Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3

 ,
,
Abstract
:Simple Summary
Abstract
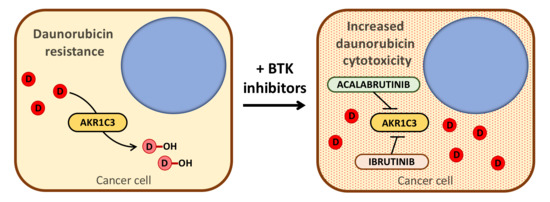
1. Introduction
2. Results
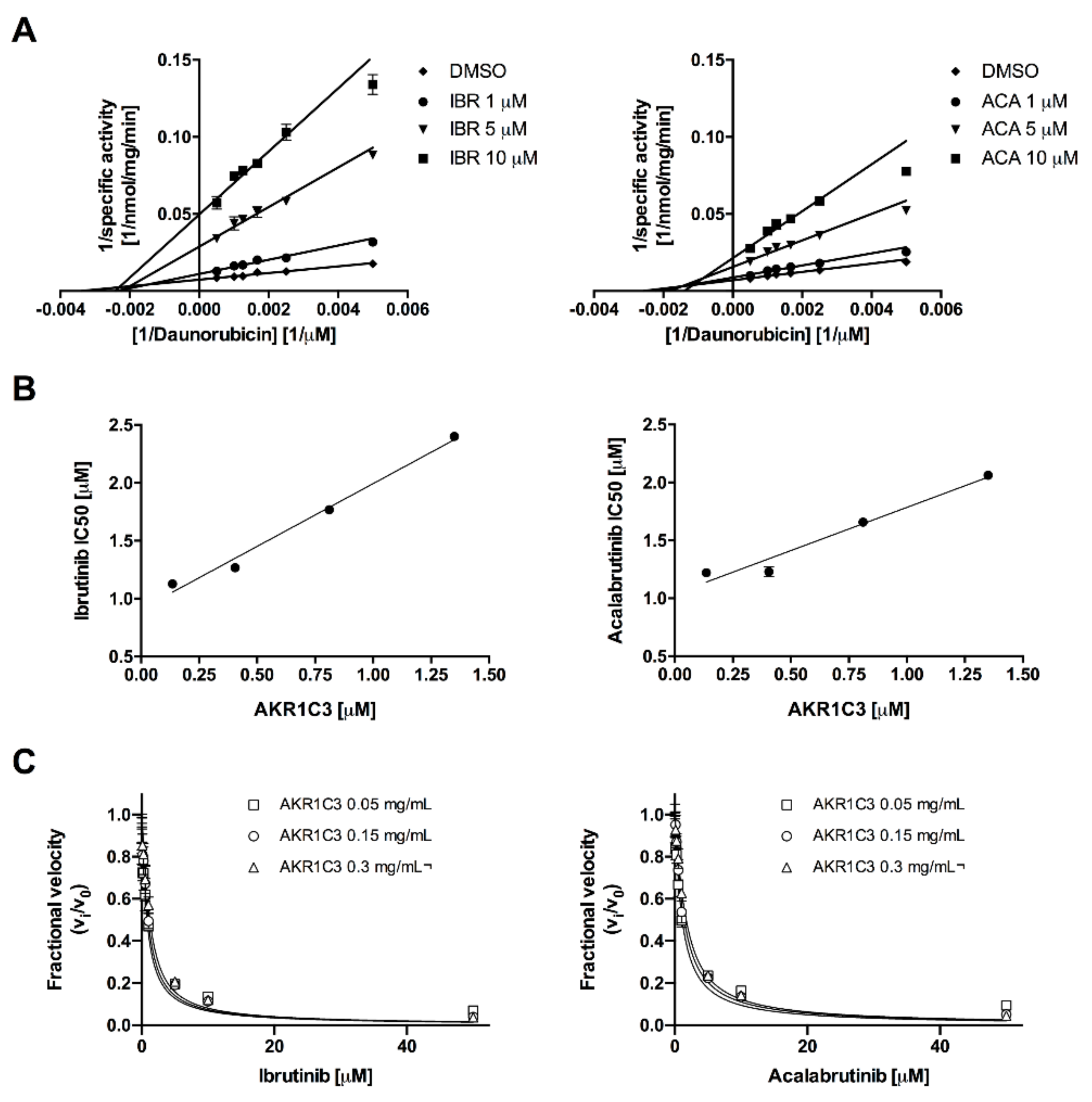
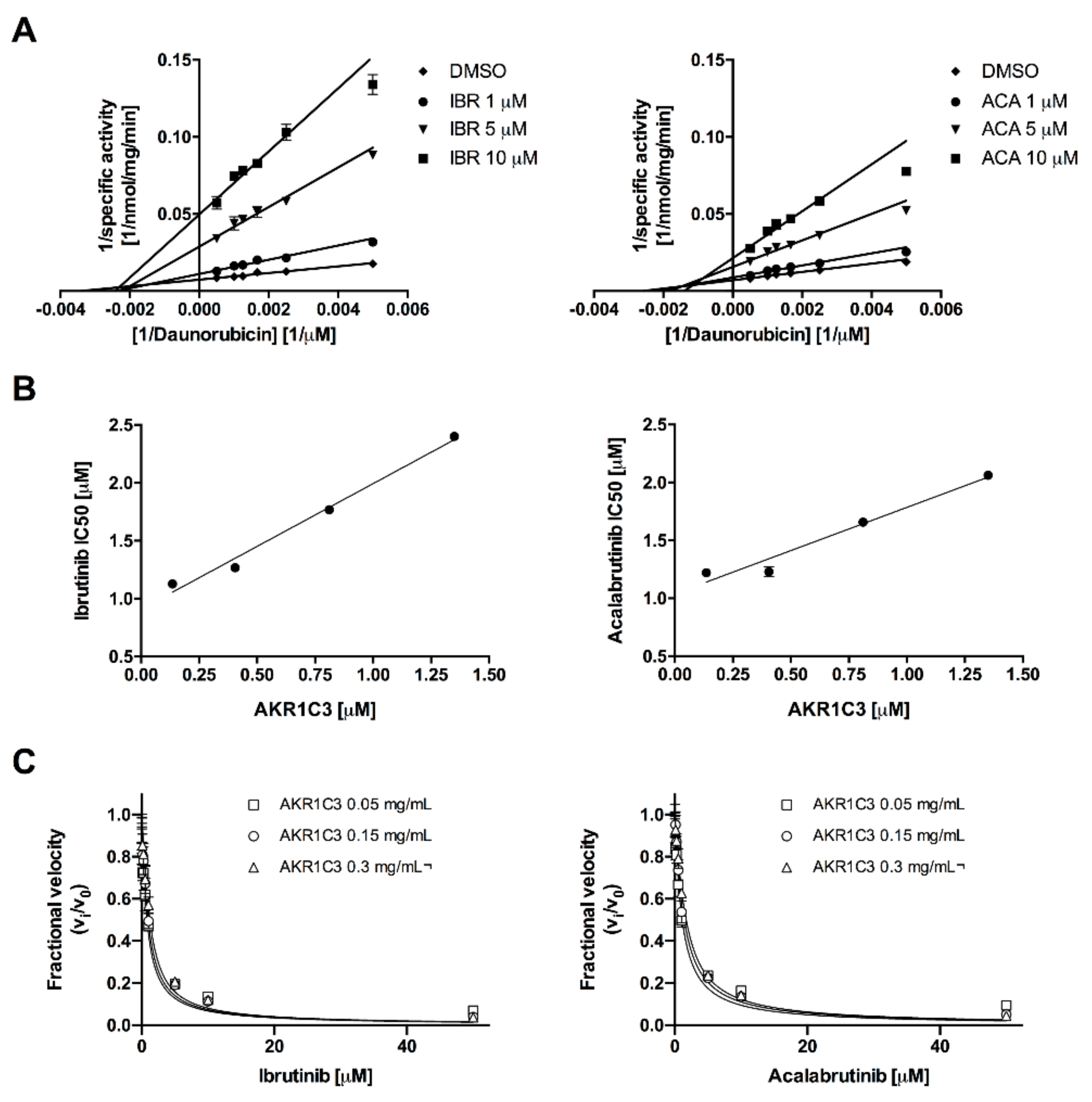
2.1. Acalabrutinib and Ibrutinib Inhibit the AKR1C3-Mediated Reduction of Daunorubicin In Vitro
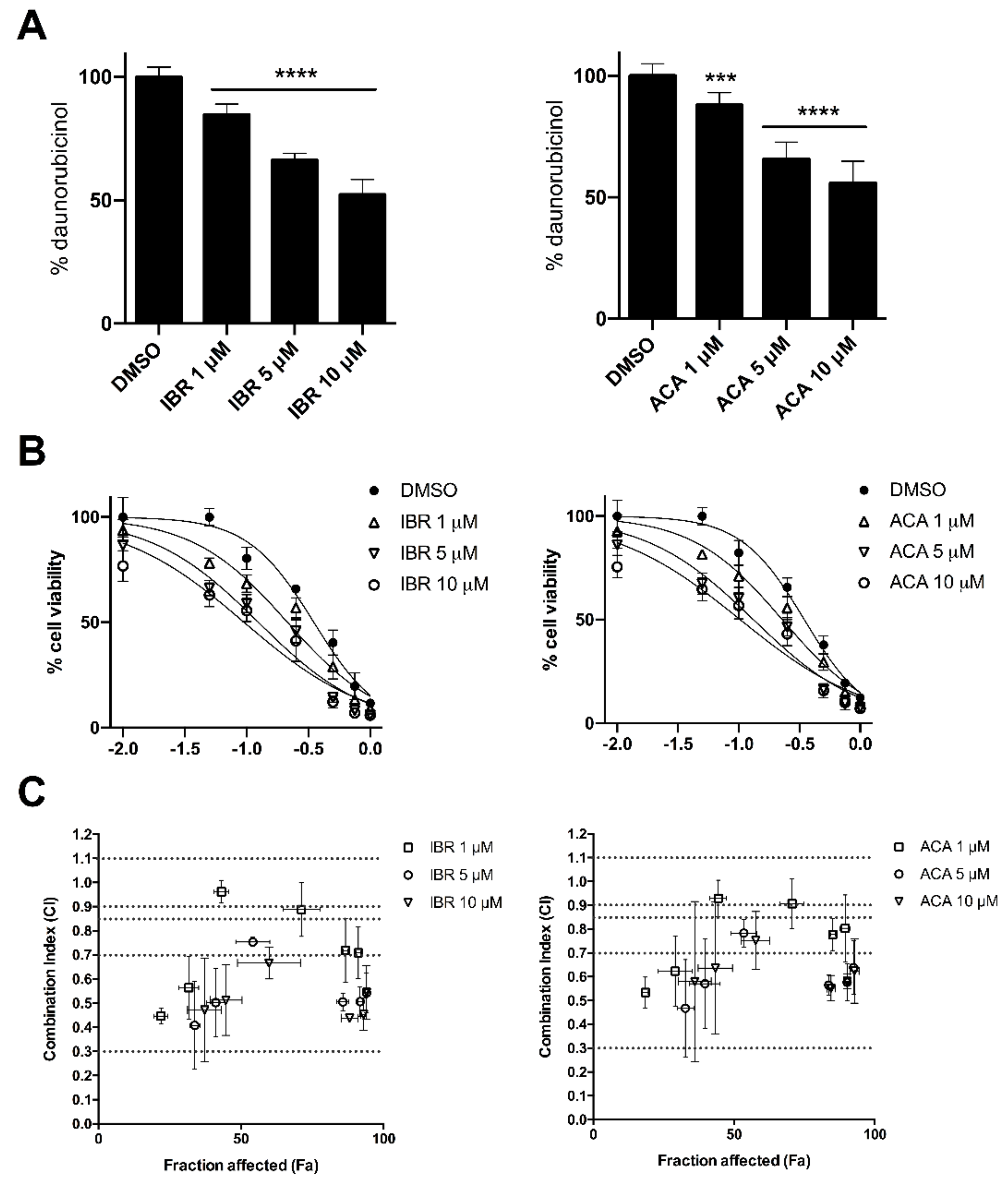
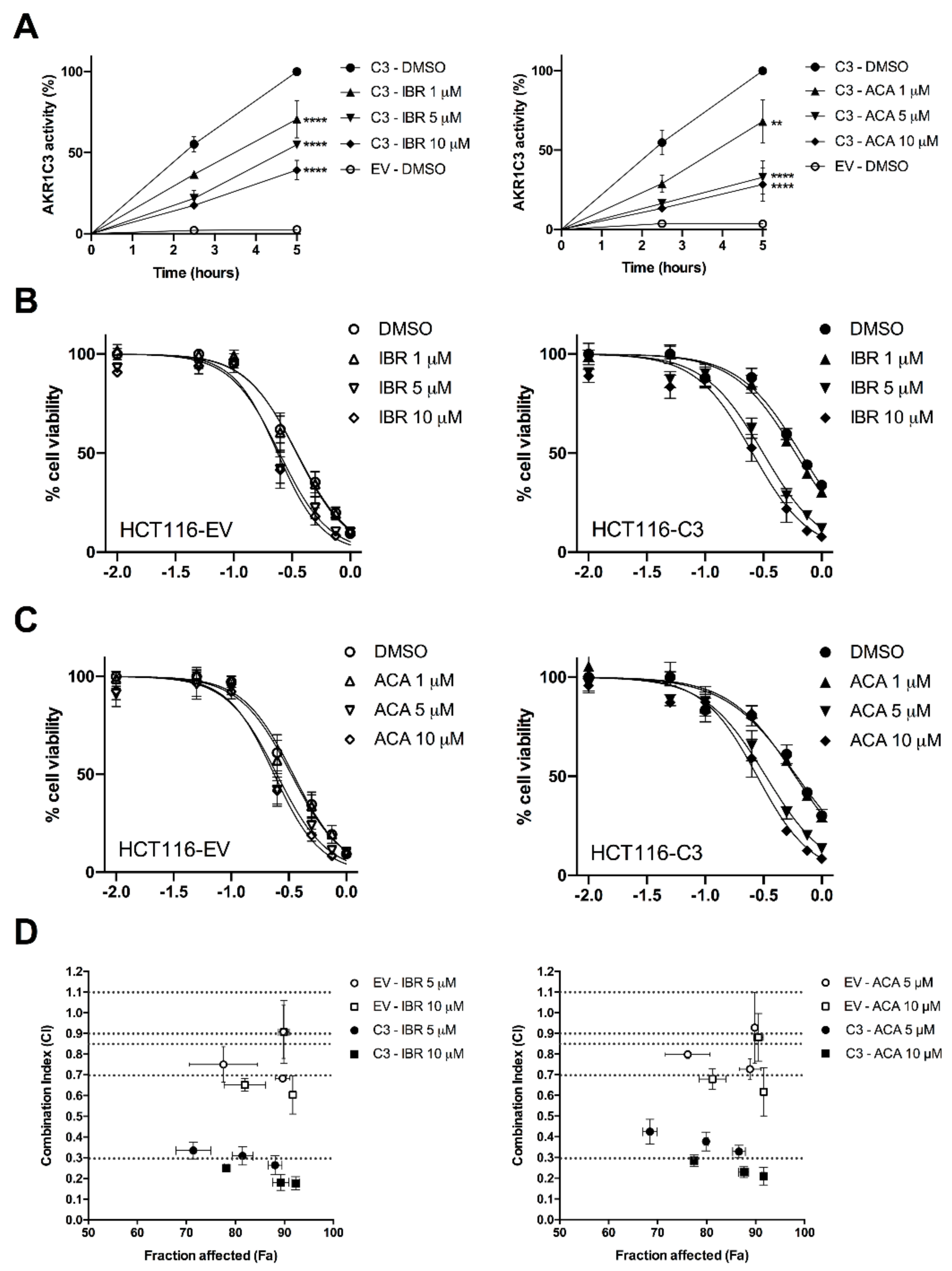
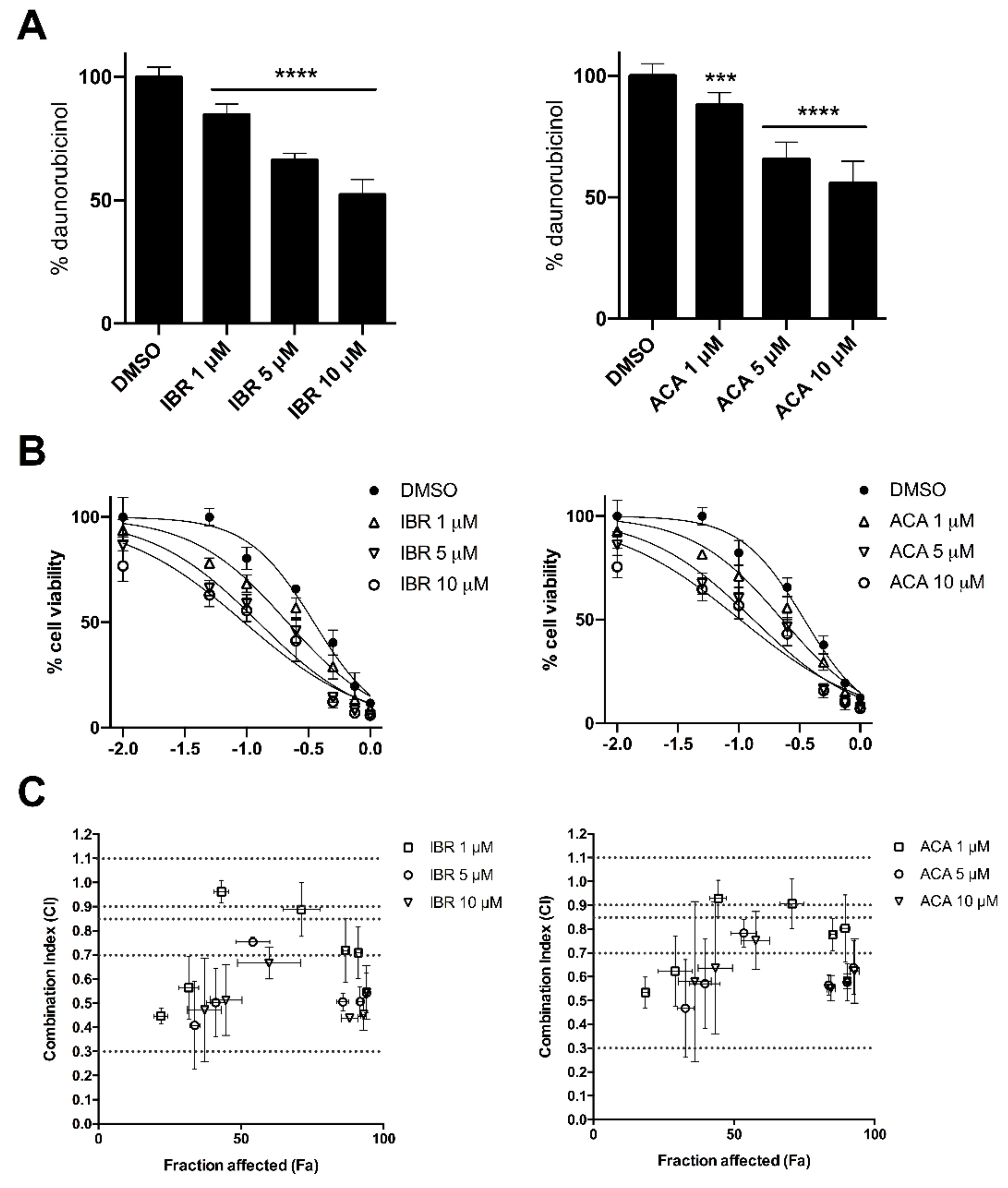
2.2. BTK Inhibitors Counteract AKR1C3-Mediated Daunorubicin Resistance
2.3. BTK Inhibitors Synergise Daunorubicin Cytotoxicity in Cancer Cells
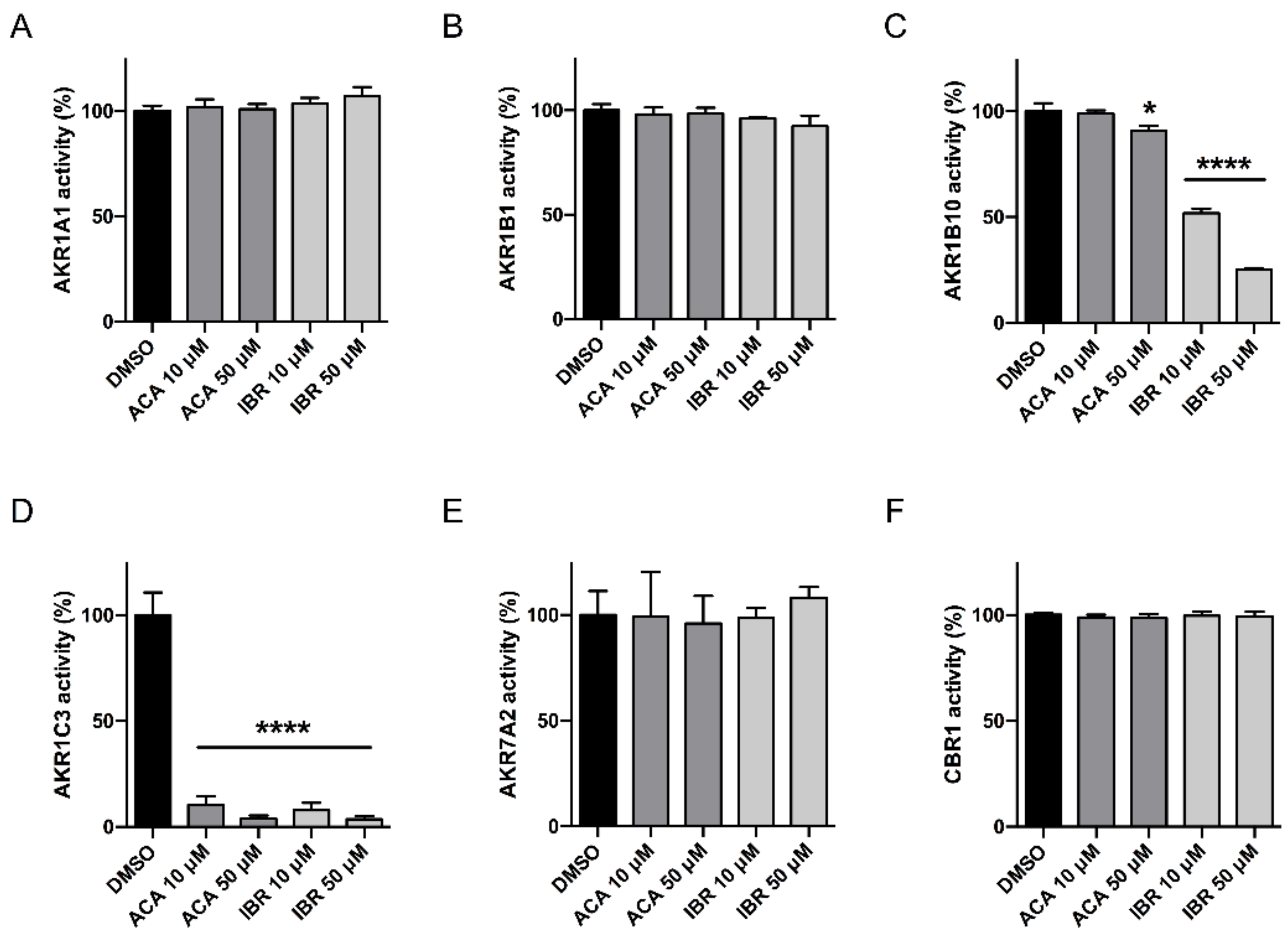
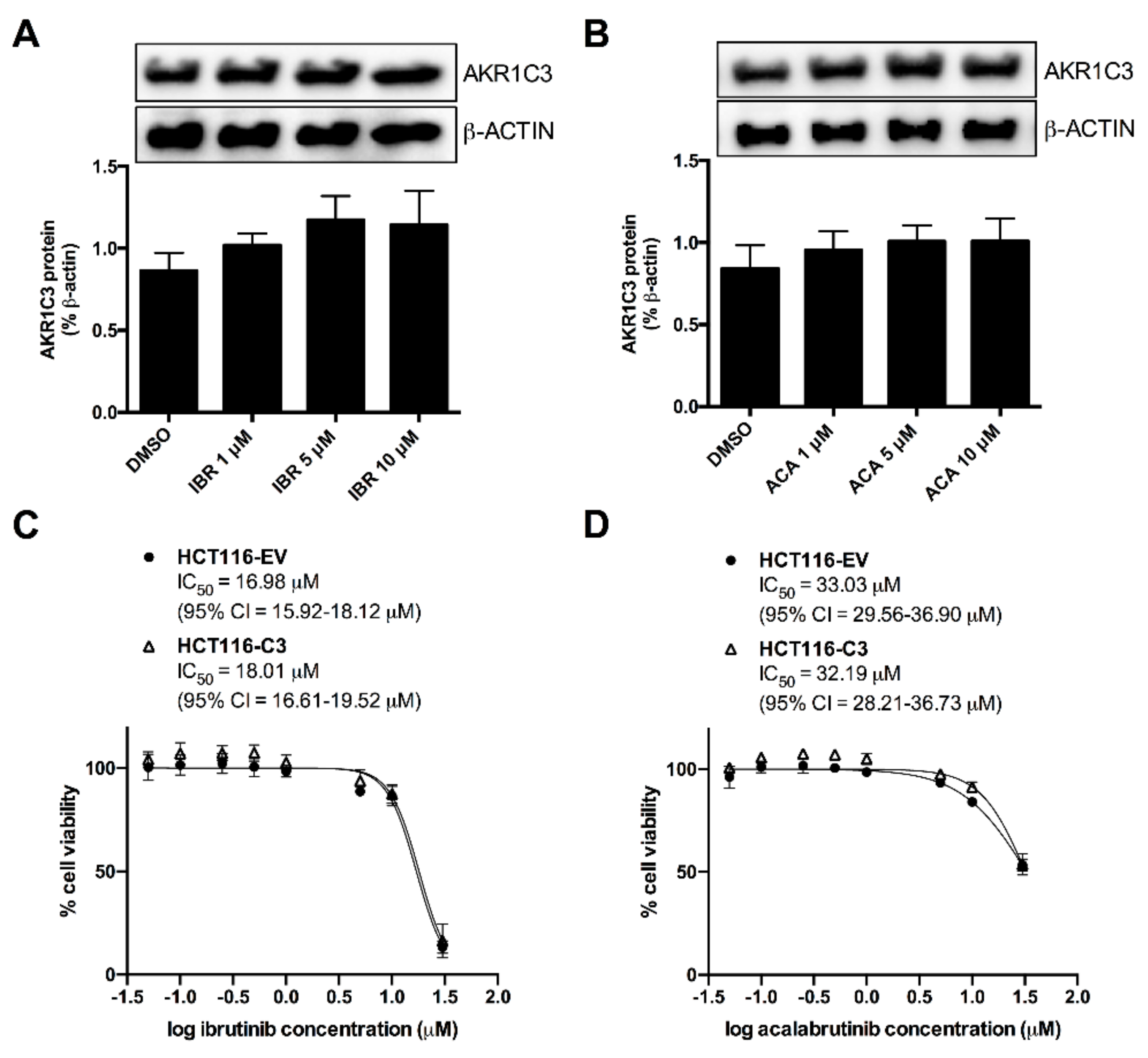
2.4. AKR1C3 Expression Is Not Correlated with the Effects of BTK Inhibitors
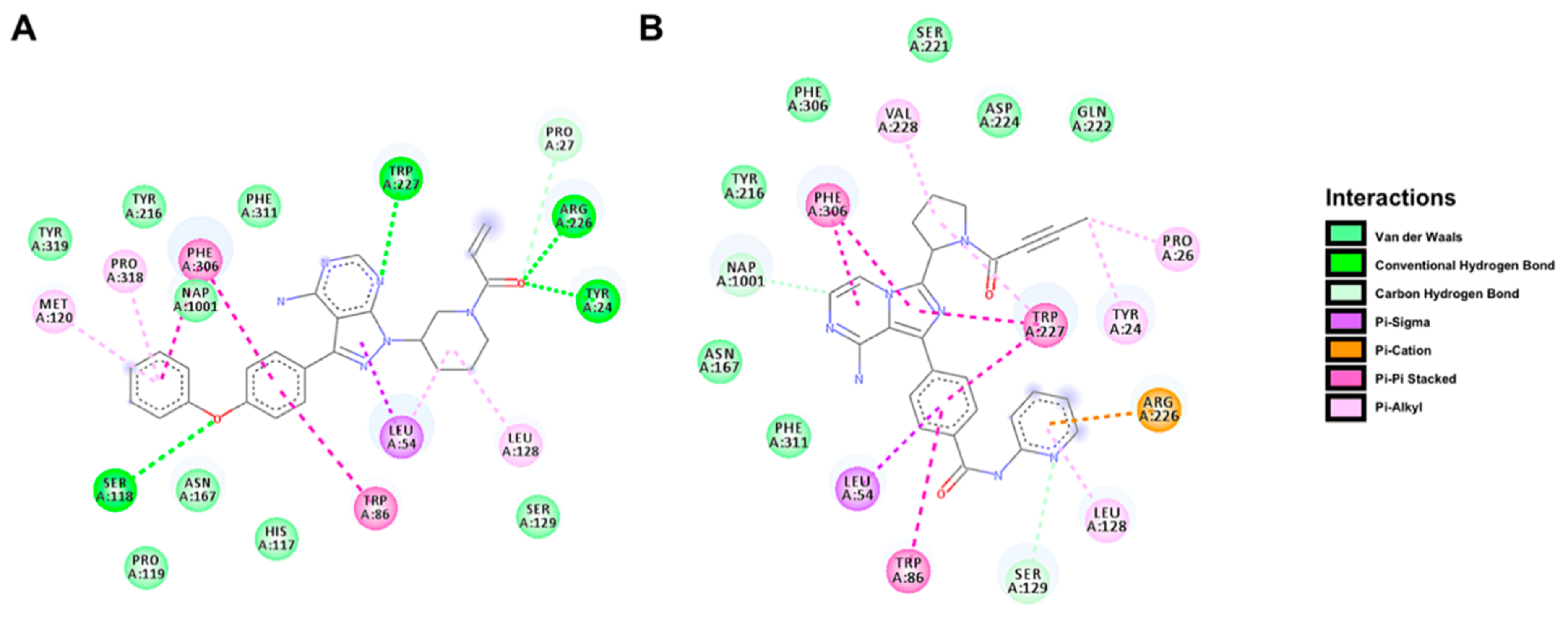
2.5. Docking Prediction for the Binding of BTK Inhibitors with AKR1C3
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Determination of Dau Metabolism by Recombinant CREs
4.3. Cell Cultures
4.4. Transient Transfection of HCT116 Cells
4.5. Determination of Dau Metabolism by Cells
4.6. Determination of Synergism
4.7. Flexible Docking of BTK Inhibitors in the Catalytic Site of AKR1C3
4.8. Statistical Analysis
5. Conclusions
Abbreviations
| ABC | ATP-binding cassette |
| AKR | Aldo-keto reductase |
| AML | Acute myeloid leukaemia |
| CI | Combination index |
| CRE | Carbonyl-reducing enzyme |
| Dau | Daunorubicin |
| Dau-ol | Daunorubicinol |
| DMSO | Dimethyl sulfoxide |
| Fa | Fraction affected |
| FLT3 | FMS-like tyrosine kinase 3 receptor |
| HCT116-AKR1C3 | Cells transfected with pCI_AKR1C3 |
| Mid | Midostaurin |
| MTT | 3-(4,5-Dimethylthiazoyl-2-yl)2,5-diphenyl tetrazolium bromide |
| MDR | Multidrug resistance |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| PGD2 | Prostaglandin D2 |
| 11β-PGF2α | 11β-Prostaglandin F2α |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| SD | Standard deviation |
| SDR | Short-chain dehydrogenase/reductase |
| TKI | Tyrosine kinase inhibitor |
| UHPLC | Ultra high-performance liquid chromatography |
