Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment


Abstract
:Simple Summary
Abstract
1. Introduction
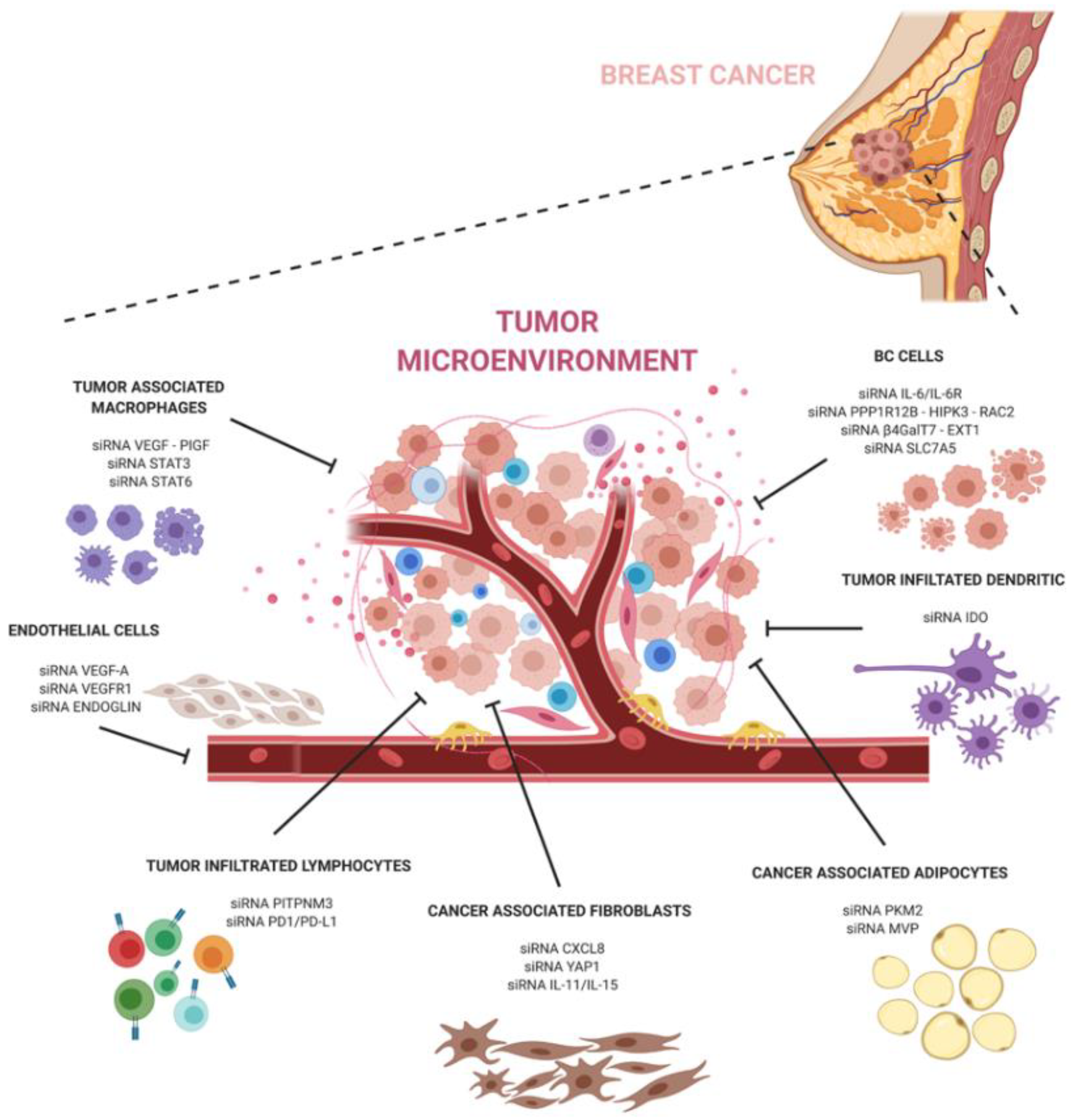
2. Tumor Microenvironment (TME) in Breast Cancer
2.1. Cancer-Associated Fibroblasts (CAFs)
2.2. Vasculature
2.3. The ECM
2.4. Immune Cells and Dendritic Cells (DCs)
2.5. Adipocytes
3. siRNA and Clinical Applications
4. siRNA Therapeutics Targeting the TME
4.1. siRNAs against CAFs
4.2. siRNAs Targeting Angiogenesis
4.3. siRNA Targeting ECM
4.4. siRNA Targeting TAMs
4.5. siRNA DCs
4.6. siRNA Targeting Immune Infiltrates
4.7. siRNA Targeting Cancer-Associated Adipocytes
5. siRNA Delivery Strategies
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reinert, T.; Barrios, C.H. Optimal management of hormone receptor positive metastatic breast cancer in 2016. Ther. Adv. Med. Oncol. 2015, 7, 304–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.; Spicer, D.; Lu, J. Overcoming endocrine resistance in metastatic hormone receptor-positive breast cancer. J. Hematol. Oncol. 2018, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, S.; Tolaney, S.M.; Winer, E.P.; Goel, S. CDK4/6 inhibition in breast cancer: Current practice and future directions. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, H.J. The Distinctive Nature of HER2-Positive Breast Cancers. N. Engl. J. Med. 2005, 353, 1652–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rexer, B.N.; Arteaga, C.L. Intrinsic and Acquired Resistance to HER2-Targeted Therapies in HER2 Gene-Amplified Breast Cancer: Mechanisms and Clinical Implications. Crit. Rev. Oncog. 2012, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Cooke, T.; Reeves, J.; Lanigan, A.; Stanton, P. HER2 as a prognostic and predictive marker for breast cancer. Ann. Oncol. 2001, 12, S23–S28. [Google Scholar] [CrossRef]
- Seidman, A.; Fornier, M.N.; Esteva, F.J.; Tan, L.; Kaptain, S.; Bach, A.; Panageas, K.S.; Arroyo, C.; Valero, V.; Currie, V.; et al. Weekly Trastuzumab and Paclitaxel Therapy for Metastatic Breast Cancer with Analysis of Efficacy by HER2 Immunophenotype and Gene Amplification. J. Clin. Oncol. 2001, 19, 2587–2595. [Google Scholar] [CrossRef]
- Saura, C.; Garcia-Saenz, J.A.; Xu, B.; Harb, W.; Moroose, R.; Pluard, T.; Cortés, J.; Kiger, C.; Germa, C.; Wang, K.; et al. Safety and Efficacy of Neratinib in Combination with Capecitabine in Patients with Metastatic Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. J. Clin. Oncol. 2014, 32, 3626–3633. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.S.; Sledge, G.; Aktan, G.; Ellis, C.; Florance, A.; Vukelja, S.; Bischoff, J.; et al. Overall Survival Benefit with Lapatinib in Combination With Trastuzumab for Patients with Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer: Final Results From the EGF104900 Study. J. Clin. Oncol. 2012, 30, 2585–2592. [Google Scholar] [CrossRef]
- Blackwell, K.L.; Burstein, H.J.; Storniolo, A.M.; Rugo, H.S.; Sledge, G.; Koehler, M.; Ellis, C.; Casey, M.; Vukelja, S.; Bischoff, J.; et al. Randomized Study of Lapatinib Alone or in Combination with Trastuzumab in Women with ErbB2-Positive, Trastuzumab-Refractory Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Cortés, J.; Kim, S.-B.; Im, S.-A.; Hegg, R.; Im, Y.-H.; Roman, L.; Pedrini, J.L.; Pienkowski, T.; Knott, A.; et al. Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer. N. Engl. J. Med. 2012, 366, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Atezolizumab Combo Approved for PD-L1–positive TNBC. Cancer Discov. 2019, 9, OF2. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Rønnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Investig. 1993, 68, 696–707. [Google Scholar]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F.; et al. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Liu, B.; DeFilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast Fibroblasts Modulate Early Dissemination, Tumorigenesis, and Metastasis through Alteration of Extracellular Matrix Characteristics. Neoplasia 2013, 15, 249–IN7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Mojares, E.; Hernández, A.E.D.R. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Jussila, T.; Kauppila, S.; Risteli, L.; Risteli, J.; Stenbäck, F. Collagen formation in extracellular matrix of transplants of human transformed keratinocyte cell lines. Anticancer Res. 2002, 22, 1705–1711. [Google Scholar]
- Huijbers, I.J.; Iravani, M.; Popov, S.; Robertson, D.; Al-Sarraj, S.; Jones, C.; Isacke, C.M. A Role for Fibrillar Collagen Deposition and the Collagen Internalization Receptor Endo180 in Glioma Invasion. PLoS ONE 2010, 5, e9808. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef]
- Evans, A.; Armstrong, S.; Whelehan, P.; Thomson, K.; Rauchhaus, P.; Purdie, C.; Jordan, L.; Jones, L.; Thompson, A.; Vinnicombe, S. Can shear-wave elastography predict response to neoadjuvant chemotherapy in women with invasive breast cancer? Br. J. Cancer 2013, 109, 2798–2802. [Google Scholar] [CrossRef] [Green Version]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J.; Toida, T. Role of Glycosaminoglycans in Cellular Communication. Accounts Chem. Res. 2004, 37, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bülow, H.E.; Hobert, O. The Molecular Diversity of Glycosaminoglycans Shapes Animal Development. Annu. Rev. Cell Dev. Biol. 2006, 22, 375–407. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.R.; Schuksz, M.; Esko, J. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nat. Cell Biol. 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Blackhall, F.H.; Merry, C.L.R.; Davies, E.J.; Jayson, G.C. Heparan sulfate proteoglycans and cancer. Br. J. Cancer 2001, 85, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjeldstad, K.; Kolset, S. Decreasing the Metastatic Potential in Cancers—Targeting the Heparan Sulfate Proteoglycans. Curr. Drug Targets 2005, 6, 665–682. [Google Scholar] [CrossRef]
- Itano, N.; Kimata, K. Altered hyaluronan biosynthesis in cancer progression. Semin. Cancer Biol. 2008, 18, 268–274. [Google Scholar] [CrossRef]
- Yip, G.W.; Smollich, M.; Götte, M. Therapeutic value of glycosaminoglycans in cancer. Mol. Cancer Ther. 2006, 5, 2139–2148. [Google Scholar] [CrossRef] [Green Version]
- Appay, V.; Douek, D.C.; A Price, D. CD8+ T cell efficacy in vaccination and disease. Nat. Med. 2008, 14, 623–628. [Google Scholar] [CrossRef]
- Burugu, S.; Asleh-Aburaya, K.; Nielsen, T.O. Immune infiltrates in the breast cancer microenvironment: Detection, characterization and clinical implication. Breast Cancer 2017, 24, 3–15. [Google Scholar] [CrossRef]
- Kim, P.S.; Ahmed, R. Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol. 2010, 22, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Joshi, K.; Wig, J.; Arora, S.K. Intratumoral FOXP3 expression in infiltrating breast carcinoma: Its association with clinicopathologic parameters and angiogenesis. Acta Oncol. 2007, 46, 792–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, J.W. Macrophages define the invasive microenvironment in breast cancer. J. Leukoc. Biol. 2008, 84, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Cruz-Merino, L.; Barco-Sánchez, A.; Carrasco, F.H.; Fernández, E.N.; Benítez, A.V.; Molina, J.B.; Peinado, A.M.; López, A.G.; Borrego, M.R.; De Villena, M.C.M.; et al. New Insights into the Role of the Immune Microenvironment in Breast Carcinoma. Clin. Dev. Immunol. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Lehuédé, C.; Laurent, V.; Dirat, B.; Dauvillier, S.; Bochet, L.; Le Gonidec, S.; Escourrou, G.; Valet, P.; Muller, C. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012, 324, 142–151. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Picon-Ruiz, M.; Pan, C.; Drews-Elger, K.; Jang, K.; Besser, A.H.; Zhao, D.; Morata-Tarifa, C.; Kim, M.; Ince, T.A.; Azzam, D.J.; et al. Interactions between Adipocytes and Breast Cancer Cells Stimulate Cytokine Production and Drive Src/Sox2/miR-302b–Mediated Malignant Progression. Cancer Res. 2016, 76, 491–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochet, L.; Meulle, A.; Imbert, S.; Salles, B.; Valet, P.; Muller, C. Cancer-associated adipocytes promotes breast tumor radioresistance. Biochem. Biophys. Res. Commun. 2011, 411, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell 1990, 2, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roscigno, G.; Cirella, A.; Affinito, A.; Quintavalle, C.; Scognamiglio, I.; Palma, F.; Ingenito, F.; Nuzzo, S.; De Micco, F.; Cuccuru, A.; et al. miR-216a Acts as a Negative Regulator of Breast Cancer by Modulating Stemness Properties and Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, F.; Affinito, A.; Nuzzo, S.; Roscigno, G.; Scognamiglio, I.; Ingenito, F.; Martinez, L.; Franzese, M.; Zanfardino, M.; Soricelli, A.; et al. miR-34c-3p targets CDK1 a synthetic lethality partner of KRAS in non-small cell lung cancer. Cancer Gene Ther. 2020, 1–14. [Google Scholar] [CrossRef]
- Roscigno, G.; Puoti, I.; Giordano, I.; Donnarumma, E.; Russo, V.; Affinito, A.; Adamo, A.; Quintavalle, C.; Todaro, M.; Vivanco, M.D.; et al. MiR-24 induces chemotherapy resistance and hypoxic advantage in breast cancer. Oncotarget 2017, 8, 19507–19521. [Google Scholar] [CrossRef] [Green Version]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Cho, W.G.; Albuquerque, R.J.C.; Kleinman, M.E.; Tarallo, V.; Greco, A.; Nozaki, M.; Green, M.G.; Baffi, J.Z.; Ambati, B.K.; De Falco, M.; et al. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc. Natl. Acad. Sci. USA 2009, 106, 7137–7142. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Sardh, E.; Harper, P.; Balwani, M.; Stein, P.; Rees, D.; Bissell, D.M.; Desnick, R.; Parker, C.; Phillips, J.; Bonkovsky, H.L.; et al. Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N. Engl. J. Med. 2019, 380, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Balwani, M.; Sardh, E.; Ventura, P.; Peiró, P.A.; Rees, D.C.; Stölzel, U.; Bissell, D.M.; Bonkovsky, H.L.; Windyga, J.; Anderson, K.E.; et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N. Engl. J. Med. 2020, 382, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Brandão, P.R.D.P.; Titze-De-Almeida, S.S.; De Almeida, R.T. Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran. Mol. Diagn. Ther. 2019, 24, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Simon, A.R.; Goel, V.; Habtemariam, B.A.; Clausen, V.A.; Kim, J.B.; Robbie, G.J. Pharmacokinetics and Pharmacodynamics of the Small Interfering Ribonucleic Acid, Givosiran, in Patients with Acute Hepatic Porphyria. Clin. Pharmacol. Ther. 2020, 108, 63–72. [Google Scholar] [CrossRef]
