Circulating Tumour DNA Sequencing Identifies a Genetic Resistance-Gap in Colorectal Cancers with Acquired Resistance to EGFR-Antibodies and Chemotherapy

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
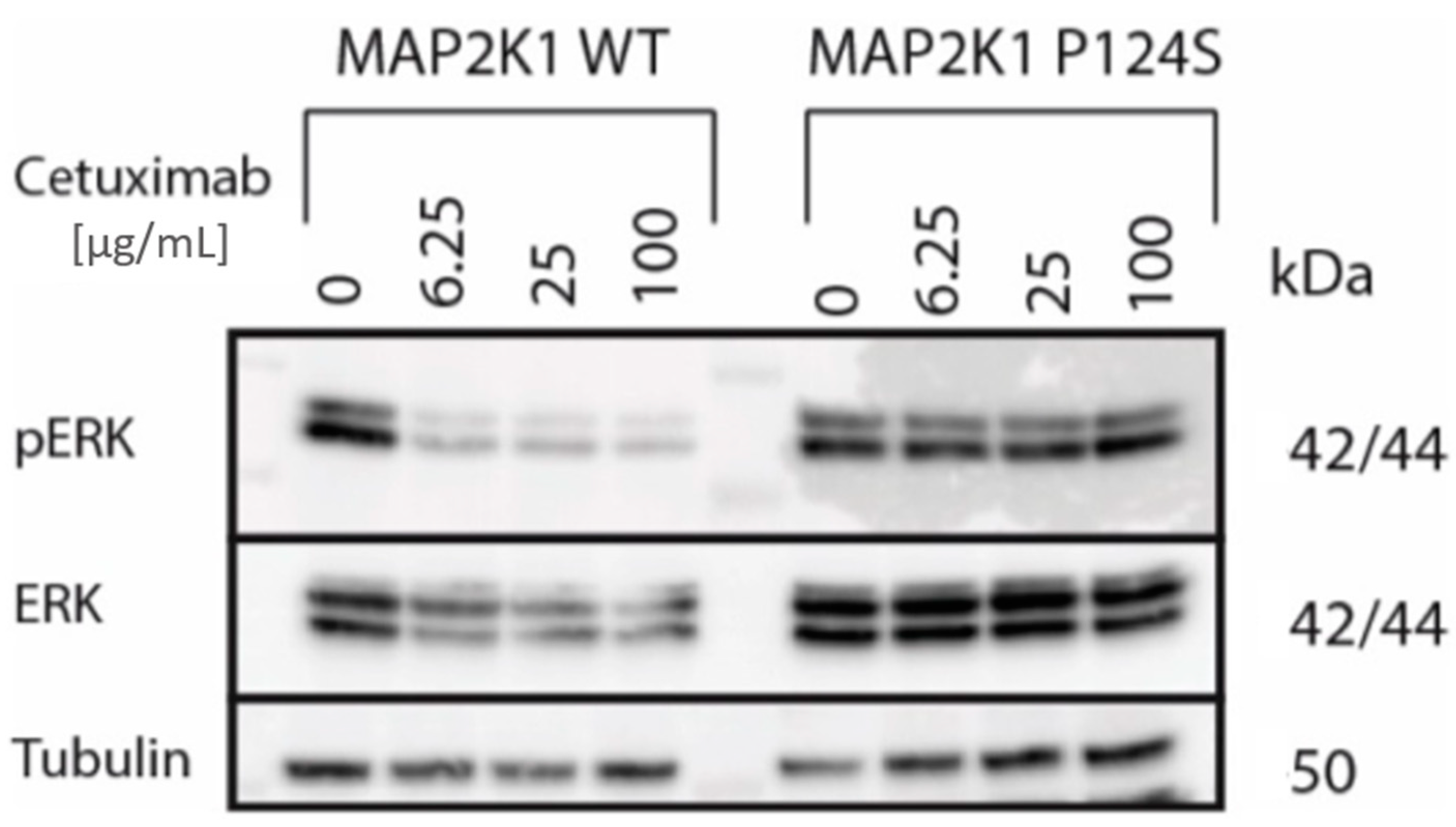
2. Results
2.1. ctDNA Sequencing Results
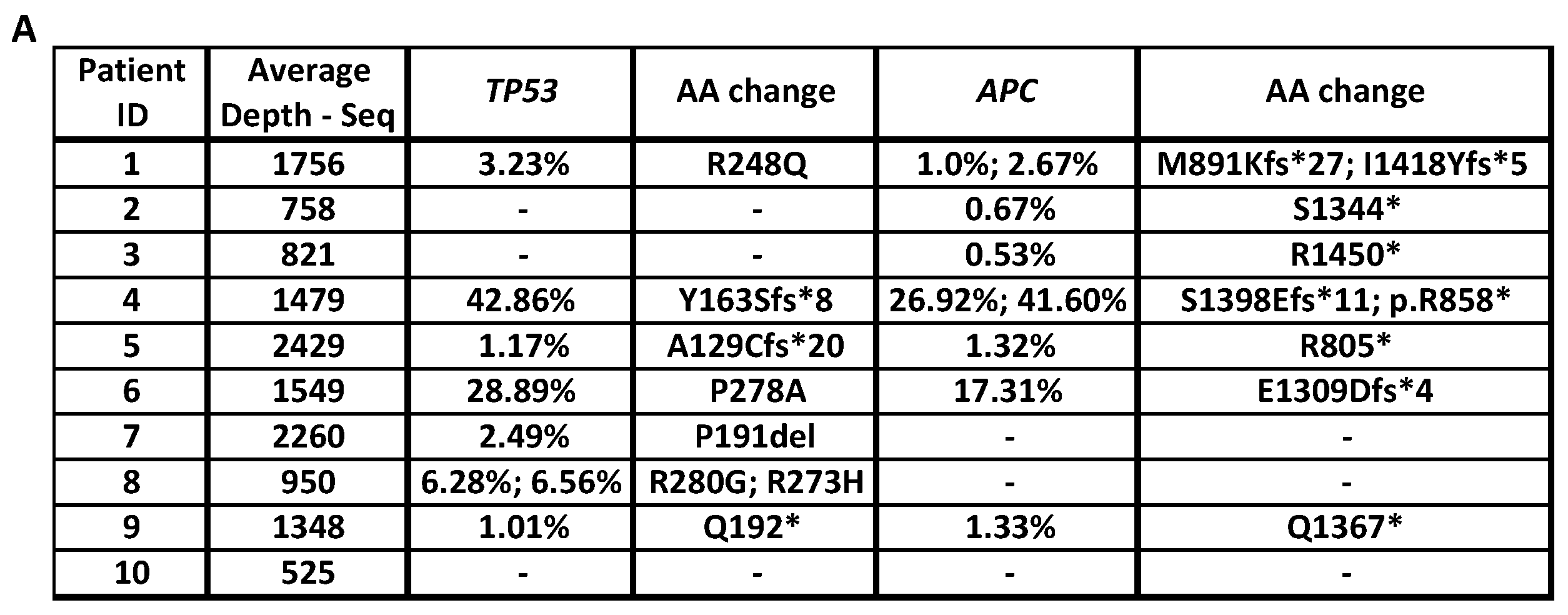
2.1.1. Identification of Drivers of Primary Resistance by ctDNA Sequencing
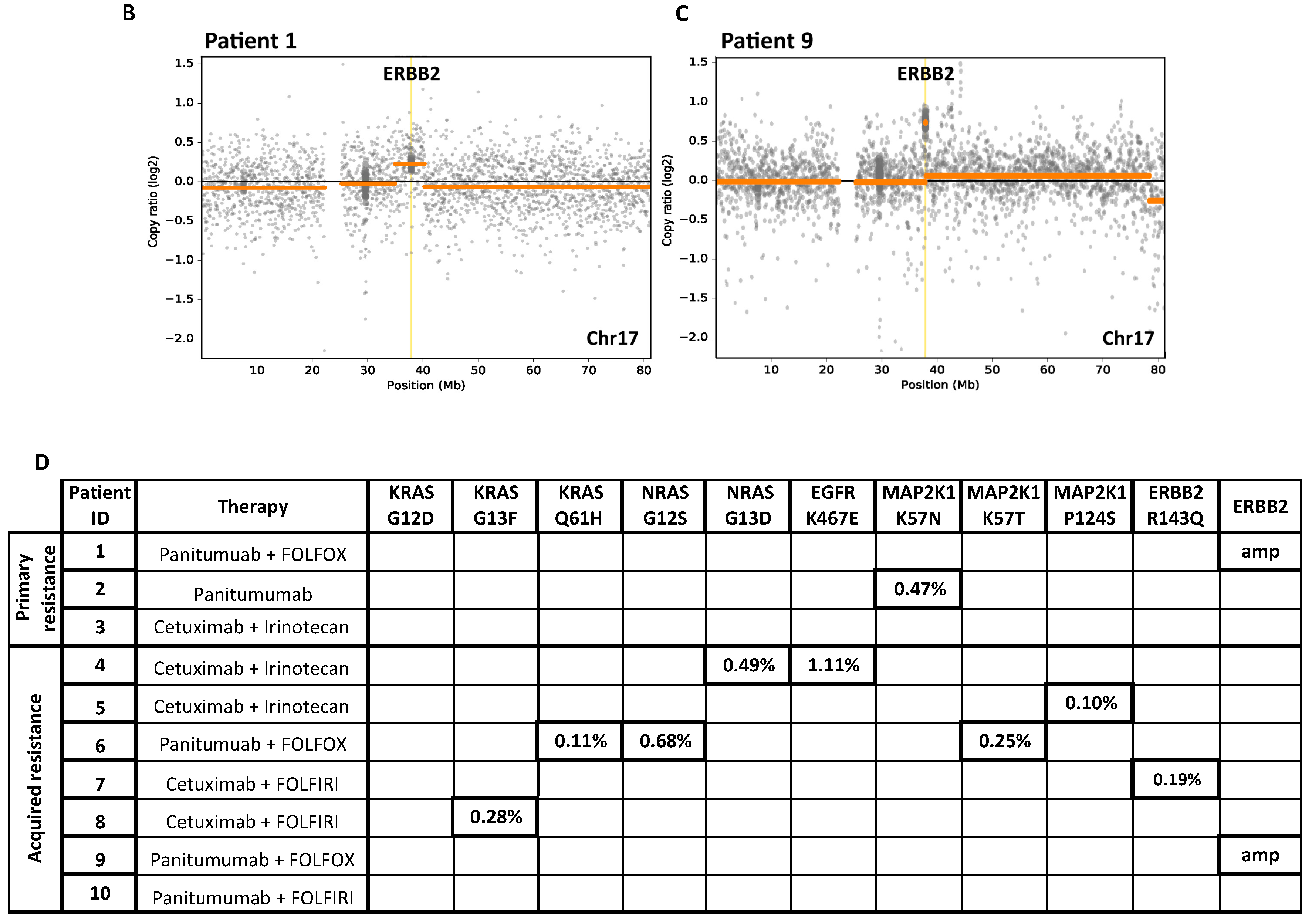
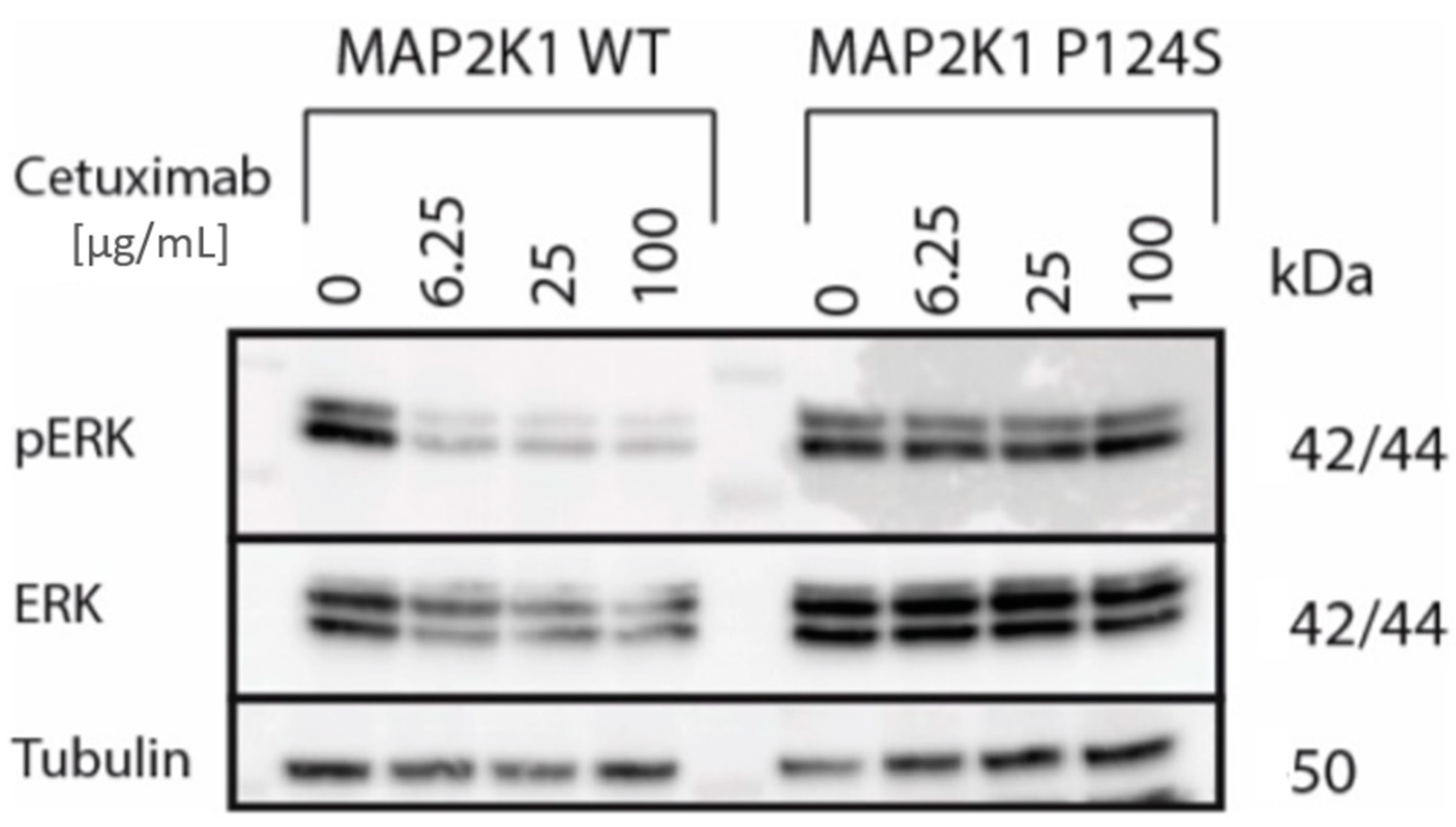
2.1.2. Identification of Drivers of Acquired Resistance by ctDNA Sequencing
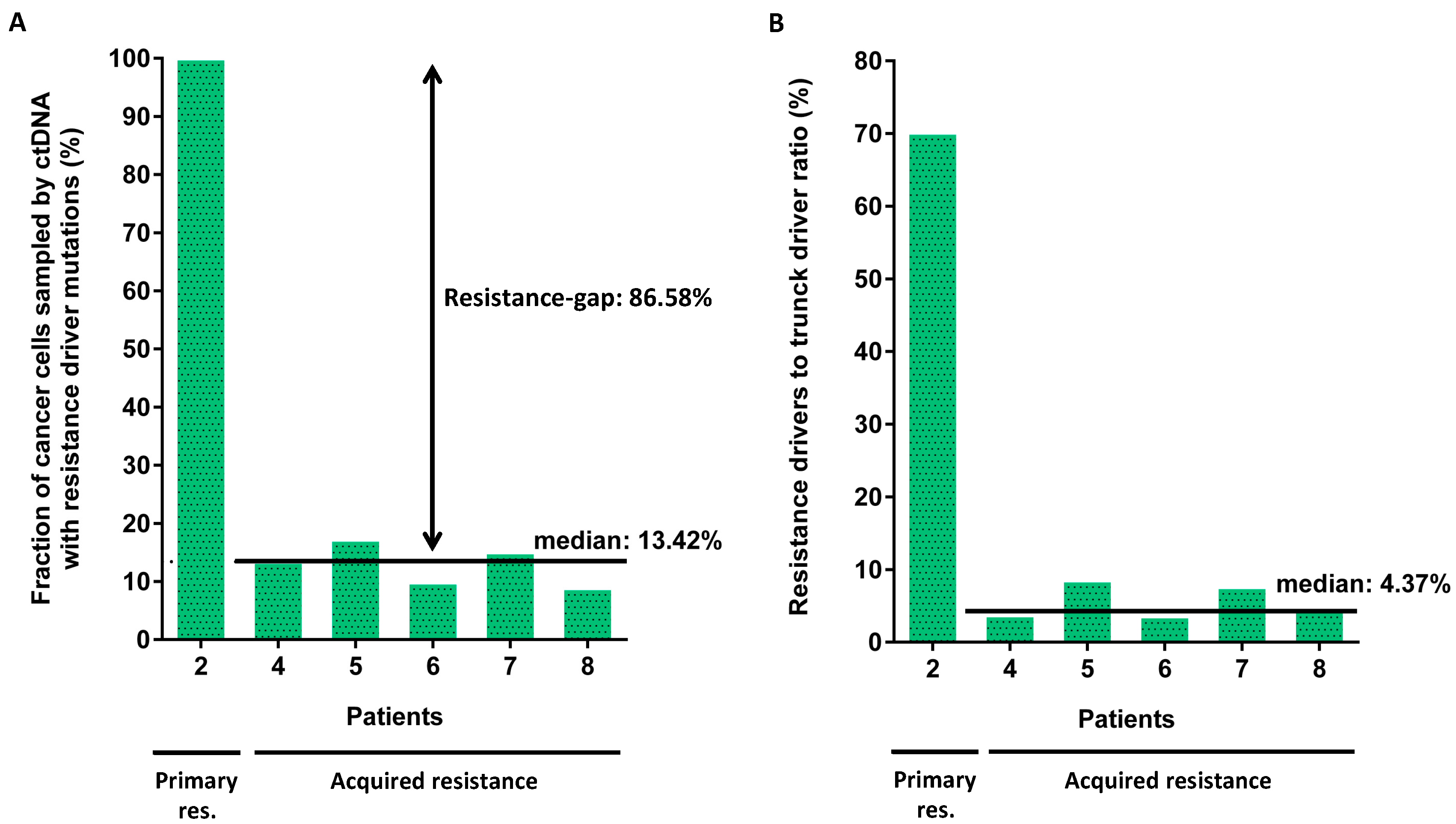
2.1.3. Clonality of Drivers of Primary and Acquired Resistance
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Plasma Samples
4.3. ctDNA-Sequencing
4.4. Variant Calling
4.5. Genome-Wide DNA Copy Number Analysis
4.6. Mutation Clonality Analysis
4.7. Generation of MAP2K1 Transgenic DiFi Cell Lines and Western Blot Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Köhne, C.H.; Hitre, E.; Zaluski, J.; Chien, C.-R.C.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 treatment and RAS Mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.; Douillard, J.