| WT | Wild type |
| 15dPGJ2 | 15-deoxyΔ12,14PGJ2 |
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. Bioinform. MicroRNA Res. 2010, 596, 47–76. [Google Scholar] [CrossRef]
- Wu, C.-P.; Hsieh, C.-H.; Wu, Y.-S. The Emergence of Drug Transporter-Mediated Multidrug Resistance to Cancer Chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Pal, D.; Mandal, A. Mechanisms of drug resistance in cancer chemotherapy: Coordinated role and regulation of efflux transporters and metabolizing enzymes. Curr. Pharm. Des. 2013, 19, 7126–7140. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.; Maare, C.; Skovsgaard, T. Cellular resistance to anthracyclines. Gen. Pharmacol. Vasc. Syst. 1996, 27, 251–255. [Google Scholar] [CrossRef]
- Chien, A.J.; Moasser, M.M. Cellular Mechanisms of Resistance to Anthracyclines and Taxanes in Cancer: Intrinsic and Acquired. Semin. Oncol. 2008, 35, S1–S14. [Google Scholar] [CrossRef]
- Kaye, S.; Merry, S. Tumour cell resistance to anthracyclines? A review. Cancer Chemother. Pharmacol. 1985, 14, 96–103. [Google Scholar] [CrossRef]
- Gianni, L. Anthracycline resistance: The problem and its current definition. Semin. Oncol. 1997, 24, 10. [Google Scholar]
- Bains, O.S.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Naturally Occurring Variants of Human Aldo-Keto Reductases with Reduced In Vitro Metabolism of Daunorubicin and Doxorubicin. J. Pharmacol. Exp. Ther. 2010, 335, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Bains, O.S.; Szeitz, A.; Lubieniecka, J.M.; Cragg, G.E.; Grigliatti, T.A.; Riggs, K.W.; Reid, R.E. A Correlation between Cytotoxicity and Reductase-Mediated Metabolism in Cell Lines Treated with Doxorubicin and Daunorubicin. J. Pharmacol. Exp. Ther. 2013, 347, 375–387. [Google Scholar] [CrossRef]
- Birtwistle, J.; Hayden, R.E.; Khanim, F.; Green, R.M.; Pearce, C.; Davies, N.J.; Wake, N.; Schrewe, H.; Ride, J.P.; Chipman, J.K.; et al. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat. Res. Mol. Mech. Mutagen. 2009, 662, 67–74. [Google Scholar] [CrossRef]