- Heidel, J.D.; Liu, J.Y.-C.; Yen, Y.; Zhou, B.; Heale, B.S.; Rossi, J.J.; Bartlett, D.W.; Davis, M.E. Potent siRNA Inhibitors of Ribonucleotide Reductase Subunit RRM2 Reduce Cell Proliferation In Vitro and In Vivo. Clin. Cancer Res. 2007, 13, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Heidel, J.D.; Yu, Z.; Liu, J.Y.-C.; Rele, S.M.; Liang, Y.; Zeidan, R.K.; Kornbrust, D.J.; Davis, M.E. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 5715–5721. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.; Mitra, R.; McArthur, M.J.; Baze, W.; Barnhart, K.; Wu, S.Y.; Rodriguez-Aguayo, C.; Zhang, X.; Coleman, R.L.; Lopez-Berestein, G.; et al. Preclinical Mammalian Safety Studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol. Cancer Ther. 2017, 16, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Verma, S.; McLeod, D.; Batist, G.; Robidoux, A.; Martins, I.R.S.; Mackey, J.R. In the End What Matters Most? A Review of Clinical Endpoints in Advanced Breast Cancer. Oncologist 2011, 16, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Egorova, A.A.; Shtykalova, S.V.; Maretina, M.A.; Sokolov, D.I.; Selkov, S.A.; Baranov, V.S.; Kiselev, A.V. Synergistic Anti-Angiogenic Effects Using Peptide-Based Combinatorial Delivery of siRNAs Targeting VEGFA, VEGFR1, and Endoglin Genes. Pharmaceutics 2019, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Tang, C.; Yin, C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials 2018, 185, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Binnemars-Postma, K.; Bansal, R.; Storm, G.; Prakash, J. Targeting the Stat6 pathway in tumor-associated macrophages reduces tumor growth and metastatic niche formation in breast cancer. FASEB J. 2018, 32, 969–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Gao, Y.; Gong, C.; Han, Z.; Qiang, L.; Tai, Z.; Tian, J.; Gao, S. Dual-Blockade Immune Checkpoint for Breast Cancer Treatment Based on a Tumor-Penetrating Peptide Assembling Nanoparticle. ACS Appl. Mater. Interfaces 2019, 11, 39513–39524. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Li, K.; Pang, X.; Guo, B.; Su, M.; Huang, Y.; Wang, N.; Ji, F.; Zhong, C.; Yang, J.; et al. Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 2016, 35, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Zheng, X.; Koropatnick, J.; Chen, D.; Velenosi, T.; Ling, H.; Zhang, X.; Jiang, N.; Navarro, B.; Ichim, T.E.; Urquhart, B.; et al. Silencing IDO in dendritic cells: A novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int. J. Cancer 2012, 132, 967–977. [Google Scholar] [CrossRef]
- Haanen, J.B.; Robert, C. Immune Checkpoint Inhibitors. Prog. Tumor Res. 2015, 42, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Lerrer, S.; Liubomirski, Y.; Bott, A.; Abnaof, K.; Oren, N.; Yousaf, A.; Körner, C.; Meshel, T.; Wiemann, S.; Ben-Baruch, A. Co-Inflammatory Roles of TGFβ1 in the Presence of TNFα Drive a Pro-inflammatory Fate in Mesenchymal Stem Cells. Front. Immunol. 2017, 8, 479. [Google Scholar] [CrossRef]
- Katanov, C.; Lerrer, S.; Liubomirski, Y.; Leider-Trejo, L.; Meshel, T.; Bar, J.; Feniger-Barish, R.; Kamer, I.; Soria, G.; Kahani, H.; et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: Prominent roles for TNF-α and the NF-κB pathway. Stem Cell Res. Ther. 2015, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Liubomirski, Y.; Lerrer, S.; Meshel, T.; Morein, D.; Rubinstein-Achiasaf, L.; Sprinzak, D.; Wiemann, S.; Körner, C.; Ehrlich, M.; Ben-Baruch, A. Notch-Mediated Tumor-Stroma-Inflammation Networks Promote Invasive Properties and CXCL8 Expression in Triple-Negative Breast Cancer. Front. Immunol. 2019, 10, 804. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.-E.; Tu, G.; Yang, G.; Li, G.; Yang, D.; Lang, L.; Xi, L.; Sun, K.; Chen, Y.; Shu, K.; et al. MiR-205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF-independent Angiogenesis through STAT3 Signaling. Theranostics 2017, 7, 3972–3988. [Google Scholar] [CrossRef] [PubMed]