-Y.; Van Cutsem, E.; Siena, S.; Zhang, K.; Williams, R.; Wiezorek, J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal Cancer: Assessment as prognostic and predictive biomarkers of response to panitumumab. J. Clin. Oncol. 2013, 31, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Woolston, A.; Khan, K.; Spain, G.; Barber, L.J.; Griffiths, B.; Gonzalez-Exposito, R.; Hornsteiner, L.; Punta, M.; Patil, Y.; Newey, A.; et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-EGFR treatment in colorectal cancer. Cancer Cell 2019, 36, 35–50.e9. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Molecular subtypes and the evolution of treatment decisions in metastatic colorectal cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 231–238. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Mansukhani, S.; Barber, L.J.; Kleftogiannis, D.; Moorcraft, S.Y.; Davidson, M.; Woolston, A.; Proszek, P.Z.; Griffiths, B.; Fenwick, K.; Herman, B.; et al. Ultra-sensitive mutation detection and genome-wide DNA copy number reconstruction by error-corrected circulating tumor DNA sequencing. Clin. Chem. 2018, 64, 1626–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [Green Version]
- De Martino, M.; Zhuang, D.; Klatte, T.; Rieken, M.; Rouprêt, M.; Xylinas, E.; Clozel, T.; Krzywinski, M.; Elemento, O.; Shariat, S.F. Impact ofERBB2mutations on in vitro sensitivity of bladder cancer to lapatinib. Cancer Biol. Ther. 2014, 15, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Korn, W.M.; Atreya, C.E.; Banks, K.C.; et al. Genomic landscape of cell-free DNA in patients with colorectal cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case–control study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting alterations in the RAF–MEK pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, E.; Nordentoft, I.; Vang, S.; Birkenkamp-Demtröder, K.; Jensen, J.B.; Agerbæk, M.; Pedersen, J.S.; Dyrskjøt, L. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci. Rep. 2018, 8, 1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilke, F.J.; Muyas, F.; Admard, J.; Kootz, B.; Nann, D.; Welz, S.; Rieß, O.; Zips, D.; Ossowski, S.; Schroeder, C.M.; et al. Dynamics of cell-free tumour DNA correlate with treatment response of head and neck cancer patients receiving radiochemotherapy. Radiother. Oncol. 