- Desmond, J.C.; Mountford, J.C.; Drayson, M.T.; Walker, A.E.; Hewison, M.; Ride, J.P.; Luong, Q.T.; Hayden, E.R.; Vanin, E.F.; Bunce, C.M. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003, 63, 505–512. [Google Scholar] [PubMed]
- Byrns, M.C.; Duan, L.; Lee, S.H.; Blair, I.A.; Penning, T.M. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J. Steroid Biochem. Mol. Biol. 2010, 118, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozmorov, M.G.; Azzarello, J.T.; Wren, J.D.; Fung, K.-M.; Yang, Q.; Davis, J.S.; Hurst, R.E.; Culkin, D.J.; Penning, T.M.; Lin, H.-K. Elevated AKR1C3 expression promotes prostate cancer cell survival and prostate cell-mediated endothelial cell tube formation: Implications for prostate cancer progressioan. BMC Cancer 2010, 10, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; He, S.; Chen, Y.; Liu, Y.; Feng, F.; Liu, W.; Guo, Q.; Zhao, L.; Sun, H. Overview of AKR1C3: Inhibitor Achievements and Disease Insights. J. Med. Chem. 2020, 63, 11305–11329. [Google Scholar] [CrossRef] [PubMed]
- Hofman, J.; Malcekova, B.; Skarka, A.; Novotna, E.; Wsól, V. Anthracycline resistance mediated by reductive metabolism in cancer cells: The role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–248. [Google Scholar] [CrossRef]
- Verma, K.; Zang, T.; Gupta, N.; Penning, T.M.; Trippier, P.C. Selective AKR1C3 Inhibitors Potentiate Chemotherapeutic Activity in Multiple Acute Myeloid Leukemia (AML) Cell Lines. ACS Med. Chem. Lett. 2016, 7, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Verma, K.; Zang, T.; Penning, T.M.; Trippier, P.C. Potent and Highly Selective Aldo–Keto Reductase 1C3 (AKR1C3) Inhibitors Act as Chemotherapeutic Potentiators in Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. J. Med. Chem. 2019, 62, 3590–3616. [Google Scholar] [CrossRef]
- Novotná, E.; Büküm, N.; Hofman, J.; Flaxová, M.; Kouklíková, E.; Louvarová, D.; Wsól, V. Roscovitine and purvalanol A effectively reverse anthracycline resistance mediated by the activity of aldo-keto reductase 1C3 (AKR1C3): A promising therapeutic target for cancer treatment. Biochem. Pharmacol. 2018, 156, 22–31. [Google Scholar] [CrossRef]
