Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy



Abstract
:Simple Summary
Abstract
1. Introduction
2. Aberrant Ubiquitination in MM
Exploiting the Ubiquitin Proteasome System Therapeutically
3. Deubiquitinating Enzymes (DUBs)
Therapeutic Targeting of DUBs
4. NEDDylation
NEDDylation as Therapeutic Target
5. SUMOylation
SUMOylation as Therapeutic Target
6. DeSUMOylation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.C.; Lonial, S.; Bladé, J.; Mateos, M.-V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V. The multiple myelomas—Current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018, 15, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Fonseca, R.; Ketterling, R.P.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Knudson, R.A.; et al. Trisomies in multiple myeloma: Impact on survival in patients with high-risk cytogenetics. Blood 2012, 119, 2100–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, R.A. Monoclonal gammopathy of undetermined significance. Am. J. Med. 1978, 64, 814–826. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.S.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Seidl, S.; Kaufmann, H.; Drach, J. New insights into the pathophysiology of multiple myeloma. Lancet Oncol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC Translocations in Multiple Myeloma Cell Lines. J. Natl. Cancer Inst. Monogr. 2008, 2008, 25–31. [Google Scholar] [CrossRef]
- Jovanović, K.K.; Escure, G.; Demonchy, J.; Willaume, A.; Van De Wyngaert, Z.; Farhat, M.; Chauvet, P.; Facon, T.; Quesnel, B.; Manier, S. Deregulation and Targeting of TP53 Pathway in Multiple Myeloma. Front. Oncol. 2019, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and biological implications of MYC activation: A common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011, 25, 1026–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, A.; Murphy, J.P.; Cro, L.; Ferrero, D.; Tarella, C.; Baldini, L.; Dalla-Favera, R. Ras oncogene mutation in multiple myeloma. J. Exp. Med. 1989, 170, 1715–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, M.; Rajkumar, S.V.; Ketterling, R.P.; Greipp, P.T.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Hayman, S.R.; Hwa, Y.L.; et al. Prognostic implications of abnormalities of chromosome 13 and the presence of multiple cytogenetic high-risk abnormalities in newly diagnosed multiple myeloma. Blood Cancer J. 2017, 7, e600. [Google Scholar] [CrossRef] [Green Version]
- Marzin, Y.; Jamet, D.; Douet-Guilbert, N.; Morel, F.; Le Bris, M.-J.; Morice, P.; Abgrall, J.-F.; Berthou, C.; De Braekeleer, M. Chromosome 1 abnormalities in multiple myeloma. Anticancer. Res. 2006, 26, 953–959. [Google Scholar]
- Lakshman, A.; Painuly, U.; Rajkumar, V.; Ketterling, R.P.; Kapoor, P.; Greipp, P.T.; Dispenzieri, A.; Gertz, M.A.; Buadi, F.K.; Lacy, M.Q.; et al. Impact of acquired del(17p) in multiple myeloma. Blood Adv. 2019, 3, 1930–1938. [Google Scholar] [CrossRef]
- Van Andel, H.; A Kocemba, K.; De Haan-Kramer, A.; Mellink, C.H.; Piwowar, M.; Broijl, A.; Van Duin, M.; Sonneveld, P.; Maurice, M.M.; Kersten, M.J.; et al. Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 2016, 36, 2105–2115. [Google Scholar] [CrossRef]
- Keats, J.J.; Fonseca, R.; Chesi, M.; Schop, R.; Baker, A.; Chng, W.-J.; Van Wier, S.; Tiedemann, R.; Shi, C.-X.; Sebag, M.; et al. Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell 2007, 12, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, C.M.; Davis, R.E.; Demchenko, Y.; Bellamy, W.; Gabrea, A.; Zhan, F.; Lenz, G.; Hanamura, I.; Wright, G.; Xiao, W.; et al. Frequent Engagement of the Classical and Alternative NF-κB Pathways by Diverse Genetic Abnormalities in Multiple Myeloma. Cancer Cell 2007, 12, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Kaiser, M.F.; Heuck, C.; Melchor, L.; Wardell, C.P.; Murison, A.; Chavan, S.S.; Johnson, D.C.; Begum, D.B.; Dahir, N.M.; et al. The Spectrum and Clinical Impact of Epigenetic Modifier Mutations in Myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, J.R.; Tian, E.; Heuck, C.J.; Johann, D.J.; Epstein, J.; Swanson, C.M.; Lukacs, J.L.; Binz, R.L.; Johnson, M.; Sammartino, G.; et al. Evidence of an epigenetic origin for high-risk 1q21 copy number aberrations in multiple myeloma. Blood 2015, 125, 3756–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agirre, X.; Castellano, G.; Pascual, M.; Heath, S.; Kulis, M.; Segura, V.; Bergmann, A.; Esteve, A.; Merkel, A.; Raineri, E.; et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 2015, 25, 478–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, B.; On Behalf of GEM (Grupo Español de MM)/PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Groups; Puig, N.; Cedena, M.T.; De Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; Alignani, D.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, K.; De Smedt, E.; Kassambara, A.; Hose, D.; Seckinger, A.; Van Valckenborgh, E.; Menu, E.; Klein, B.; Vanderkerken, K.; Moreaux, J.; et al. In vivotreatment with epigenetic modulating agents induces transcriptional alterations associated with prognosis and immunomodulation in multiple myeloma. Oncotarget 2014, 6, 3319–3334. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Leotta, M.; Bellizzi, D.; Di Martino, M.T.; D’Aquila, P.; Lionetti, M.; Fabiani, F.; Leone, E.; Gullà, A.M.; Passarino, G.; et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012, 3, 1246–1258. [Google Scholar] [CrossRef] [Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Sci. 2008, 319, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Cenci, S.; Sitia, R. Managing and exploiting stress in the antibody factory. FEBS Lett. 2007, 581, 3652–3657. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- El Kennani, S.; Crespo, M.; Govin, J.; Pflieger, D. Proteomic Analysis of Histone Variants and Their PTMs: Strategies and Pitfalls. Proteomes 2018, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Chen, Y.; Zhang, F.; Yang, C.-Y.; Wang, S.; Yu, X. RNF111-dependent neddylation activates DNA damage-induced ubiquitination. Mol. Cell 2013, 49, 897–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-κB Activating Pathways in Multiple Myeloma. Biomedicine 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A Monoubiquitination Represses Transcription by Inhibiting RNA Polymerase II Transcriptional Elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, S.; Chahwan, R.; Nepal, R.M.; Frieder, D.; Panier, S.; Roa, S.; Zaheen, A.; Durocher, D.; Scharff, M.D.; Pezo, R.C. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc. Natl. Acad. Sci. USA 2009, 107, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; Van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [Green Version]
- Chng, W.J.; Rajkumar, V.; VanWier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.-H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular Dissection of Hyperdiploid Multiple Myeloma by Gene Expression Profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef] [Green Version]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.-M.; Lengauer, C.; et al. The Polycomb Group Protein Bmi-1 Is Essential for the Growth of Multiple Myeloma Cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Muñoz, I.; Lund, A.H.; Van Der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D.A.; Panning, B.; Marahrens, Y.; Van Lohuizen, M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7635–7640. [Google Scholar] [CrossRef] [Green Version]