2020, 151, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.N.; Zhou, Y.C.; Chan, Y.M.; Foo, C.M.; Lee, P.Y.; Mok, W.Y.; Wong, W.S.; Fung, Y.Y.; Wong, K.Y.; Huang, J.Y.; et al. Comparison of target enrichment platforms for circulating tumor DNA detection. Sci. Rep. 2020, 10, 4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Hobor, S.; Van Emburgh, B.O.; Crowley, E.; Misale, S.; Di Nicolantonio, F.; Bardelli, A. TGFα and amphiregulin paracrine network promotes resistance to EGFR blockade in colorectal cancer cells. Clin. Cancer Res. 2014, 20, 6429–6438. [Google Scholar] [CrossRef] [Green Version]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age (Years) | Gender | Histology | Primary Location | Differentiation Grade | EGFR-Ab Therapy | Line of Therapy for Metastatic Disease | Time on EGFR-Ab Therapy | Resistance |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 80 | Male | Adenocarcinoma | Right-colon | Moderate | Panitumumab + FOLFOX | 2nd | 2 weeks | Primary |
| 2 | 79 | Male | Adenocarcinoma | Rectum | Moderate | Panitumumab | 3rd | 9 weeks | Primary |
| 3 | 57 | Male | Adenocarcinoma | Sigmoid | Well | Cetuximab + Irinotecan (rechallenge with EGFR-Ab) | 3rd | 10 weeks | Primary |
| 4 | 58 | Female | Adenocarcinoma | Rectum | Well | Cetuximab + Irinotecan (rechallenge with EGFR-Ab) | 3rd | 16 weeks | Acquired |