- Novotná, E.; Büküm, N.; Hofman, J.; Flaxová, M.; Kouklíková, E.; Louvarová, D.; Wsól, V. Aldo-keto reductase 1C3 (AKR1C3): A missing piece of the puzzle in the dinaciclib interaction profile. Arch. Toxicol. 2018, 92, 2845–2857. [Google Scholar] [CrossRef]
- Novotná, E.; Morell, A.; Büküm, N.; Hofman, J.; Danielisová, P.; Wsól, V. Interactions of antileukemic drugs with daunorubicin reductases: Could reductases affect the clinical efficacy of daunorubicin chemoregimens? Arch. Toxicol. 2020, 94, 3059–3068. [Google Scholar] [CrossRef]
- Lindvall, J.M.; Blomberg, K.E.M.; Väliaho, J.; Vargas, L.; Heinonen, J.E.; Berglof, A.; Mohamed, A.J.; Nore, B.F.; Vihinen, M.; Smith, C.I.E. Bruton’s tyrosine kinase: Cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol. Rev. 2005, 203, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, S.; Saffran, D.C.; Rawlings, D.J.; Parolini, O.; Allen, R.; Klisak, I.; Sparkes, R.S.; Kubagawa, H.; Mohandas, T.; Quan, S.; et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993, 72, 279–290. [Google Scholar] [CrossRef]
- Di Paolo, A.J.; Huang, T.; Balazs, M.; Barbosa, J.; Barck, K.H.; Bravo, B.J.; Carano, R.A.D.; Darrow, J.W.; Davies, D.R.; DeForge, E.L.; et al. Specific Btk inhibition suppresses B cell– and myeloid cell–mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Deepali, S.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Rotin, L.E.; Gronda, M.; Hurren, R.; Wang, X.; Minden, M.D.; Slassi, M.; Schimmer, A.D. Investigating the synergistic mechanism between ibrutinib and daunorubicin in acute myeloid leukemia cells. Leuk. Lymphoma 2016, 57, 2432–2436. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; Wiley: Hobokin, NJ, USA, 2013. [Google Scholar]
