Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance

 , ,
, ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
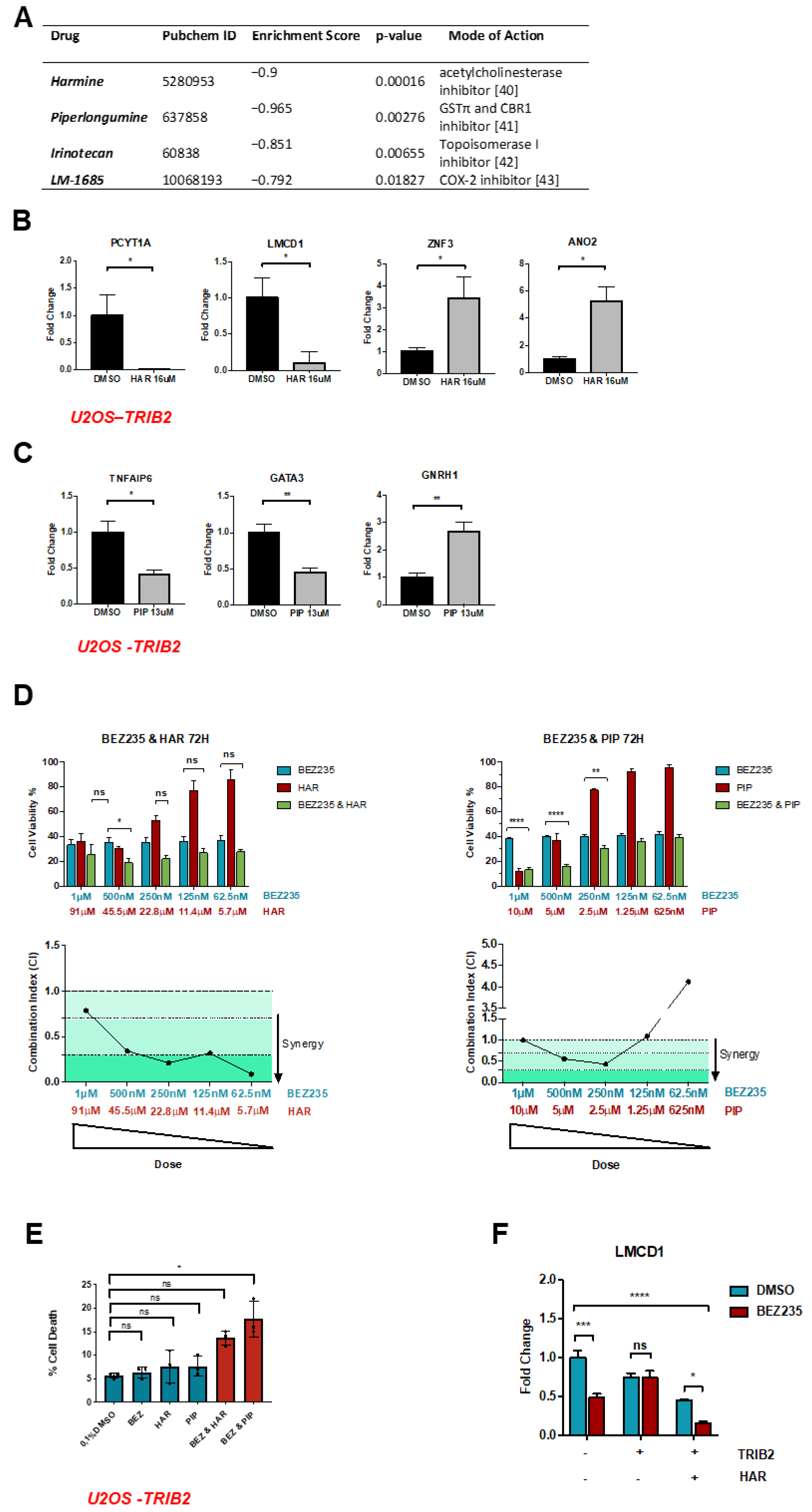
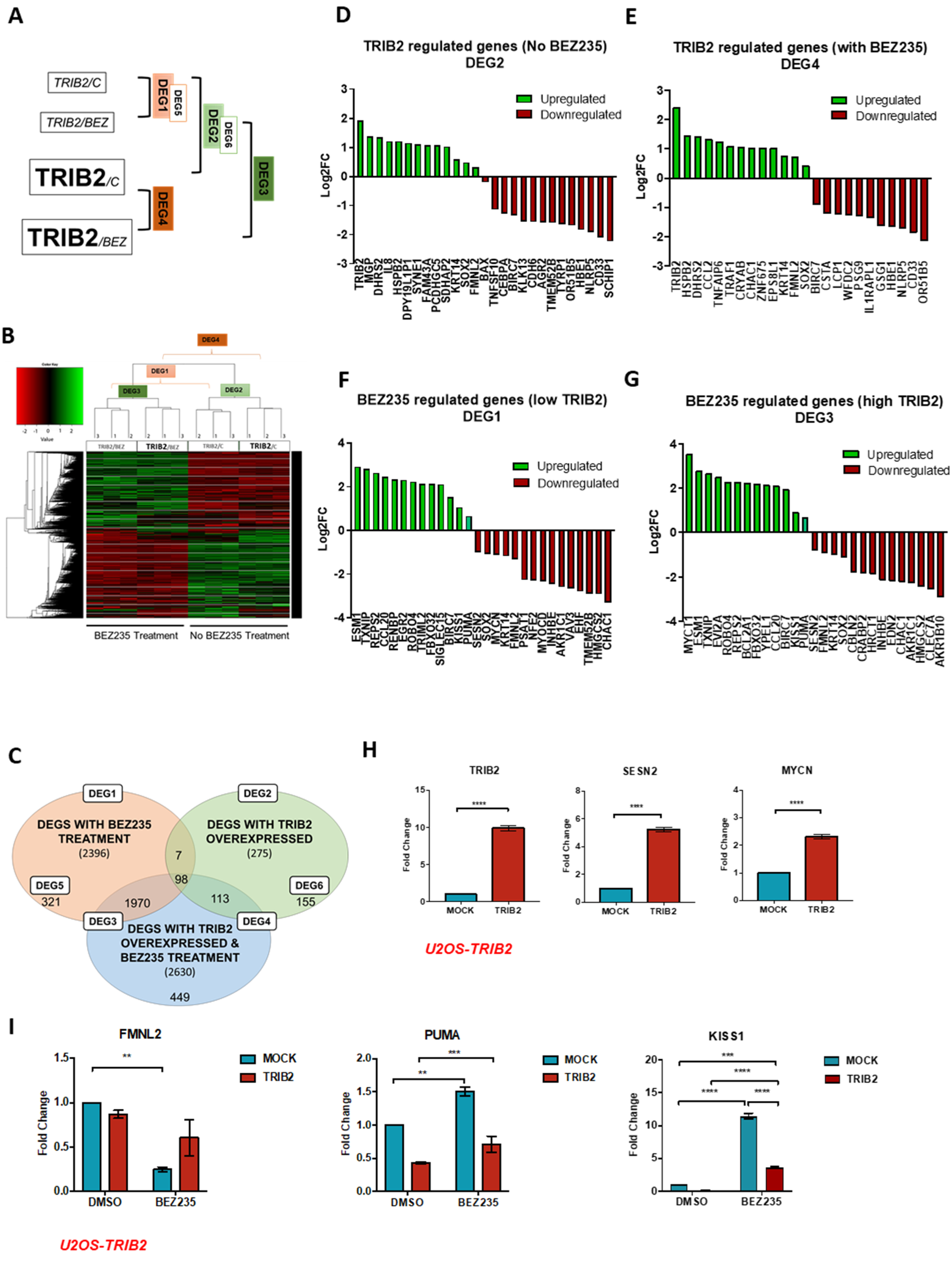
2.1. TRIB2-Induced Transcriptional Signature
2.2. HAR and PIP Reverse TRIB2-Induced Expression Profiles
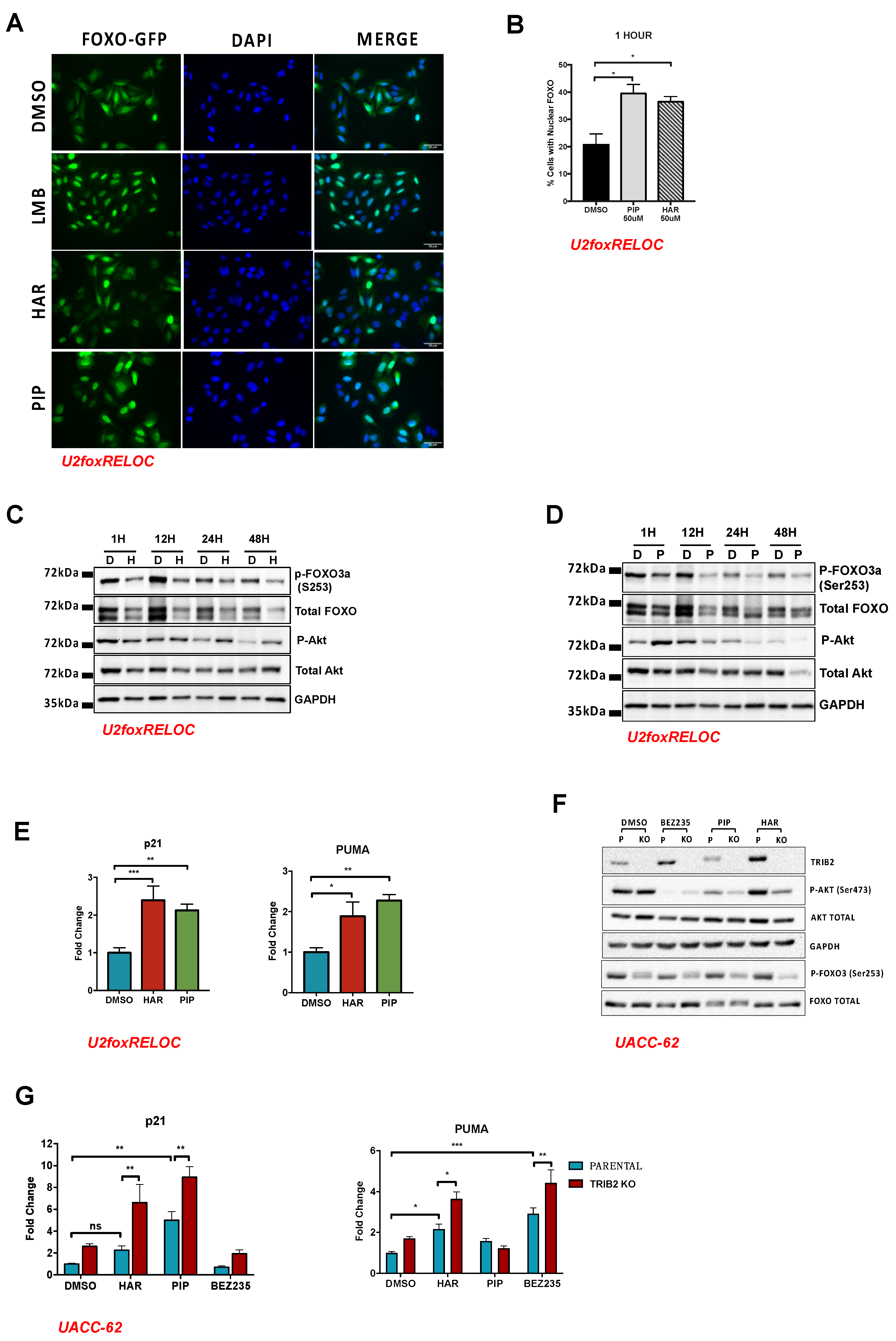
2.3. HAR and PIP Promote FOXO Nuclear Translocation
2.4. Validation Using CRISPR/Cas9 TRIB2 KO Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. hTRIB2 KO Cell Generation
4.3. Cell Transfection and Selection
4.4. RNA Sequencing
4.5. Mapping and Determination of DEGs and Gene Enrichment Analysis
4.6. Quantitative Real-Time PCR
4.7. MTT Assay
4.8. Drug Synergy Assay
4.9. FOXO Translocation Assay
4.10. Image and Data Analysis
4.11. Western Blot
4.12. Trypan Blue Exclusion Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaco, L.; Dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef] [PubMed]
- Grosshans, J.; Wieschaus, E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell 2000, 101, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Hegedus, Z.; Czibula, A.; Kiss-Toth, E. Tribbles: A family of kinase-like proteins with potent signalling regulatory function. Cell Signal 2007, 19, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, L.; Brand, A.H. Cell cycle heterogeneity directs the timing of neural stem cell activation from quiescence. Science 2018, 360, 99–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, W. Tribbles breaking bad: TRIB2 suppresses FOXO and acts as an oncogenic protein in melanoma. Biochem. Soc. Trans. 2015, 43, 1085–1088. [Google Scholar] [CrossRef]
- Zanella, F.; Renner, O.; Garcia, B.; Callejas, S.; Dopazo, A.; Peregrina, S.; Carnero, A.; Link, W. Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene 2010, 29, 2973–2982. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Kalathur, R.K.; Callejas, S.; Colaco, L.; Brandao, R.; Serelde, B.; Cebria, A.; Blanco-Aparicio, C.; Pastor, J.; Futschik, M.; et al. A novel phosphatidylinositol 3-kinase (PI3K) inhibitor directs a potent FOXO-dependent, p53-independent cell cycle arrest phenotype characterized by the differential induction of a subset of FOXO-regulated genes. Breast Cancer Res. 2014, 16, 482. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Kalathur, R.K.; Colaco, L.; Brandao, R.; Ugurel, S.; Futschik, M.; Link, W. TRIB2 as a biomarker for diagnosis and progression of melanoma. Carcinogenesis 2015, 36, 469–477. [Google Scholar] [CrossRef]
- Hou, Z.; Guo, K.; Sun, X.; Hu, F.; Chen, Q.; Luo, X.; Wang, G.; Hu, J.; Sun, L. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol. Cancer 2018, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Keeshan, K.; Bailis, W.; Dedhia, P.H.; Vega, M.E.; Shestova, O.; Xu, L.; Toscano, K.; Uljon, S.N.; Blacklow, S.C.; Pear, W.S. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood 2010, 116, 4948–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Yu, D.; Perez-Soler, R.; Klostergaard, J.; Zou, Y. TRIB2 contributes to cisplatin resistance in small cell lung cancer. Oncotarget 2017, 8, 109596–109608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhou, X.; Qu, H.; Ma, Y.; Yue, Z.; Shang, W.; Wang, P.; Xie, S.; Li, Y.; Sun, Y. TRIB2 knockdown as a regulator of chemotherapy resistance and proliferation via the ERK/STAT3 signaling pathway in human chronic myelogenous leukemia K562/ADM cells. Oncol. Rep. 2018, 39, 1910–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.; Yalla, K.; Salome, M.; Moka, H.A.; Castaneda, E.G.; Eyers, P.A.; Keeshan, K. Trib2 expression in granulocyte-monocyte progenitors drives a highly drug resistant acute myeloid leukaemia linked to elevated Bcl2. Oncotarget 2018, 9, 14977–14992. [Google Scholar] [CrossRef]
- Salome, M.; Magee, A.; Yalla, K.; Chaudhury, S.; Sarrou, E.; Carmody, R.J.; Keeshan, K. A Trib2-p38 axis controls myeloid leukaemia cell cycle and stress response signalling. Cell Death Dis. 2018, 9, 443. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zuo, J.; Wahafu, A.; Wang, M.D.; Li, R.C.; Xie, W.F. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci. Ther. 2020, 26, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Wang, L.; Bi, K.; Jiang, G. Regulation of the C/EBPα signaling pathway in acute myeloid leukemia (Review). Oncol. Rep. 2015, 33, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Lohan, F.; Campos, J.; Ohlsson, E.; Salomè, M.; Forde, C.; Artschwager, R.; Liskamp, R.M.; Cahill, M.R.; Kiely, P.A.; et al. The presence of C/EBPα and its degradation are both required for TRIB2-mediated leukaemia. Oncogene 2016, 35, 5272–5281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Han, L.; Shan, S.; Sun, Y.; Mao, Y. KRT14 promoting invasion and migration of lung cancer cells through ROCK-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 795–803. [Google Scholar]
- Schaefer, T.; Steiner, R.; Lengerke, C. SOX2 and p53 Expression Control Converges in PI3K/AKT Signaling with Versatile Implications for Stemness and Cancer. Int. J. Mol. Sci. 2020, 21, 4902. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Liu, F.; Zhang, Z.; Zou, Y.; Yang, B.; Luo, Y.; Wang, L.; Huang, O. Inhibition of formin like 2 promotes the transition of ectopic endometrial stromal cells to epithelial cells in adenomyosis through a MET-like process. Gene 2019, 710, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ding, Y.; Ye, N.; Wild, C.; Chen, H.; Zhou, J. Direct Activation of Bax Protein for Cancer Therapy. Med. Res. Rev. 2016, 36, 313–341. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Kong, W.Y.; Fang, C.M.; Loh, H.S.; Chuah, L.H.; Abdullah, S.; Ngai, S.C. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit. Rev. Oncol. Hematol. 2019, 143, 81–94. [Google Scholar] [CrossRef]
- Wilhelmson, A.S.; Porse, B.T. CCAAT enhancer binding protein alpha (CEBPA) biallelic acute myeloid leukaemia: Cooperating lesions, molecular mechanisms and clinical relevance. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Yu, Q.; Li, H.; Xie, C.; Wu, Y.; Ma, D.; Sheng, P.; Dai, W.; Jiang, H. BIRC7 promotes epithelial-mesenchymal transition and metastasis in papillary thyroid carcinoma through restraining autophagy. Am. J. Cancer Res. 2020, 10, 78–94. [Google Scholar]