| 5 | 52 | Male | Adenocarcinoma | Sigmoid | Moderate | Cetuximab + Irinotecan | 2nd | 20 weeks | Acquired |
| 6 | 64 | Male | Adenocarcinoma | Rectum | Moderate | Panitumumab + FOLFOX | 1st | 12 weeks | Acquired |
| 7 | 41 | Female | Adenocarcinoma | Sigmoid | Poor | Cetuximab + FOLFIRI | 1st | 27 weeks | Acquired |
| 8 | 53 | Female | Adenocarcinoma | Right-colon | Poor | Cetuximab + FOLFIRI | 2nd | 27 weeks | Acquired |
| 9 | 46 | Female | Adenocarcinoma | Sigmoid | Moderate | Panitumumab + FOLFOX | 2nd | 29 weeks | Acquired |
| 10 | 30 | Male | Adenocarcinoma | Rectum | Moderate | Panitumumab + FOLFIRI | 5th | 26 weeks | Acquired |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knebel, F.H.; Barber, L.J.; Newey, A.; Kleftogiannis, D.; Woolston, A.; Griffiths, B.; Fenwick, K.; Bettoni, F.; Ribeiro, M.F.S.A.; da Fonseca, L.; et al. Circulating Tumour DNA Sequencing Identifies a Genetic Resistance-Gap in Colorectal Cancers with Acquired Resistance to EGFR-Antibodies and Chemotherapy. Cancers 2020, 12, 3736. https://doi.org/10.3390/cancers12123736
