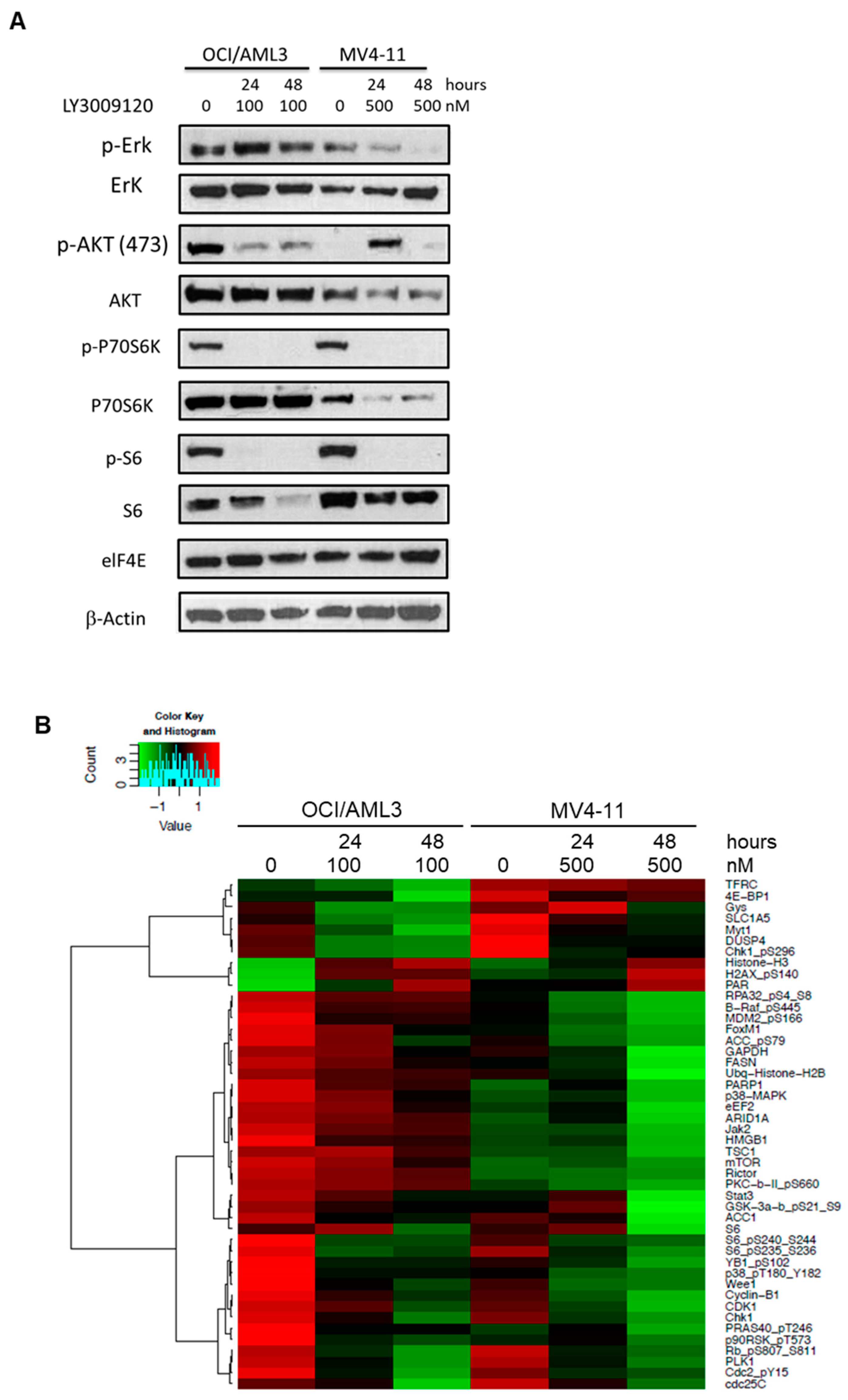
3.2. LY3009120 Impacts the RAS-Mediated Signaling Pathway in a Leukemia Cell-Specific Manner
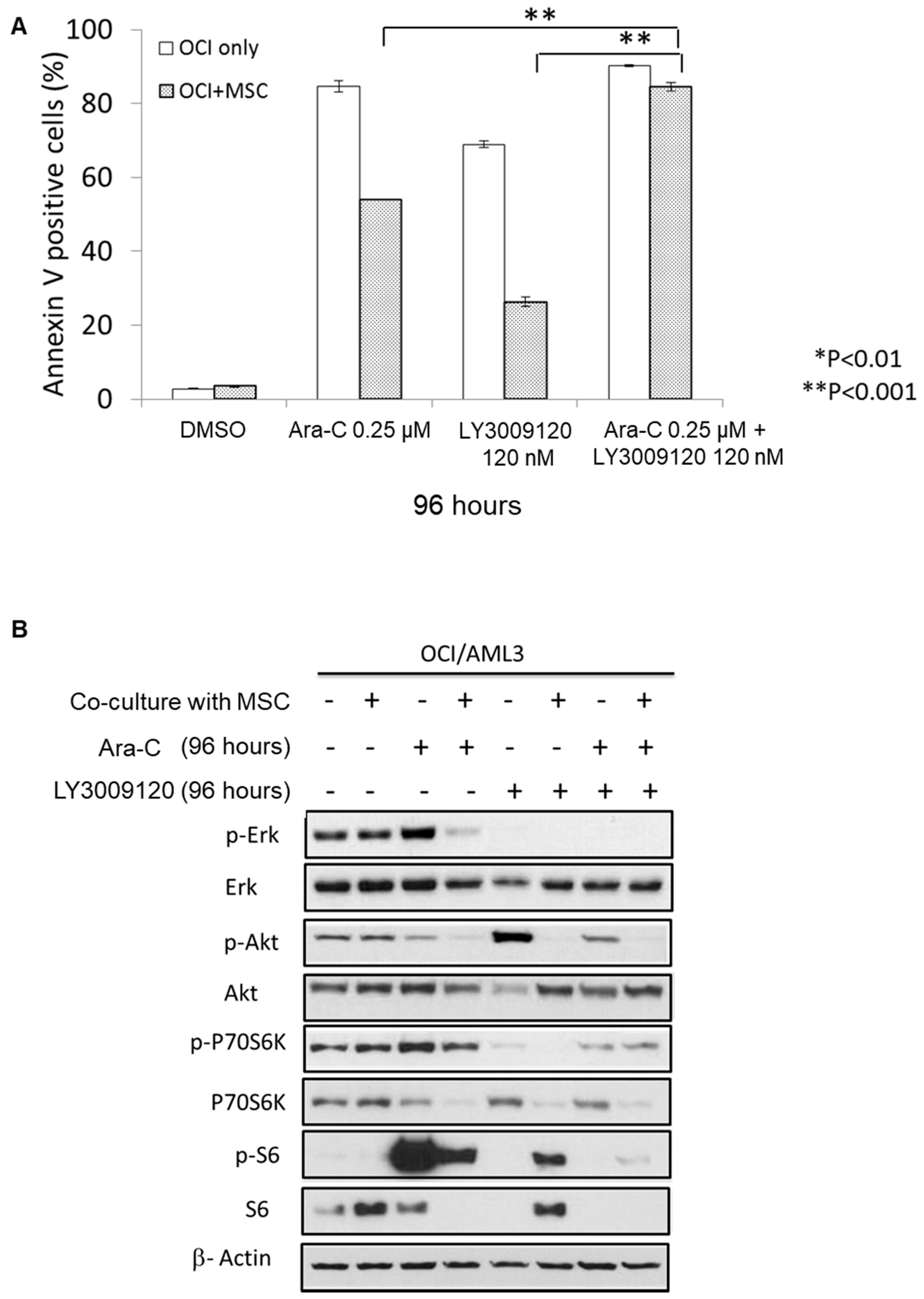
3.3. Combining LY3009120 with Ara-C Overcomes Bone Marrow Stroma-Mediated Chemoresistance
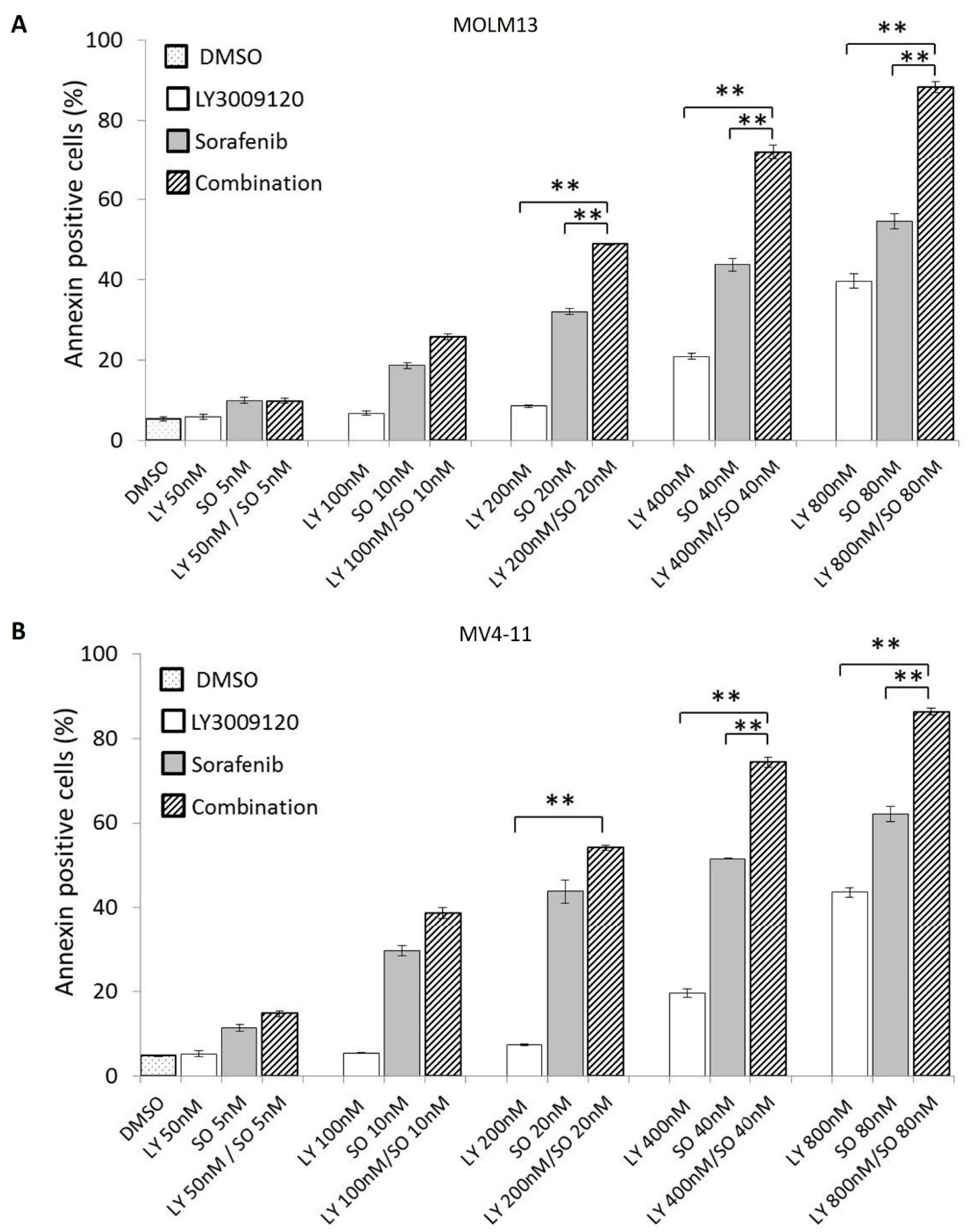
3.4. Concurrent Pan-RAF and FLT3 Inhibition Exerts a Synergistic Antiapoptotic Effect in Sorafenib-Sensitive AML Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Matallanas, D.; Birtwistle, M.; Romano, D.; Zebisch, A.; Rauch, J.; von Kriegsheim, A.; Kolch, W. Raf Family Kinases: Old Dogs Have Learned New Tricks. Genes Cancer 2011, 2, 232–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacher, U.; Haferlach, T.; Schoch, C.; Kern, W.; Schnittger, S. Implications of NRAS mutations in AML: A study of 2502 patients. Blood 2006, 107, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.T.; Frew, M.E.; Hills, R.; Gale, R.E.; Wheatley, K.; Groves, M.J.; Langabeer, S.E.; Kottaridis, P.D.; Moorman, A.V.; Burnett, A.K.; et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005, 106, 2113–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisfeld, A.K.; Kohlschmidt, J.; Mrozek, K.; Blachly, J.S.; Walker, C.J.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Carroll, A.J.; Stone, R.M.; et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: An analysis of Alliance studies. Leukemia 2018, 32, 1338–1348. [Google Scholar] [CrossRef]
- Kadia, T.M.; Kantarjian, H.; Kornblau, S.; Borthakur, G.; Faderl, S.; Freireich, E.J.; Luthra, R.; Garcia-Manero, G.; Pierce, S.; Cortes, J.; et al. Clinical and Proteomic Characterization of Acute Myeloid Leukemia with Mutated RAS. Cancer 2012, 118, 5550–5559. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Gao, C.; Konopleva, M.; Chen, Y.; Jacamo, R.O.; Borthakur, G.; Cortes, J.E.; Ravandi, F.; Ramachandran, A.; Andreeff, M. Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin. Cancer Res. 2014, 20, 2363–2374. [Google Scholar] [CrossRef] [Green Version]
- Tallis, E.; Borthakur, G. Novel treatments for relapsed/refractory acute myeloid leukemia with FLT3 mutations. Expert Rev. Hematol. 2019, 12, 621–640. [Google Scholar] [CrossRef]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Ragon, B.K.; Odenike, O.; Baer, M.R.; Stock, W.; Borthakur, G.; Patel, K.; Han, L.; Chen, H.; Ma, H.; Joseph, L.; et al. Oral MEK 1/2 Inhibitor Trametinib in Combination with AKT Inhibitor GSK2141795 in Patients With Acute Myeloid Leukemia With RAS Mutations: A Phase II Study. Clin. Lymphoma Myeloma Leuk. 2019, 19, 431–440.e13. [Google Scholar] [CrossRef]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.; Ravandi, F.; Daver, N. Emerging treatment paradigms with FLT3 inhibitors in acute myeloid leukemia. Ther. Adv. Hematol. 2019, 10, 2040620719827310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Henry, J.R.; Kaufman, M.D.; Peng, S.B.; Ahn, Y.M.; Caldwell, T.M.; Vogeti, L.; Telikepalli, H.; Lu, W.P.; Hood, M.M.; Rutkoski, T.J.; et al. Discovery of 1-(3,3-Dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3-d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a Pan-RAF Inhibitor with Minimal Paradoxical Activation and Activity against BRAF or RAS Mutant Tumor Cells. J. Med. Chem. 2015, 58, 4165–4179. [Google Scholar] [CrossRef]