- Lovering, A.L.; Ride, J.P.; Bunce, C.M.; Desmond, J.C.; Cummings, S.M.; White, S.A. Crystal Structures of Prostaglandin D211-Ketoreductase (AKR1C3) in Complex with the Nonsteroidal Anti-Inflammatory Drugs Flufenamic Acid and Indomethacin. Cancer Res. 2004, 64, 1802–1810. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Murray, M.Y.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J. Identification of Bruton’s tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood 2014, 123, 1229–1238. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Patel, A.; Ma, S.-L.; Li, X.J.; Zhang, Y.-K.; Yang, P.-Q.; Kathawala, R.J.; Wang, Y.-J.; Anreddy, N.; Fu, L.-W.; et al. In vitro, in vivoandex vivocharacterization of ibrutinib: A potent inhibitor of the efflux function of the transporter MRP1. Br. J. Pharmacol. 2014, 171, 5845–5857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berglof, A.; Hamasy, A.; Meinke, S.; Palma, M.; Krstić, A.; Månsson, R.; Kimby, E.; Österborg, A.; Smith, C.I.E. Targets for Ibrutinib Beyond B Cell Malignancies. Scand. J. Immunol. 2015, 82, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Griner, L.A.M.; Guha, R.; Shinn, P.; Young, R.M.; Keller, J.M.; Liu, D.; Goldlust, I.S.; Yasgar, A.; McKnight, C.; Boxer, M.B.; et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell–like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2349–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oellerich, T.; Mohr, S.; Corso, J.; Beck, J.; Döbele, C.; Braun, H.; Cremer, A.; Münch, S.; Wicht, J.; Oellerich, M.F.; et al. FLT3-ITD and TLR9 use Bruton tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood 2015, 125, 1936–1947. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Yamaguchi, A.; Morikawa, Y.; Kezuka, C.; Takazawa, H.; Endo, S.; El-Kabbani, O.; Tajima, K.; Ikari, A.; Hara, A. Induction of aldo-keto reductases (AKR1C1 and AKR1C3) abolishes the efficacy of daunorubicin chemotherapy for leukemic U937 cells. Anti-Cancer Drugs 2014, 25, 868–877. [Google Scholar] [CrossRef]
- Bukum, N.; Novotna, E.; Morell, A.; Hofman, J.; Wsól, V. Buparlisib is a novel inhibitor of daunorubicin reduction mediated by aldo-keto reductase 1C3. Chem. Interact. 2019, 302, 101–107. [Google Scholar] [CrossRef]