- Gagliano, T.; Shah, K.; Gargani, S.; Lao, L.; Alsaleem, M.; Chen, J.; Ntafis, V.; Huang, P.; Ditsiou, A.; Vella, V.; et al. PIK3Cδ expression by fibroblasts promotes triple-negative breast cancer progression. J. Clin. Investig. 2020, 130, 3188–3204. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.S.; Yeung, T.-L.; Yip, K.-P.; Wong, K.-K.; Ho, S.Y.; Mangala, L.S.; Sood, A.K.; Lopez-Berestein, G.; Sheng, J.; Wong, S.T.; et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J. Clin. Investig. 2017, 128, 589–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngan, E.; Stoletov, K.; Smith, H.W.; Common, J.; Muller, W.J.; Lewis, J.D.; Siegel, P.M. LPP is a Src substrate required for invadopodia formation and efficient breast cancer lung metastasis. Nat. Commun. 2017, 8, 15059. [Google Scholar] [CrossRef]
- Jung, Y.D.; Ahmad, S.A.; Liu, W.; Reinmuthb, N.; Parikha, A.; Stoeltzing, O.; Fan, F.; Ellis, L.M. The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin. Cancer Biol. 2002, 12, 105–112. [Google Scholar] [CrossRef]
- Yang, B.; Jiang, J.; Du, H.; Geng, G.; Jiang, Z.; Yao, C.; Zhang, Q.; Jin, L. Decreased monoamine oxidase (MAO) activity and MAO-A expression as diagnostic indicators of human esophageal cancers. Biomarkers 2009, 14, 624–629. [Google Scholar] [CrossRef]
- Rybaczyk, L.A.; Bashaw, M.J.; Pathak, D.R.; Huang, K. An indicator of cancer: Downregulation of Monoamine Oxidase-A in multiple organs and species. BMC Genom. 2008, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Hwang, H.J.; Lee, Y.-R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.-K.; Kim, Y.-N.; Ko, Y.-G.; Park, H.J.; Lee, D. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Fokkelman, M.; Balcıoğlu, H.E.; Klip, J.E.; Yan, K.; Verbeek, F.J.; Danen, E.H.J.; Van De Water, B. Cellular adhesome screen identifies critical modulators of focal adhesion dynamics, cellular traction forces and cell migration behaviour. Sci. Rep. 2016, 6, 31707. [Google Scholar] [CrossRef] [PubMed]
- Beauvais, D.M.; Rapraeger, A.C. Syndecan-1-mediated cell spreading requires signaling by αvβ3 integrins in human breast carcinoma cells. Exp. Cell Res. 2003, 286, 219–232. [Google Scholar] [CrossRef]
- Soares, M.A.; Teixeira, F.C.O.B.; Fontes, M.; Arêas, A.L.; Leal, M.G.; Pavão, M.S.G.; Stelling, M.P. Heparan Sulfate Proteoglycans May Promote or Inhibit Cancer Progression by Interacting with Integrins and Affecting Cell Migration. BioMed Res. Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage Polarization: Anti-Cancer Strategies to Target Tumor-Associated Macrophage in Breast Cancer. J. Cell. Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef]
- Lin, E.Y.; Pollard, J.W. Tumor-Associated Macrophages Press the Angiogenic Switch in Breast Cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [Green Version]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Groner, B.; Von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Yang, J.; Liao, D.; Chen, C.; Liu, Y.; Chuang, T.-H.; Xiang, R.; Markowitz, D.; Reisfeld, R.A.; Luo, Y. Tumor-Associated Macrophages Regulate Murine Breast Cancer Stem Cells Through a Novel Paracrine EGFR/Stat3/Sox-2 Signaling Pathway. Stem Cells 2013, 31, 248–258. [Google Scholar] [CrossRef]
- Cerundolo, V.; Hermans, I.F.; Salio, M. Dendritic cells: A journey from laboratory to clinic. Nat. Immunol. 2004, 5, 7–10. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, D.W.; Adams, S.; Bhardwaj, N. Manipulating dendritic cell biology for the active immunotherapy of cancer. Blood 2004, 104, 2235–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grohmann, U.; Fallarino, F.; Puccetti, P. Tolerance, DCs and tryptophan: Much ado about IDO. Trends Immunol. 2003, 24, 242–248. [Google Scholar] [CrossRef]