- Chen, S.H.; Zhang, Y.Y.; van Horn, R.D.; Yin, T.G.; Buchanan, S.; Yadav, V.; Mochalkin, I.; Wong, S.S.; Yue, Y.G.; Huber, L.; et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016, 6, 300–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.B.; Henry, J.R.; Kaufman, M.D.; Lu, W.P.; Smith, B.D.; Vogeti, S.; Rutkoski, T.J.; Wise, S.; Chun, L.; Zhang, Y.; et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell 2015, 28, 384–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ruvolo, V.R.; Gao, C.; Zhou, L.; Bornmann, W.; Tsao, T.; Schober, W.D.; Smith, P.; Guichard, S.; Konopleva, M.; et al. Evaluation of apoptosis induction by concomitant inhibition of MEK, mTOR, and Bcl-2 in human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolova, O.; Samudio, I.; Benito, J.M.; Jacamo, R.; Kornblau, S.M.; Markovic, A.; Schober, W.; Lu, H.; Qiu, Y.H.; Buglio, D.; et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol. Ther. 2014, 13, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Borthakur, G.; Gao, C.; Chen, Y.; Mu, H.; Ruvolo, V.R.; Nomoto, K.; Zhao, N.; Konopleva, M.; Andreeff, M. The Dual MEK/FLT3 Inhibitor E6201 Exerts Cytotoxic Activity against Acute Myeloid Leukemia Cells Harboring Resistance-Conferring FLT3 Mutations. Cancer Res. 2016, 76, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, M.R.; McQueen, T.; Chism, D.; Milella, M.; Estey, E.; Kaldjian, E.; Sebolt-Leopold, J.; Konopleva, M.; Andreeff, M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 2005, 19, 1543–1549. [Google Scholar] [CrossRef] [Green Version]
- Kornblau, S.M.; Womble, M.; Qiu, Y.H.; Jackson, C.E.; Chen, W.; Konopleva, M.; Estey, E.H.; Andreeff, M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 2006, 108, 2358–2365. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.J.; Luo, L.S.