- Doyen, C.-M.; An, W.; Angelov, D.; Bondarenko, V.; Mietton, F.; Studitsky, V.M.; Hamiche, A.; Roeder, R.G.; Bouvet, P.; Dimitrov, S. Mechanism of Polymerase II Transcription Repression by the Histone Variant macroH2A. Mol. Cell. Biol. 2006, 26, 1156–1164. [Google Scholar] [CrossRef] [Green Version]
- Angelov, D.; Molla, A.; Perche, P.-Y.; Hans, F.; Côté, J.; Khochbin, S.; Bouvet, P.; Dimitrov, S. The Histone Variant MacroH2A Interferes with Transcription Factor Binding and SWI/SNF Nucleosome Remodeling. Mol. Cell 2003, 11, 1033–1041. [Google Scholar] [CrossRef]
- Zhu, K.; Lei, P.-J.; Ju, L.-G.; Wang, X.; Huang, K.; Yang, B.; Shao, C.; Zhu, Y.; Changwei, S.; Fu, X.-D.; et al. SPOP-containing complex regulates SETD2 stability and H3K36me3-coupled alternative splicing. Nucleic Acids Res. 2017, 45, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ci, Y.; Dai, X.; Wu, F.; Guo, J.; Liu, D.; North, B.J.; Huo, J.; Zhang, J. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget 2017, 8, 47890–47901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Bao, Y.-C.; Ji, X.-X.; Chen, B.; Deng, Q.-F.; Zhou, S.-W. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochem. Biophys. Res. Commun. 2017, 483, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Sá, M.J.N.; El Tekle, G.; De Brouwer, A.P.; Sawyer, S.L.; Del Gaudio, D.; Parker, M.J.; Kanani, F.; Boogaard, M.-J.H.V.D.; Van Gassen, K.; Van Allen, M.I.; et al. De Novo Variants in SPOP Cause Two Clinically Distinct Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 405–411. [Google Scholar] [CrossRef]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer–associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Janouskova, H.; El Tekle, G.; Bellini, E.; Udeshi, N.D.; Rinaldi, A.; Ulbricht, A.; Bernasocchi, T.; Civenni, G.; Losa, M.; Svinkina, T.; et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat. Med. 2017, 23, 1046–1054. [Google Scholar] [CrossRef]
- Liu, J.; Ghanim, M.; Xue, L.; Brown, C.D.; Iossifov, I.; Angeletti, C.; Hua, S.; Nègre, N.; Ludwig, M.; Stricker, T.; et al. Analysis of Drosophila Segmentation Network Identifies a JNK Pathway Factor Overexpressed in Kidney Cancer. Science 2009, 323, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Agnelli, L.; Mosca, L.; Fabris, S.; Lionetti, M.; Andronache, A.; Kwee, I.; Todoerti, K.; Verdelli, D.; Battaglia, C.; Bertoni, F.; et al. A SNP microarray and FISH-based procedure to detect allelic imbalances in multiple myeloma: An integrated genomics approach reveals a wide gene dosage effect. Genes Chromosom. Cancer 2009, 48, 603–614. [Google Scholar] [CrossRef]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.-W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 Is a Nucleosomal Histone H3-Selective Methyltransferase That Mediates Transcriptional Repression. Mol. Cell. Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, S.; Vítor, A.C.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.M.P.; Ferreira, J.; De Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. eLife 2014, 3, e02482. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Vikova, V.; Jourdan, M.; Robert, N.; Requirand, G.; Boireau, S.; Bruyer, A.; Vincent, L.; Cartron, G.; Klein, B.; Elemento, O.; et al. Comprehensive characterization of the mutational landscape in multiple myeloma cell lines reveals potential drivers and pathways associated with tumor progression and drug resistance. Theranostics 2019, 9, 540–553. [Google Scholar] [CrossRef]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Wirth, M.; Stojanovic, N.; Christian, J.; Paul, M.C.; Stauber, R.H.; Schmid, R.M.; Häcker, G.; Krämer, O.H.; Saur, D.; Schneider, G. MYC and EGR1 synergize to trigger tumor cell death by controlling NOXA and BIM transcription upon treatment with the proteasome inhibitor bortezomib. Nucleic Acids Res. 2014, 42, 10433–10447. [Google Scholar] [CrossRef] [Green Version]