- Hira, S.K.; Manna, P.P. Down regulation of CD24 and HER-2/neu in breast carcinoma cells by activated human dendritic cell. Role of STAT3. Cell. Immunol. 2012, 275, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Aigner, S.; Sthoeger, Z.M.; Fogel, M.; Weber, E.; Zarn, J.; Ruppert, M.; Zeller, Y.; Vestweber, D.; Stahel, R.; Sammar, M.; et al. CD24, a Mucin-Type Glycoprotein, Is a Ligand for P-Selectin on Human Tumor Cells. Blood 1997, 89, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Mass, R. The role of HER-2 expression in predicting response to therapy in breast cancer. Semin. Oncol. 2000, 27, 46–52. [Google Scholar]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Jego, G.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, M. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Liu, W.; Yabu, J.M.; Laszik, Z.G.; Putnam, A.; Belingheri, M.; Gross, D.M.; Townsend, R.M.; Vincenti, F. The Effect of Costimulatory and Interleukin 2 Receptor Blockade on Regulatory T Cells in Renal Transplantation. Arab. Archaeol. Epigr. 2008, 8, 2086–2096. [Google Scholar] [CrossRef] [Green Version]
- Rech, A.J.; Mick, R.; Martin, S.; Recio, A.; Aqui, N.A.; Powell, D.J.; Colligon, T.A.; Trosko, J.A.; Leinbach, L.I.; Pletcher, C.H.; et al. CD25 Blockade Depletes and Selectively Reprograms Regulatory T Cells in Concert with Immunotherapy in Cancer Patients. Sci. Transl. Med. 2012, 4, 134ra62. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Liao, J.; Liu, J.; Huang, D.; He, C.; Chen, F.; Yang, L.; Wu, W.; Chen, J.; Lin, L.; et al. Blocking the recruitment of naive CD4+ T cells reverses immunosuppression in breast cancer. Cell Res. 2017, 27, 461–482. [Google Scholar] [CrossRef]
- Nanda, R.R.; Chow, L.Q.M.L.; Dees, E.C.; Berger, R.R.; Gupta, S.S.; Geva, R.R.; Pusztai, L.L.; Pathiraja, K.K.; Aktan, G.; Cheng, J.D.J.; et al. Pembrolizumab in Patients with Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.-M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updat. 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; Craze, M.L.; Althobiti, M.; Alfarsi, L.; Ellis, I.O.; Rakha, E.A.; Green, A.R. Enhanced glutamine uptake influences composition of immune cell infiltrates in breast cancer. Br. J. Cancer 2019, 122, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Lehuédé, C.; Li, X.; Dauvillier, S.; Vaysse, C.; Franchet, C.; Clement, E.; Esteve, D.; Longué, M.; Chaltiel, L.; Le Gonidec, S.; et al. Adipocytes promote breast cancer resistance to chemotherapy, a process amplified by obesity: Role of the major vault protein (MVP). Breast Cancer Res. 2019, 21, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Scheper, R.J.; Broxterman, H.J.; Scheffer, G.L.; Kaaijk, P.; Dalton, W.S.; Van Heijningen, T.H.; Van Kalken, C.K.; Slovak, M.L.; De Vries, E.G.; Van Der Valk, P. Overexpression of a M(r) 110,000 vesicular protein in non-P-glycoprotein-mediated multidrug resistance. Cancer Res. 1993, 53, 1475–1479. [Google Scholar]
- Mossink, M.H.; Van Zon, A.; Scheper, R.J.; Sonneveld, P.; Wiemer, E.A.C. Vaults: A ribonucleoprotein particle involved in drug resistance? Oncogene 2003, 22, 7458–7467. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol. Metab. 2012, 23, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Young, S.W.S.; Stenzel, M.; Jia-Lin, Y. Nanoparticle-siRNA: A potential cancer therapy? Crit. Rev. Oncol. 2016, 98, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, J.; Zheng, S.; Ding, Y.; Guo, S.; Zhang, H.; Zhang, X.; Du, Q.; Liang, Z. Elimination Pathways of Systemically Delivered siRNA. Mol. Ther. 2011, 19, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Lu, M. RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Front. Immunol. 2017, 8, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, S.; Pitard, B.; Benoit, J.-P.; Passirani, C. Non-viral nanosystems for systemic siRNA delivery. Pharmacol. Res. 2010, 62, 100–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassannia, H.; Chaleshtari, M.G.; Atyabi, F.; Nosouhian, M.; Masjedi, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Mohammadi, H.; Ghalamfarsa, G.; et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology 2019, 159, 75–87. [Google Scholar] [CrossRef]