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambe, M.; Karjalainen, E.; Vaha-Koskela, M.; Bulanova, D.; Gjertsen, B.T.; Kontro, M.; Porkka, K.; Heckman, C.A.; Wennerberg, K. Pan-RAF inhibition induces apoptosis in acute myeloid leukemia cells and synergizes with BCL2 inhibition. Leukemia 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, W.H.; Steelman, L.S.; Long, J.M.; Kempf, R.C.; Abrams, S.L.; Franklin, R.A.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Inhibitors: Rationale and Importance to Inhibiting These Pathways in Human Health. Oncotarget 2011, 2, 135–164. [Google Scholar] [CrossRef] [Green Version]
- Di Tullio, A.; Rouault-Pierre, K.; Abarrategi, A.; Mian, S.; Grey, W.; Gribben, J.; Stewart, A.; Blackwood, E.; Bonnet, D. The combination of CHK1 inhibitor with G-CSF overrides cytarabine resistance in human acute myeloid leukemia. Nat. Commun. 2017, 8, 1679. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Jordan, C.T. Leukemia stem cells and microenvironment: Biology and therapeutic targeting. J. Clin. Oncol. 2011, 29, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updates 2012, 15, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Konopleva, M.; Konoplev, S.; Hu, W.; Zaritskey, A.Y.; Afanasiev, B.V.; Andreeff, M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia 2002, 16, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Tabe, Y.; Jin, L.H.; Tsutsumi-Ishii, Y.; Xu, Y.Y.; McQueen, T.; Priebe, W.; Mills, G.B.; Ohsaka, A.; Nagaoka, I.; Andreeff, M.; et al. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer Res. 2007, 67, 684–694. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Liu, W.; Tsao, T.; Qiu, Y.; Zhao, Y.; Samudio, I.; Sarbassov, D.D.; Kornblau, S.M.; Baggerly, K.A.; Kantarjian, H.M.; et al. High-throughput profiling of signaling networks identifies mechanism-based combination therapy to eliminate microenvironmental resistance in acute myeloid leukemia. Haematologica 2017, 102, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Mak, P.Y.; Mak, D.H.; Mu, H.; Shi, Y.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.; Burks, J.K.; Wei, W.; Huang, X.; et al. Apoptosis repressor with caspase recruitment domain is regulated by MAPK/PI3K and confers drug resistance and survival advantage to AML. Apoptosis 2014, 19, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.Z.; Mak, P.Y.; Wang, X.; Tao, W.; Ruvolo, V.; Mak, D.; Mu, H.; Burks, J.K.; Andreeff, M. An ARC-Regulated IL1beta/Cox-2/PGE2/beta-Catenin/ARC Circuit Controls Leukemia-Microenvironment Interactions and Confers Drug Resistance in AML. Cancer Res. 2019, 79, 1165–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoury, J.D.; Tashakori, M.; Yang, H.; Loghavi, S.; Wang, Y.; Wang, J.; Piya, S.; Borthakur, G. Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation. Cancers 2020, 12, 3511. https://doi.org/10.3390/cancers12123511
Khoury JD, Tashakori M, Yang H, Loghavi S, Wang Y, Wang J, Piya S, Borthakur G. Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation. Cancers. 2020; 12(12):3511. https://doi.org/10.3390/cancers12123511
Chicago/Turabian StyleKhoury, Joseph D., Mehrnoosh Tashakori, Hong Yang, Sanam Loghavi, Ying Wang, Jing Wang, Sujan Piya, and Gautam Borthakur. 2020. "Pan-RAF Inhibition Shows Anti-Leukemic Activity in RAS-Mutant Acute Myeloid Leukemia Cells and Potentiates the Effect of Sorafenib in Cells with FLT3 Mutation" Cancers 12, no. 12: 3511. https://doi.org/10.3390/cancers12123511