- Lankes, K.; Hassan, Z.; Doffo, M.J.; Schneeweis, C.; Lier, S.; Öllinger, R.; Rad, R.; Krämer, O.H.; Keller, U.; Saur, D.; et al. Targeting the ubiquitin-proteasome system in a pancreatic cancer subtype with hyperactive MYC. Mol. Oncol. 2020, 14, 3048–3064. [Google Scholar] [CrossRef]
- Wirth, M.; Mahboobi, S.; Krämer, O.H.; Schneider, G. Concepts to Target MYC in Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting Proteotoxic Stress in Cancer: A Review of the Role that Protein Quality Control Pathways Play in Oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [Green Version]
- Naymagon, L.; Abdul-Hay, M. Novel agents in the treatment of multiple myeloma: A review about the future. J. Hematol. Oncol. 2016, 9, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; MacLean, K.H.; Courage, J.F.; Huang, P.; Houghton, J.A.; Cleveland, J.L.; Giles, F.J.; McConkey, D.J. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood 2008, 112, 2917–2926. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, U.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.; Irwin, D.; Stadtmauer, E.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or High-Dose Dexamethasone for Relapsed Multiple Myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R.; Velcade, R.U.S. FDA Approval for the Treatment of Multiple Myeloma Progressing on Prior Therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Yong, K.; Gonzalez-McQuire, S.; Szabo, Z.; Schoen, P.; Hajek, R. The start of a new wave: Developments in proteasome inhibition in multiple myeloma. Eur. J. Haematol. 2018, 101, 220–236. [Google Scholar] [CrossRef]
- Guerrero-Garcia, T.A.; Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Mitsiades, C.; Anderson, K.C.; Richardson, P.G. The power of proteasome inhibition in multiple myeloma. Expert Rev. Proteom. 2018, 15, 1033–1052. [Google Scholar] [CrossRef]
- Dimopoulos, M.-A.; Goldschmidt, H.; Niesvizky, R.; Joshua, D.; Chng, W.-J.; Oriol, A.; Orlowski, R.Z.; Ludwig, H.; Facon, T.; Hajek, R.; et al. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): An interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1327–1337. [Google Scholar] [CrossRef]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuki, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P. Oral therapy for multiple myeloma: Ixazomib arriving soon. Blood 2014, 124, 986–987. [Google Scholar] [CrossRef] [Green Version]
- Raza, S.; Safyan, R.; Suzanne, L. Immunomodulatory Drugs (IMiDs) in Multiple Myeloma. Curr. Cancer Drug Targets 2017, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.-C.; Miller, K.; Fang, W.; Wang, N.-Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nat. Cell Biol. 2016, 535, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Słabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.-D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nat. Cell Biol. 2020, 585, 293–297. [Google Scholar] [CrossRef]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Phthalimide conjugation as a strategy for in vivo target protein degradation. Sciience 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.L.; Damnernsawad, A.; Shyamsunder, P.; Chng, W.J.; Han, B.C.; Xu, L.; Pan, J.; Pravin, D.P.; Alkan, S.; Tyner, J.W.; et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: Efficacy, biomarker and drug combinations. Haematology 2019, 104, 1209–1220. [Google Scholar] [CrossRef]