- Morell, A.; Novotná, E.; Milan, J.; Danielisová, P.; Büküm, N.; Wsól, V. Selective inhibition of aldo-keto reductase 1C3: A novel mechanism involved in midostaurin and daunorubicin synergism. Arch. Toxicol. 2020, 1–12. [Google Scholar] [CrossRef]
- Cihalova, D.; Ceckova, M.; Kučera, R.; Klimes, J.; Staud, F. Dinaciclib, a cyclin-dependent kinase inhibitor, is a substrate of human ABCB1 and ABCG2 and an inhibitor of human ABCC1 in vitro. Biochem. Pharmacol. 2015, 98, 465–472. [Google Scholar] [CrossRef]
- Hsiao, S.-H.; Lusvarghi, S.; Huang, Y.-H.; Ambudkar, S.V.; Hsu, S.-C.; Wu, C.-P. The FLT3 inhibitor midostaurin selectively resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to conventional chemotherapeutic agents. Cancer Lett. 2019, 445, 34–44. [Google Scholar] [CrossRef]
- Ji, N.; Yang, Y.; Cai, C.-Y.; Wang, J.-Q.; Lei, Z.-N.; Wu, Z.-X.; Cui, Q.; Yang, D.-H.; Chen, Z.-S.; Kong, D. Midostaurin Reverses ABCB1-Mediated Multidrug Resistance, an in vitro Study. Front. Oncol. 2019, 9, 514. [Google Scholar] [CrossRef]
- Van Der Kolk, D.M.; De Vries, E.G.E.; Müller, M.; Vellenga, E. The Role of Drug Efflux Pumps in Acute Myeloid Leukemia. Leuk. Lymphoma 2002, 43, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Bailly, J.D.; Muller, C.; Jaffrézou, J.P.; Demur, C.; Gassar, G.; Bordier, C.; Laurent, G. Lack of correlation between expression and function of P-glycoprotein in acute myeloid leukemia cell lines. Leukemia 1995, 9, 799–807. [Google Scholar] [PubMed]
- Scheers, E.; Leclercq, L.; De Jong, J.; Bode, N.; Bockx, M.; Laenen, A.; Cuyckens, F.; Skee, D.; Murphy, J.; Sukbuntherng, J.; et al. Absorption, Metabolism, and Excretion of Oral 14C Radiolabeled Ibrutinib: An Open-Label, Phase I, Single-Dose Study in Healthy Men. Drug Metab. Dispos. 2015, 43, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, V.K.; Lamothe, B.; Ayres, M.L.; Gay, J.; Cheung, J.; Balakrishnan, K.; Ivan, C.; Morse, J.; Nelson, M.; Keating, M.J.; et al. Pharmacodynamics and proteomic analysis of acalabrutinib therapy: Similarity of on-target effects to ibrutinib and rationale for combination therapy. Leukemia 2018, 32, 920–930. [Google Scholar] [CrossRef]