- Ly, T.; Harihar, S.; Welch, D.R. KISS1 in metastatic cancer research and treatment: Potential and paradoxes. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Pasha, M.; Eid, A.H.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxid. Med. Cell Longev. 2017, 2017, 3296294. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Beyond the Warburg Effect: N-Myc Contributes to Metabolic Reprogramming in Cancer Cells. Front. Oncol. 2020, 10, 791. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27 (Suppl. S1), S71–S83. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Callow, M.; Speed, T.; Dudoit, S. Statistical Methods for Identifying Differentially Expressed Genes in Replicated cDNA Microarray Experiments. Stat. Sin. 2002, 12, 111–139. [Google Scholar]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Brierley, D.I.; Davidson, C. Developments in harmine pharmacology—Implications for ayahuasca use and drug-dependence treatment. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 39, 263–272. [Google Scholar] [CrossRef]
- Prejanò, M.; Marino, T.; Russo, N. On the Inhibition Mechanism of Glutathione Transferase P1 by Piperlongumine. Insight from Theory. Front. Chem. 2018, 6, 606. [Google Scholar] [CrossRef] [PubMed]
- Piska, K.; Gunia-Krzyzak, A.; Koczurkiewicz, P.; Wojcik-Pszczola, K.; Pekala, E. Piperlongumine (piplartine) as a lead compound for anticancer agents—Synthesis and properties of analogues: A mini-review. Eur. J. Med. Chem. 2018, 156, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Senba, M.; Mori, N. Effects of NVP-BEZ235, a dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, on HTLV-1-infected T-cell lines. Oncol. Lett. 2018, 15, 5311–5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhl, K.L.; Schultz, C.R.; Geerts, D.; Bachmann, A.S. Harmine, a dual-specificity tyrosine phosphorylation-regulated kinase (DYRK) inhibitor induces caspase-mediated apoptosis in neuroblastoma. Cancer Cell Int. 2018, 18, 82. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kasukabe, T.; Kumakura, S. Piperlongumine rapidly induces the death of human pancreatic cancer cells mainly through the induction of ferroptosis. Int. J. Oncol. 2018, 52, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Zanella, F.; Rosado, A.; Garcia, B.; Carnero, A.; Link, W. Chemical genetic analysis of FOXO nuclear-cytoplasmic shuttling by using image-based cell screening. Chembiochem Eur. J. Chem. Biol. 2008, 9, 2229–2237. [Google Scholar] [CrossRef]