- Chan, C.-H.; Morrow, J.K.; Li, C.-F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.P.; Ioannidis, S.; Krajewski, W.W.; Pinto-Fernandez, A.; Heride, C.; Martin, A.C.L.; Tonkin, L.M.; Townsend, E.C.; Buker, S.M.; Lancia, D.R.; et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 2017, 550, 481–486. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nat. Cell Biol. 2017, 550, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Lamberto, I.; Liu, X.; Seo, H.-S.; Schauer, N.J.; Iacob, R.E.; Hu, W.; Das, D.; Mikhailova, T.; Weisberg, E.L.; Engen, J.R.; et al. Structure-Guided Development of a Potent and Selective Non-covalent Active-Site Inhibitor of USP7. Cell Chem. Biol. 2017, 24, 1490–1500.e11. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.-K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, C.; Muraoka, H.; Ochiiwa, H.; Tsuji, S.; Hashimoto, A.; Kazuno, H.; Nakagawa, F.; Komiya, Y.; Suzuki, S.; Takenaka, T.; et al. TAS4464, A Highly Potent and Selective Inhibitor of NEDD8-Activating Enzyme, Suppresses Neddylation and Shows Antitumor Activity in Diverse Cancer Models. Mol. Cancer Ther. 2019, 18, 1205–1216. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, S.V.; Hayman, S.R.; Lacy, M.Q.; Dispenzieri, A.; Geyer, S.M.; Kabat, B.; Zeldenrust, S.R.; Kumar, S.; Greipp, P.R.; Fonseca, R.; et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005, 106, 4050–4053. [Google Scholar] [CrossRef] [Green Version]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the Myelodysplastic Syndrome with Chromosome 5q Deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krönke, J.; Fink, E.C.; Hollenbach, P.W.; Macbeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nat. Cell Biol. 2015, 523, 183–188. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018, 14, 981–987. [Google Scholar] [CrossRef]
- Donovan, K.A.; An, J.; Nowak, R.P.; Yuan, J.C.; Fink, E.C.; Berry, B.C.; Ebert, B.L.; Fischer, E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, K.; Bigby, M.; Wang, J.-H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A. The ikaros gene is required for the development of all lymphoid lineages. Cell 1994, 79, 143–156. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors I karos and A iolos via modulation of the E 3 ubiquitin ligase complex CRL 4 CRBN. Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, M.G.E.C.D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef]
- Steinebach, C.; Ng, Y.L.D.; Sosič, I.; Lee, C.-S.; Chen, S.; Lindner, S.; Vu, L.P.; Bricelj, A.; Haschemi, R.; Monschke, M.; et al. Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e9. [Google Scholar] [CrossRef]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell Chem. Biol. 2018, 25, 67–77.e3. [Google Scholar] [CrossRef]
- Yang, K.; Song, Y.; Xie, H.; Wu, H.; Wu, Y.-T.; Leisten, E.D.; Tang, W. Development of the first small molecule histone deacetylase 6 (HDAC6) degraders. Bioorganic Med. Chem. Lett. 2018, 28, 2493–2497. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Verma, R.; Ransick, A.; Stein, B.; Crews, C.M.; Deshaies, R.J. Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Mol. Cell. Proteom. 2003, 2, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinebach, C.; Lindner, S.; Udeshi, N.D.; Mani, D.C.; Kehm, H.; Köpff, S.; Carr, S.A.; Gütschow, M.; Krönke, J. Homo-PROTACs for the Chemical Knockdown of Cereblon. ACS Chem. Biol. 2018, 13, 2771–2782. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Mofers, A.; Pellegrini, P.; Linder, S.; D’Arcy, P. Proteasome-associated deubiquitinases and cancer. Cancer Metastasis Rev. 2017, 36, 635–653. [Google Scholar] [CrossRef] [Green Version]
- Heideker, J.; Wertz, I.E. DUBs, the regulation of cell identity and disease. Biochem. J. 2014, 465, 1–26. [Google Scholar] [CrossRef]