Knebel FH, Barber LJ, Newey A, Kleftogiannis D, Woolston A, Griffiths B, Fenwick K, Bettoni F, Ribeiro MFSA, da Fonseca L, et al. Circulating Tumour DNA Sequencing Identifies a Genetic Resistance-Gap in Colorectal Cancers with Acquired Resistance to EGFR-Antibodies and Chemotherapy. Cancers. 2020; 12(12):3736. https://doi.org/10.3390/cancers12123736
Chicago/Turabian StyleKnebel, Franciele H., Louise J. Barber, Alice Newey, Dimitrios Kleftogiannis, Andrew Woolston, Beatrice Griffiths, Kerry Fenwick, Fabiana Bettoni, Maurício Fernando Silva Almeida Ribeiro, Leonardo da Fonseca, and et al. 2020. "Circulating Tumour DNA Sequencing Identifies a Genetic Resistance-Gap in Colorectal Cancers with Acquired Resistance to EGFR-Antibodies and Chemotherapy" Cancers 12, no. 12: 3736. https://doi.org/10.3390/cancers12123736
APA StyleKnebel, F. H., Barber, L. J., Newey, A., Kleftogiannis, D., Woolston, A., Griffiths, B., Fenwick, K., Bettoni, F., Ribeiro, M. F. S. A., da Fonseca, L., Costa, F., Capareli, F. C., Hoff, P. M., Sabbaga, J., Camargo, A. A., & Gerlinger, M. (2020). Circulating Tumour DNA Sequencing Identifies a Genetic Resistance-Gap in Colorectal Cancers with Acquired Resistance to EGFR-Antibodies and Chemotherapy. Cancers, 12(12), 3736. https://doi.org/10.3390/cancers12123736