- Zanella, F.; Rosado, A.; Blanco, F.; Henderson, B.R.; Carnero, A.; Link, W. An HTS approach to screen for antagonists of the nuclear export machinery using high content cell-based assays. Assay Drug Dev. Technol. 2007, 5, 333–341. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [Green Version]
- You, H.; Pellegrini, M.; Tsuchihara, K.; Yamamoto, K.; Hacker, G.; Erlacher, M.; Villunger, A.; Mak, T.W. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 2006, 203, 1657–1663. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosich, L.; Montraveta, A.; Xargay-Torrent, S.; López-Guerra, M.; Roldán, J.; Aymerich, M.; Salaverria, I.; Beà, S.; Campo, E.; Pérez-Galán, P.; et al. Dual PI3K/mTOR inhibition is required to effectively impair microenvironment survival signals in mantle cell lymphoma. Oncotarget 2014, 5, 6788–6800. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, L.F.; Bhasin, M.K.; de Vasconcellos, J.F.; Paccez, J.D.; Gu, X.; Kung, A.L.; Libermann, T.A. Computational Repositioning and Preclinical Validation of Pentamidine for Renal Cell Cancer. Mol. Cancer Ther. 2014, 13, 1929–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Noort, V.; Schölch, S.; Iskar, M.; Zeller, G.; Ostertag, K.; Schweitzer, C.; Werner, K.; Weitz, J.; Koch, M.; Bork, P. Novel drug candidates for the treatment of metastatic colorectal cancer through global inverse gene-expression profiling. Cancer Res. 2014, 74, 5690–5699. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e1417. [Google Scholar] [CrossRef]
- Shrivastava, S.; Kulkarni, P.; Thummuri, D.; Jeengar, M.K.; Naidu, V.G.M.; Alvala, M.; Redddy, G.B.; Ramakrishna, S. Piperlongumine, an alkaloid causes inhibition of PI3 K/Akt/mTOR signaling axis to induce caspase-dependent apoptosis in human triple-negative breast cancer cells. Apoptosis Int. J. Program. Cell Death 2014, 19, 1148–1164. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, Z.; Lin, J.; Zhao, S.; Hao, M.; Xu, J.; Li, Y.; Zhao, Q.; Tao, L.; Diao, A. Piperlongumine-induced nuclear translocation of the FOXO3A transcription factor triggers BIM-mediated apoptosis in cancer cells. Biochem. Pharmacol. 2019, 163, 101–110. [Google Scholar] [CrossRef]