- Kim, H.T.; Goldberg, A.L. UBL domain of Usp14 and other proteins stimulates proteasome activities and protein degradation in cells. Proc. Natl. Acad. Sci. USA 2018, 115, E11642–E11650. [Google Scholar] [CrossRef] [Green Version]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Amerik, A.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. et Biophys. Acta (BBA) Bioenerg. 2004, 1695, 189–207. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Yan, Q. Histone Ubiquitination and Deubiquitination in Transcription, DNA Damage Response, and Cancer. Front. Oncol. 2011, 2, 26. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.-X.; Nguyen, Q.; Chou, Y.; Wang, T.; Nandakumar, V.; Yates, P.; Jones, L.; Wang, L.; Won, H.-J.; Lee, H.-R.; et al. Control of B Cell Development by the Histone H2A Deubiquitinase MYSM1. Immun. 2011, 35, 883–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, H.; Nandakumar, V.; Yates, P.; Sanchez, S.; Jones, L.; Huang, X.F.; Chen, S.-Y. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 2014, 124, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijnik, A.; Clare, S.; Hale, C.; Raisen, C.; McIntyre, R.E.; Yusa, K.; Everitt, A.R.; Mottram, L.; Podrini, C.; Lucas, M.; et al. The critical role of histone H2A-deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood 2012, 119, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nandakumar, V.; Jiang, X.-X.; Jones, L.; Yang, A.-G.; Huang, X.F.; Chen, S.-Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, T.; Touzot, F.; André-Schmutz, I.; Lagresle-Peyrou, C.; France, B.; Kermasson, L.; Lambert, N.C.; Picard, C.; Nitschke, P.; Carpentier, W.; et al. An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J. Allergy Clin. Immunol. 2015, 136, 1619–1626.e5. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, S.; Tong, J.; Jiang, S.; Yang, Y.; Zhang, Z.; Xu, Y.; Zeng, Y.-Y.; Cao, B.; Moran, M.F.; et al. The deubiquitinase USP7 stabilizes Maf proteins to promote myeloma cell survival. J. Biol. Chem. 2020, 295, 2084–2096. [Google Scholar] [CrossRef]
- Qi, S.-M.; Cheng, G.; Cheng, X.-D.; Xu, Z.; Xu, B.; Zhang, W.-D.; Qin, J.-J. Targeting USP7-Mediated Deubiquitination of MDM2/MDMX-p53 Pathway for Cancer Therapy: Are We There Yet? Front. Cell Dev. Biol. 2020, 8, 233. [Google Scholar] [CrossRef] [Green Version]
- Van Der Knaap, J.A.; Kumar, B.P.; Moshkin, Y.M.; Langenberg, K.; Krijgsveld, J.; Heck, A.J.; Karch, F.; Verrijzer, C.P. GMP Synthetase Stimulates Histone H2B Deubiquitylation by the Epigenetic Silencer USP7. Mol. Cell 2005, 17, 695–707. [Google Scholar] [CrossRef]
- Inoue, D.; Nishimura, K.; Kozuka-Hata, H.; Oyama, M.; Kitamura, T. The stability of epigenetic factor ASXL1 is regulated through ubiquitination and USP7-mediated deubiquitination. Leukemia 2015, 29, 2257–2260. [Google Scholar] [CrossRef]
- Lecona, E.; Rodriguez-Acebes, S.; Specks, J.; Lopez-Contreras, A.J.; Ruppen, I.; Murga, M.; Muñoz, I.R.J.; Mendez, S.R.-A.J.; Fernandez-Capetillo, E.L.J.S.M.M.O. USP7 is a SUMO deubiquitinase essential for DNA replication. Nat. Struct. Mol. Biol. 2016, 23, 270–277. [Google Scholar] [CrossRef]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jiang, S.; Li, M.; Xiong, X.; Zhu, M.; Li, D.; Zhao, L.; Qian, L.; Zhai, L.; Li, J.; et al. Proteome-wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Lee, B.-H.; Hanna, J.; King, R.W.; Finley, D. Trimming of Ubiquitin Chains by Proteasome-associated Deubiquitinating Enzymes. Mol. Cell. Proteom. 2010, 10, 110-003871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.A.; Dimova, N.; Hanna, J.V.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nat. Cell Biol. 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Yan, Y.; Wang, D.; Ding, D.; Ma, T.; Ye, Z.; Jimenez, R.; Wang, L.; Wu, H.; Huang, H. DUB3 Promotes BET Inhibitor Resistance and Cancer Progression by Deubiquitinating BRD4. Mol. Cell 2018, 71, 592–605.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.; Deshaies, R.J. Multimodal Activation of the Ubiquitin Ligase SCF by Nedd8 Conjugation. Mol. Cell 2008, 32, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.; Krist, D.T.; Prabu, J.R.; Hill, S.; Klügel, M.; Neumaier, L.-M.; Von Gronau, S.; Kleiger, G.; A Schulman, B. NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly. Nat. Cell Biol. 2020, 578, 461–466. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Chauhan, D.; Schlossman, R.; Richardson, P.; Anderson, K.C. The role of tumor necrosis factor α in the pathophysiology of human multiple myeloma: Therapeutic applications. Oncogene 2001, 20, 4519–4527. [Google Scholar] [CrossRef] [Green Version]
