The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
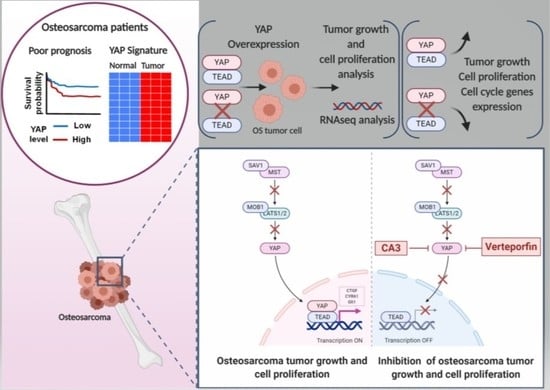
1. Introduction
2. Results
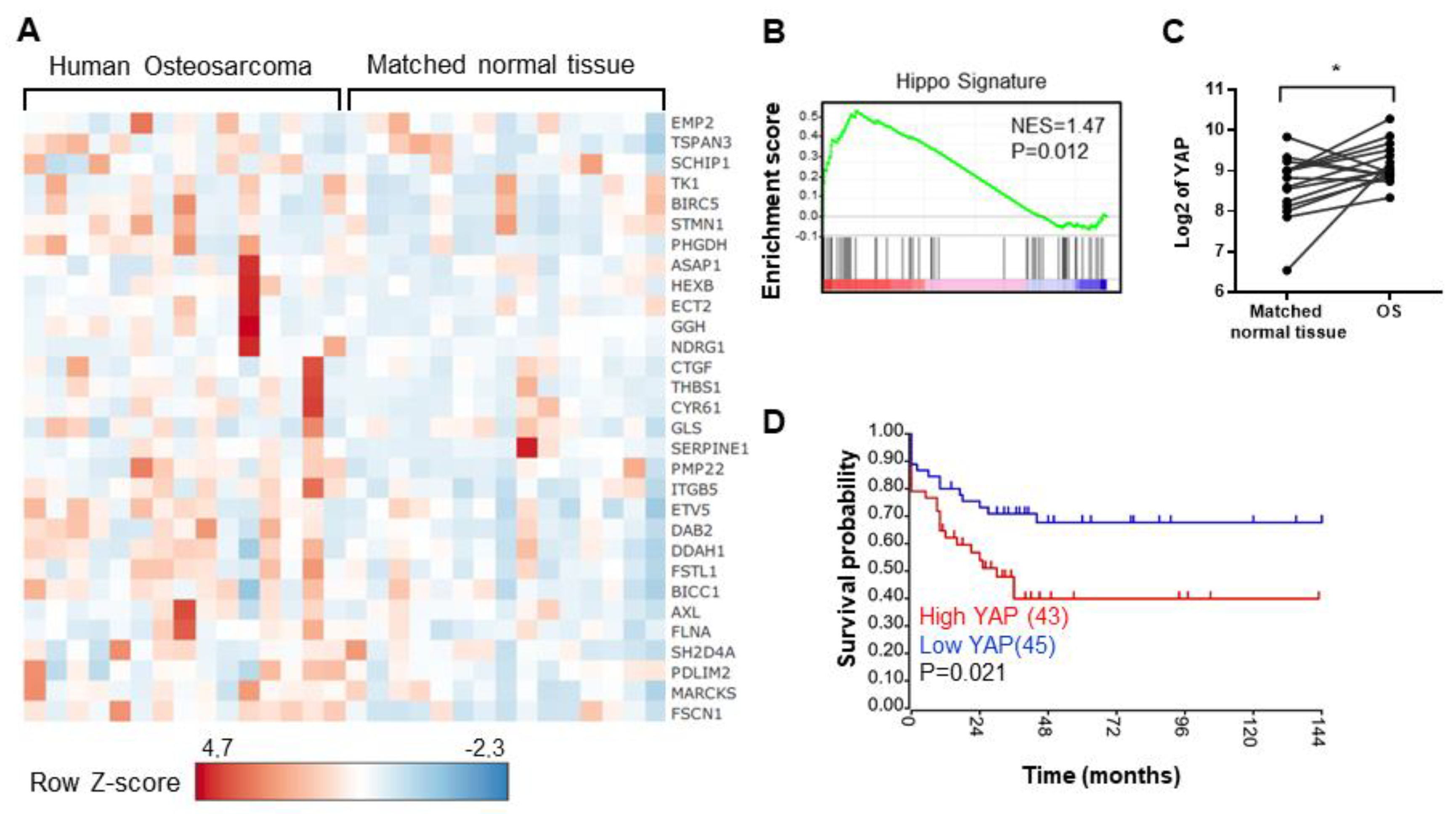