- Niu, M.; Xu, X.; Shen, Y.; Yao, Y.; Qiao, J.; Zhu, F.; Zeng, L.; Liu, X.; Xu, K. Piperlongumine is a novel nuclear export inhibitor with potent anticancer activity. Chemico-Biol. Interact. 2015, 237, 66–72. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Park, J.Y.; Kim, J.W.; Kwon, M.; Lee, B.H. Piperlongumine selectively kills cancer cells and increases cisplatin antitumor activity in head and neck cancer. Oncotarget 2014, 5, 9227–9238. [Google Scholar] [CrossRef] [Green Version]
- Kang, Q.; Yan, S. Piperlongumine reverses doxorubicin resistance through the PI3K/Akt signaling pathway in K562/A02 human leukemia cells. Exp. Ther. Med. 2015, 9, 1345–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.; Chowdhury, N.; Doddapaneni, R.; Boakye, C.H.A.; Godugu, C.; Singh, M. Piperlongumine for Enhancing Oral Bioavailability and Cytotoxicity of Docetaxel in Triple-Negative Breast Cancer. J. Pharm. Sci. 2015, 104, 4417–4426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-Q.; Wang, Y.-C.; Guo, Y.-T.; Tang, X. Effect of piperlongumine on drug resistance reversal in human retinoblastoma HXO-RB44/VCR and SO-Rb50/CBP cell lines. Int. J. Clin. Exp. Pathol. 2015, 8, 2525–2534. [Google Scholar] [PubMed]
- Zhang, C.; He, L.J.; Zhu, Y.B.; Fan, Q.Z.; Miao, D.D.; Zhang, S.P.; Zhao, W.Y.; Liu, X.P. Piperlongumine Inhibits Akt Phosphorylation to Reverse Resistance to Cisplatin in Human Non-Small Cell Lung Cancer Cells via ROS Regulation. Front. Pharmacol. 2019, 10, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Sablin, S.O.; Ramsay, R.R. Inhibition of Monoamine Oxidase A by β-Carboline Derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [Green Version]