- Zhong, L.; Shen, H.; Huang, C.; Jing, H.; Cao, D. AKR1B10 induces cell resistance to daunorubicin and idarubicin by reducing C13 ketonic group. Toxicol. Appl. Pharmacol. 2011, 255, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Wada, Y.; Endo, S.; Soda, M.; El-Kabbani, O.; Hara, A. Aldo–Keto Reductase 1B10 and Its Role in Proliferation Capacity of Drug-Resistant Cancers. Front. Pharmacol. 2012, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zu, X.; Ma, J.; Liu, Z.; Adeyanju, M.; Cao, D. Aldo–keto reductase family 1 B10 gene silencing results in growth inhibition of colorectal cancer cells: Implication for cancer intervention. Int. J. Cancer 2007, 121, 2301–2306. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhao, Y.; Gu, L.; Niu, X.; Lu, S. Inhibiting proliferation and migration of lung cancer using small interfering RNA targeting on Aldo-keto reductase family 1 member B10. Mol. Med. Rep. 2017, 17, 2153–2160. [Google Scholar] [CrossRef]
- Laffin, B.; Petrash, J.M. Expression of the Aldo-Ketoreductases AKR1B1 and AKR1B10 in Human Cancers. Front. Pharmacol. 2012, 3, 104. [Google Scholar] [CrossRef] [Green Version]
- Škarydová, L.; Nobilis, M.; Wsól, V. Role of carbonyl reducing enzymes in the phase I biotransformation of the non-steroidal anti-inflammatory drug nabumetone in vitro. Xenobiotica 2012, 43, 346–354. [Google Scholar] [CrossRef]
- Škarydová, L.; Živná, L.; Xiong, G.; Maser, E.; Wsól, V. AKR1C3 as a potential target for the inhibitory effect of dietary flavonoids. Chem. Interactions 2009, 178, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, W.P.; Brylinski, M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Chemin. 2015, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. Dassault Systèmes, Discovery Studio Visualizer, v.20.1.0.19295; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BTK-Inhibitor | AKR1C3 (mg/mL) | AKR1C3 (μM) | Kiapp (μM) | 95% CI (μM) | R2 |
|---|---|---|---|---|---|
| Ibrutinib | 0.005 | 0.135 | 0.312 | 0.242–0.382 | 0.881 |
| 0.015 | 0.405 | 0.332 | 0.268–0.396 | 0.944 | |