- Chen, J.; Yao, Y.; Gong, C.; Yu, F.; Su, S.; Chen, J.; Liu, B.; Deng, H.; Wang, F.; Lin, L.; et al. CCL18 from Tumor-Associated Macrophages Promotes Breast Cancer Metastasis via PITPNM3. Cancer Cell 2011, 19, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Zheng, J.; Wu, W.; Li, Q.; Saw, P.E.; Chen, J.; Xu, X.; Yao, H.; Yao, Y. A Robust Nanoparticle Platform for RNA Interference in Macrophages to Suppress Tumor Cell Migration. Front. Pharmacol. 2018, 9, 1465. [Google Scholar] [CrossRef] [Green Version]
- Champion, J.A.; Walker, A.; Mitragotri, S. Role of Particle Size in Phagocytosis of Polymeric Microspheres. Pharm. Res. 2008, 25, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Mylona, E.; Alexandrou, P.; Giannopoulou, I.; Liapis, G.; Sofia, M.; Keramopoulos, A.; Nakopoulou, L. The prognostic value of vascular endothelial growth factors (VEGFs)-A and -B and their receptor, VEGFR-1, in invasive breast carcinoma. Gynecol. Oncol. 2007, 104, 557–563. [Google Scholar] [CrossRef]
- Cao, Y. VEGF-Targeted Cancer Therapeutics—Paradoxical Effects in Endocrine Organs. Nat. Rev. Endocrinol. 2014, 10, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Parr, C.; Watkins, G.; Boulton, M.; Cai, J.; Jiang, W.G. Placenta growth factor is over-expressed and has prognostic value in human breast cancer. Eur. J. Cancer 2005, 41, 2819–2827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, C.-F.; Iqbal, S.; Yang, X.-Z.; Wang, J. Responsive Nanocarriers as an Emerging Platform for Cascaded Delivery of Nucleic Acids to Cancer. Adv. Drug Deliv. Rev. 2017, 115, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Cheng, W.-P.; Liu, J.; Lo, S.-Y.; Smith, D.; Qu, X.; Yang, Z. pH-Triggered Reversible “Stealth” Polycationic Micelles. Biomacromolecules 2008, 9, 255–262. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Tang, C.; Yin, C. pH-Responsive Core–Shell Structured Nanoparticles for Triple-Stage Targeted Delivery of Doxorubicin to Tumors. ACS Appl. Mater. Interfaces 2016, 8, 23498–23508. [Google Scholar] [CrossRef]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current Development of siRNA Bioconjugates: From Research to the Clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. VEGF-VEGFR Signals in Health and Disease. Biomol. Ther. 2014, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wolfert, M.A.; Dash, P.R.; Nazarova, O.; Oupický, D.; Seymour, L.W.; Smart, S.; Strohalm, J.; Ulbrich, K. Polyelectrolyte vectors for gene delivery: Influence of cationic polymer on biophysical properties of complexes formed with DNA. Bioconjugate Chem. 1999, 10, 993–1004. [Google Scholar] [CrossRef]
- Chiou, H.C.; Tangco, M.V.; Kormis, K.; Levine, S.M.; Robertson, D.; Wu, C.H.; Wu, G.Y. Enhanced resistance to nuclease degradation of nucleic acids complexed to asialoglycoprotein-polylysine carriers. Nucleic Acids Res. 1994, 22, 5439–5446. [Google Scholar] [CrossRef] [Green Version]