- Woods, Y.L.; Rena, G.; Morrice, N.; Barthel, A.; Becker, W.; Guo, S.; Unterman, T.G.; Cohen, P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 2001, 355, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, A.; Nanjappa, V.; Raja, R.; Sathe, G.; Puttamallesh, V.N.; Jain, A.P.; Pinto, S.M.; Balaji, S.A.; Chavan, S.; Sahasrabuddhe, N.A.; et al. A dual specificity kinase, DYRK1A, as a potential therapeutic target for head and neck squamous cell carcinoma. Sci. Rep. 2016, 6, 36132. [Google Scholar] [CrossRef] [Green Version]
- Zanella, F.; Rosado, A.; Garcia, B.; Carnero, A.; Link, W. Using multiplexed regulation of luciferase activity and GFP translocation to screen for FOXO modulators. BMC Cell Biol. 2009, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrado-Castellarnau, M.; Cortés, R.; Zanuy, M.; Tarragó-Celada, J.; Polat, I.H.; Hill, R.; Fan, T.W.M.; Link, W.; Cascante, M. Methylseleninic acid promotes antitumour effects via nuclear FOXO3a translocation through Akt inhibition. Pharmacol. Res. 2015, 102, 218–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, S.; Silva, A.; De Sousa-Coelho, A.L.; Duarte, I.; Grenho, I.; Santos, B.; Mayoral-Varo, V.; Megias, D.; Sánchez-Cabo, F.; Dopazo, A.; et al. Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance. Cancers 2020, 12, 3689. https://doi.org/10.3390/cancers12123689
Machado S, Silva A, De Sousa-Coelho AL, Duarte I, Grenho I, Santos B, Mayoral-Varo V, Megias D, Sánchez-Cabo F, Dopazo A, et al. Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance. Cancers. 2020; 12(12):3689. https://doi.org/10.3390/cancers12123689
Chicago/Turabian StyleMachado, Susana, Andreia Silva, Ana Luísa De Sousa-Coelho, Isabel Duarte, Inês Grenho, Bruno Santos, Victor Mayoral-Varo, Diego Megias, Fátima Sánchez-Cabo, Ana Dopazo, and et al. 2020. "Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance" Cancers 12, no. 12: 3689. https://doi.org/10.3390/cancers12123689
APA StyleMachado, S., Silva, A., De Sousa-Coelho, A. L., Duarte, I., Grenho, I., Santos, B., Mayoral-Varo, V., Megias, D., Sánchez-Cabo, F., Dopazo, A., Ferreira, B. I., & Link, W. (2020). Harmine and Piperlongumine Revert TRIB2-Mediated Drug Resistance. Cancers, 12(12), 3689. https://doi.org/10.3390/cancers12123689