| 0.030 | 0.811 | 0.354 | 0.278–0.431 | 0.951 | |
| Acalabrutinib | 0.005 | 0.135 | 0.451 | 0.356–0.546 | 0.946 |
| 0.015 | 0.405 | 0.521 | 0.454–0.589 | 0.983 | |
| 0.030 | 0.811 | 0.566 | 0.496–0.636 | 0.987 |
| BTK-Inhibitor | Concentration (μM) | IC50 (95% CI) [μM] | |
|---|---|---|---|
| HCT116-EV | HCT116-C3 | ||
| Ibrutinib | 0 | 0.35 (0.33–0.37) | 0.66 (0.62–0.70) |
| 1 | 0.35 (0.32–0.37) | 0.59 (0.56–0.63) | |
| 5 | 0.25 (0.22–0.27) | 0.31 (0.29–0.34) ** | |
| 10 | 0.24 (0.22–0.26) | 0.25 (0.23–0.28) ** | |
| Acalabrutinib | 0 | 0.34 (0.32–0.37) | 0.61 (0.57–0.66) |
| 1 | 0.33 (0.31–0.36) | 0.59 (0.55–0.64) | |
| 5 | 0.25 (0.23–0.27) | 0.33 (0.31–0.36) ** | |
| 10 | 0.23 (0.22–0.26) | 0.28 (0.26–0.30) ** | |
| BTK-Inhibitor | Concentration (μM) | IC50 (95% CI) [μM] |
|---|---|---|
| Ibrutinib | 0 | 0.35 (0.33–0.37) |
| 1 | 0.21 (0.19–0.23) ** | |
| 5 | 0.12 (0.11–0.14) *** | |
| 10 | 0.09 (0.08–0.11) *** | |
| Acalabrutinib | 0 | 0.35 (0.33–0.37) |
| 1 | 0.23 (0.21–0.25) ** | |
| 5 | 0.13 (0.12–0.15) *** | |
| 10 | 0.10 (0.09–0.12) *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morell, A.; Čermáková, L.; Novotná, E.; Laštovičková, L.; Haddad, M.; Haddad, A.; Portillo, R.; Wsól, V. Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3. Cancers 2020, 12, 3731. https://doi.org/10.3390/cancers12123731
Morell A, Čermáková L, Novotná E, Laštovičková L, Haddad M, Haddad A, Portillo R, Wsól V. Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3. Cancers. 2020; 12(12):3731. https://doi.org/10.3390/cancers12123731
Chicago/Turabian StyleMorell, Anselm, Lucie Čermáková, Eva Novotná, Lenka Laštovičková, Melodie Haddad, Andrew Haddad, Ramon Portillo, and Vladimír Wsól. 2020. "Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3" Cancers 12, no. 12: 3731. https://doi.org/10.3390/cancers12123731
APA StyleMorell, A., Čermáková, L., Novotná, E., Laštovičková, L., Haddad, M., Haddad, A., Portillo, R., & Wsól, V. (2020). Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3. Cancers, 12(12), 3731. https://doi.org/10.3390/cancers12123731