- Milhollen, M.A.; Traore, T.; Adams-Duffy, J.; Thomas, M.P.; Berger, A.; Dang, L.; Dick, L.R.; Garnsey, J.J.; Koenig, E.; Langston, S.P.; et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-κB–dependent lymphoma. Blood 2010, 116, 1515–1523. [Google Scholar] [CrossRef]
- Li, T.; Guan, J.; Huang, Z.; Hu, X.; Zheng, X. RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J. Cell Sci. 2014, 127, 2238–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, W.J.; Braggio, E.; Mulligan, G.; Bryant, B.; Remstein, E.; Valdez, R.; Dogan, A.; Fonseca, R. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 2008, 111, 1603–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, J.J.; Jakubowiak, A.J.; O’Connor, O.A.; Orlowski, R.Z.; Harvey, R.D.; Smith, M.R.; Lebovic, D.; Diefenbach, C.; Kelly, K.; Hua, Z.; et al. Phase I Study of the Novel Investigational NEDD8-Activating Enzyme Inhibitor Pevonedistat (MLN4924) in Patients with Relapsed/Refractory Multiple Myeloma or Lymphoma. Clin. Cancer Res. 2016, 22, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Kaufman, J.L.; Bernal, L.; Torre, C.; Matulis, S.M.; Harvey, R.D.; Chen, J.; Sun, S.-Y.; Boise, L.H.; Lonial, S. MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 2014, 123, 3269–3276. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.-P.; Hu, Y.-C.; Xie, Y.; Jin, F.; Song, Z.-Q.; Liu, X.-D.; Ma, T.; Zhou, P.-K. MLN4924 suppresses the BRCA1 complex and synergizes with PARP inhibition in NSCLC cells. Biochem. Biophys. Res. Commun. 2017, 483, 223–229. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A Regulatory Protein Modification in Health and Disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef]
- Eifler, K.; Vertegaal, A.C. Mapping the SUMOylated landscape. FEBS J. 2015, 282, 3669–3680. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Schneeweis, C.; Hassan, Z.; Schick, M.; Keller, U.; Schneider, G. The SUMO pathway in pancreatic cancer: Insights and inhibition. Br. J. Cancer 2020, 1–8. [Google Scholar] [CrossRef]
- Biederstädt, A.; Hassan, Z.; Schneeweis, C.; Schick, M.; Schneider, L.; Muckenhuber, A.; Hong, Y.; Siegers, G.; Nilsson, L.; Wirth, M.; et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 2020, 69, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-Dependent Transcriptional Subprogram Is Required for Myc-Driven Tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriramachandran, A.M.; Dohmen, R.J. SUMO-targeted ubiquitin ligases. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2014, 1843, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, R.C.; Kumar, R.; Clermont, F.; Helfricht, A.; Kalev, P.; Sotiropoulou, P.A.; A Hendriks, I.; Radaelli, E.; Hochepied, T.; Blanpain, C.; et al. RNF4 is required for DNA double-strand break repair in vivo. Cell Death Differ. 2013, 20, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Millas, S.; Maul, G.G.; Yeh, E.T. Differential Regulation of Sentrinized Proteins by a Novel Sentrin-specific Protease. J. Biol. Chem. 2000, 275, 3355–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Ji, W.; Zhang, H.; Renda, M.J.; He, Y.; Lin, S.; Cheng, E.-C.; Chen, H.; Krause, D.S.; Min, W. SENP1-mediated GATA1 deSUMOylation is critical for definitive erythropoiesis. J. Exp. Med. 2010, 207, 1183–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Sun, H.-Y.; Xiao, F.-J.; Wang, H.; Yang, Y.; Wang, L.; Gao, C.-J.; Guo, Z.-K.; Wu, C.-T.; Wang, L.-S. SENP1 inhibition induces apoptosis and growth arrest of multiple myeloma cells through modulation of NF-κB signaling. Biochem. Biophys. Res. Commun. 2015, 460, 409–415. [Google Scholar] [CrossRef]