- Egorova, A.; Bogacheva, M.; Shubina, A.; Baranov, V.; Kiselev, A. Development of a receptor-targeted gene delivery system using CXCR4 ligand-conjugated cross-linking peptides. J. Gene Med. 2014, 16, 336–351. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, S.; Roscigno, G.; Affinito, A.; Ingenito, F.; Quintavalle, C.; Condorelli, G. Potential and Challenges of Aptamers as Specific Carriers of Therapeutic Oligonucleotides for Precision Medicine in Cancer. Cancers 2019, 11, 1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affinito, A.; Quintavalle, C.; Esposito, C.L.; Roscigno, G.; Vilardo, C.; Nuzzo, S.; Ricci-Vitiani, L.; De Luca, G.; Pallini, R.; Kichkailo, A.S.; et al. The Discovery of RNA Aptamers that Selectively Bind Glioblastoma Stem Cells. Mol. Ther. Nucleic Acids 2019, 18, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, V.; Paciocco, A.; Affinito, A.; Roscigno, G.; Fiore, D.; Palma, F.; Galasso, M.; Volinia, S.; Fiorelli, A.; Esposito, C.L.; et al. Aptamer-miR-34c Conjugate Affects Cell Proliferation of Non-Small-Cell Lung Cancer Cells. Mol. Ther. Nucleic Acids 2018, 13, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Iaboni, M.; Russo, V.; Fontanella, R.; Roscigno, G.; Fiore, D.; Donnarumma, E.; Esposito, C.L.; Quintavalle, C.; Giangrande, P.H.; De Franciscis, V.; et al. Aptamer-miRNA-212 Conjugate Sensitizes NSCLC Cells to TRAIL. Mol. Ther. Nucleic Acids 2016, 5, e289. [Google Scholar] [CrossRef] [Green Version]
- Catuogno, S.; Esposito, C.L.; Condorelli, G.; De Franciscis, V. Nucleic acids delivering nucleic acids. Adv. Drug Deliv. Rev. 2018, 134, 79–93. [Google Scholar] [CrossRef]
- Rajagopalan, A.; Berezhnoy, A.; Schrand, B.; Puplampu-Dove, Y.; Gilboa, E. Aptamer-Targeted Attenuation of IL-2 Signaling in CD8+ T Cells Enhances Antitumor Immunity. Mol. Ther. 2017, 25, 54–61. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| siRNA Target | TME Component | Effects | Reference Number |
|---|---|---|---|
| CXCL8 | CAFs | ↓ Tumor migration and invasion potentialities. | [67] |
| YAP1 | CAFs | ↓ CAF’s contraction ability and angiogenesis in vivo | [68] |
| IL-11, IL-15 | CAFs | ↓ Vascular endothelial cells’ angiogenic ability | [69] |
| VEGF-A VEGFR1, Endoglin | Endothelial cells | ↓ Endothelial cell migration and proliferation abilities. | [69,70] |
| VEGF-PIGF | M2-TAMs | ↓ M2-associated molecular signature, ↓ breast cancer growth and angiogenesis. | [71] |
| STAT6 | M2-TAMs | ↓ ECM remodeling and metastasis spreading. | [72] |
| PD1 | T-cells | ↓ Tumor growth in breast cancer cells bearing mice. | [73] |
| PKM2 | CAAs | ↓ EMT-related markers, inhibition of leptin-induced breast cancer cell invasion and migration in vitro. | [74] |
| IDO | DCs | ↓ T CD4+ and Treg cells within TME. | [75] |
| PITPNM3 | T-CD4+ cells | ↓ T reg recruitment and ↑ T CD8+ populations within T cells ↑ Apoptosis in cancer cells ↓ Tumor growth and lung metastasis formation. | [76] |
| MVP | CAAs | ↑ Accumulation of antineoplastic compounds within cancerous cells. | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roscigno, G.; Scognamiglio, I.; Ingenito, F.; Chianese, R.V.; Palma, F.; Chan, A.; Condorelli, G. Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment. Cancers 2020, 12, 3744. https://doi.org/10.3390/cancers12123744
Roscigno G, Scognamiglio I, Ingenito F, Chianese RV, Palma F, Chan A, Condorelli G. Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment. Cancers. 2020; 12(12):3744. https://doi.org/10.3390/cancers12123744
Chicago/Turabian StyleRoscigno, Giuseppina, Iolanda Scognamiglio, Francesco Ingenito, Rosario Vincenzo Chianese, Francesco Palma, Alan Chan, and Gerolama Condorelli. 2020. "Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment" Cancers 12, no. 12: 3744. https://doi.org/10.3390/cancers12123744
APA StyleRoscigno, G., Scognamiglio, I., Ingenito, F., Chianese, R. V., Palma, F., Chan, A., & Condorelli, G. (2020). Modulating the Crosstalk between the Tumor and the Microenvironment Using SiRNA: A Flexible Strategy for Breast Cancer Treatment. Cancers, 12(12), 3744. https://doi.org/10.3390/cancers12123744