- Maciejewska-Rodrigues, H.; Karouzakis, E.; Strietholt, S.; Hemmatazad, H.; Neidhart, M.; Ospelt, C.; Gay, R.E.; Michel, B.A.; Pap, T.; Gay, S.; et al. Epigenetics and rheumatoid arthritis: The role of SENP1 in the regulation of MMP-1 expression. J. Autoimmun. 2010, 35, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Mendes, A.; Grou, C.P.; Azevedo, J.E.; Pinto, M.P. Evaluation of the activity and substrate specificity of the human SENP family of SUMO proteases. Biochim. et Biophys. Acta (BBA) Bioenerg. 2016, 1863, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Gu, Y.; Wang, W.; Wang, X.; Ye, X.; Xin, C.; Lu, M.; Reddy, B.A.; Shu, P. Silencing of SENP2 in Multiple Myeloma Induces Bortezomib Resistance by Activating NF-κB Through the Modulation of IκBα Sumoylation. Sci. Rep. 2020, 10, 766. [Google Scholar] [CrossRef]
- Egodwin, P.; Baird, A.-M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
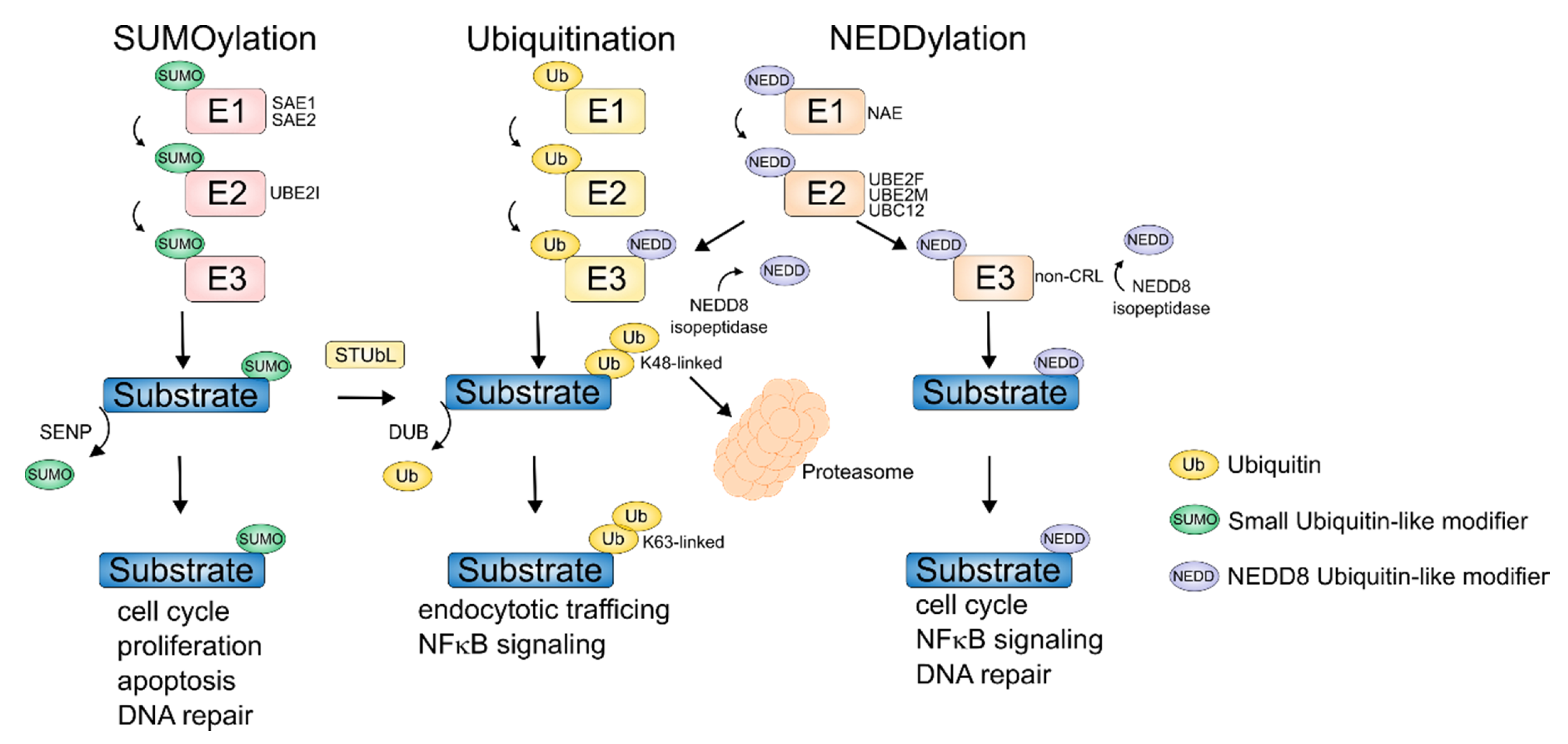


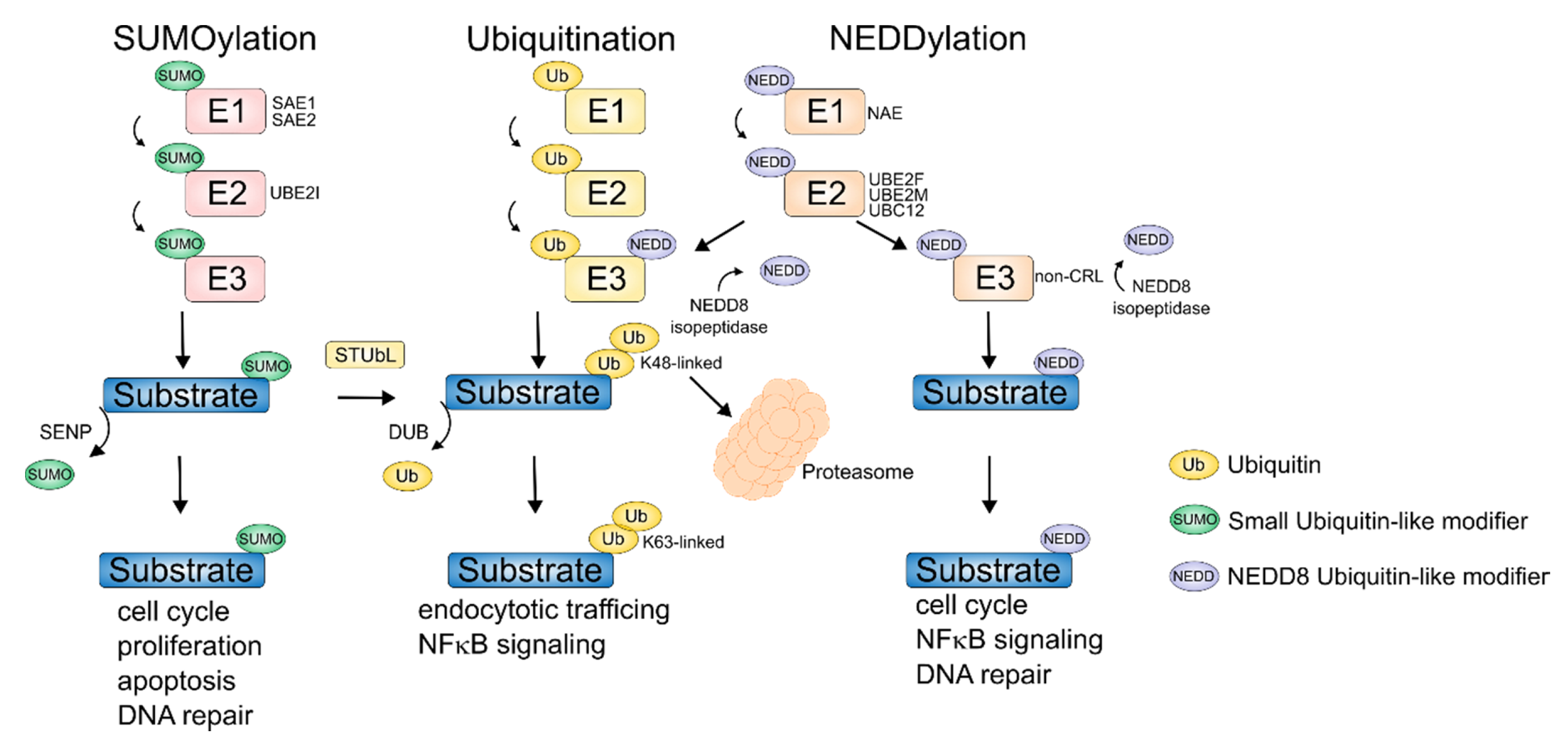{kind=link}
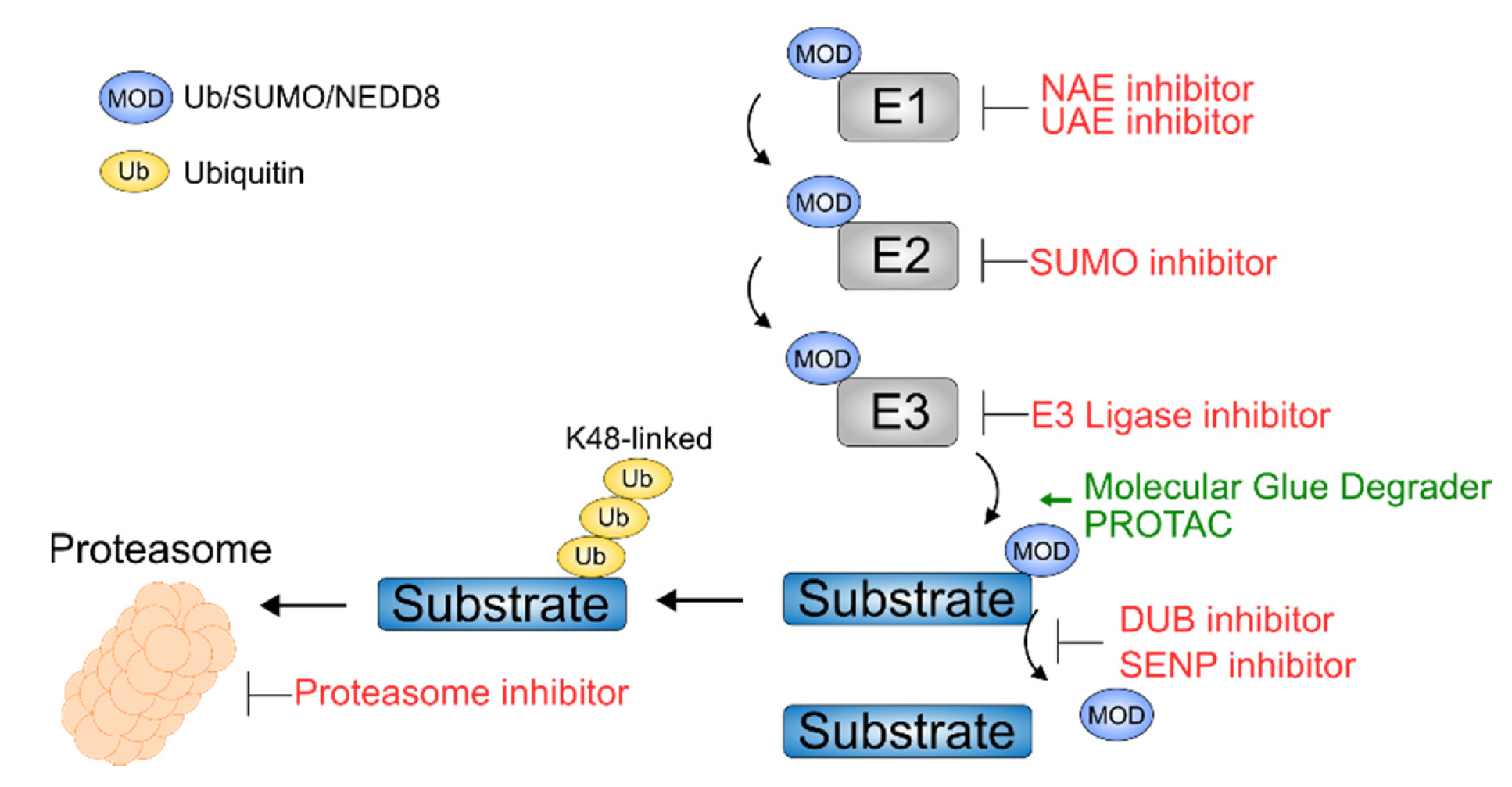{kind=link}
| Drug/Compound | Target | Main Substrates | Development | Reference/ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| Proteasome inhibitor | ||||
| Bortezomib, Carfilzomib, Ixazomib | Proteasome | many | Approved | [75,78,80] |
| Marizomib | Proteasome | many | Clinical trials | NCT03345095, NCT00461045 |
| Oprozomib | Proteasome | many | Clinical trials | NCT02939183 |
| Molecular glue degrader | ||||
| Thalidomide, Lenalidomide, Pomalidomide | CRBN | IKZF1/3 CK1 a SALL4 | Approved | [81] |
| Iberdomide, Avadomide | CRBN | IKZF1/3 | Clinical trials | NCT04564703, NCT04392037, NCT03283202, NCT03834623 |
| CC-885, CC-90009 | CRBN | GSPT1 | Clinical trials | [82], NCT02848001 |
| Indisulam | DCAF15 | RBM39 | Clinical trials | NCT01692197 |
| CR8 | DDB1 | Cyclin K | Pre-clinical | [83] |
| PROTAC | ||||
| ARV-110 | VHL | AR | Clinical trials | NCT03888612 |
| ARV-825, dBET1 | CRBN | BRD4 | Pre-clinical | [84,85] |
| E1 ubiquitin ligase inhibitor | ||||
| TAK-243/ MLN7243 | UAE1 | many | Clinical trials | NCT03816319 |
| E3 ubiquitin ligase inhibitor | ||||
| AMG-232, ALRN-6924 | MDM2 | TP53 | Clinical trials | NCT01723020, NCT03031730, NCT03654716 |
| SZL-P1–41 | SKP2 | p21Cip1, p27Kip1, p57Kip2 | Pre-clinical | [86] |
| SUMOylation inhibitor | ||||
| TAK-981 | UBE2 I | many | Clinical trials | NCT03648372, NCT04074330 |
| DUB inhibitor | ||||
| P5091, FT-671, GNE-6776, XL188 | USP7 | MDM2, TP53 | Pre-clinical | [87,88,89,90] |
| VLX1570 | USP14 | many | Pre-clinical | [91] |
| NEDDylation inhibitor | ||||
| Pevonedistat | NAE1 | CRLs | Clinical trials | NCT03770260, NCT00722488 |
| TAS4464 | NAE1 | CRLs | Pre-clinical | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wirth, M.; Schick, M.; Keller, U.; Krönke, J. Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers 2020, 12, 3764. https://doi.org/10.3390/cancers12123764
Wirth M, Schick M, Keller U, Krönke J. Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers. 2020; 12(12):3764. https://doi.org/10.3390/cancers12123764
Chicago/Turabian StyleWirth, Matthias, Markus Schick, Ulrich Keller, and Jan Krönke. 2020. "Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy" Cancers 12, no. 12: 3764. https://doi.org/10.3390/cancers12123764
APA StyleWirth, M., Schick, M., Keller, U., & Krönke, J. (2020). Ubiquitination and Ubiquitin-Like Modifications in Multiple Myeloma: Biology and Therapy. Cancers, 12(12), 3764. https://doi.org/10.3390/cancers12123764