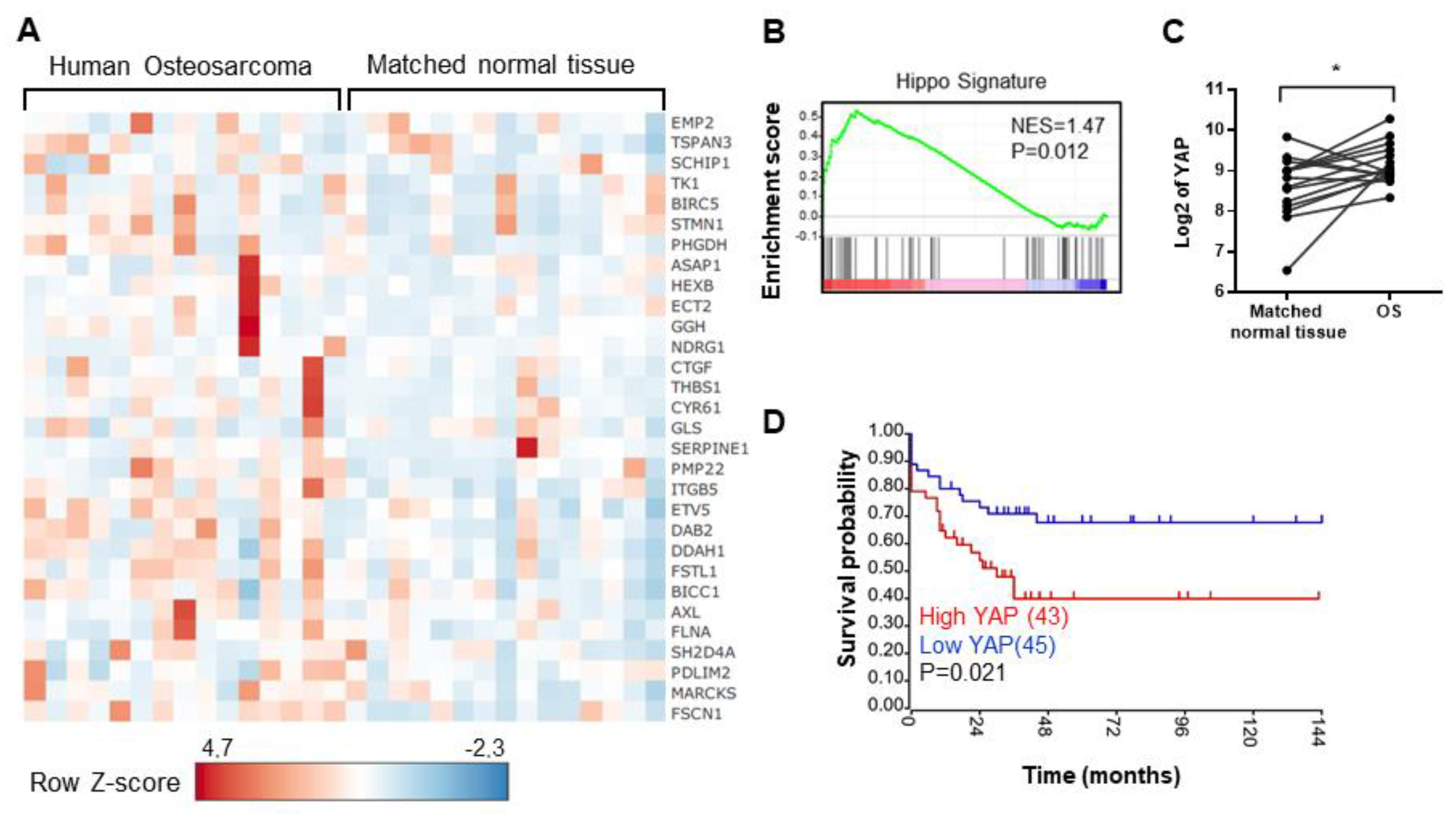
2.1. The Elevation of Hippo Target Genes Expression in OS Patients Is Associated with the Overall Survival of Patients
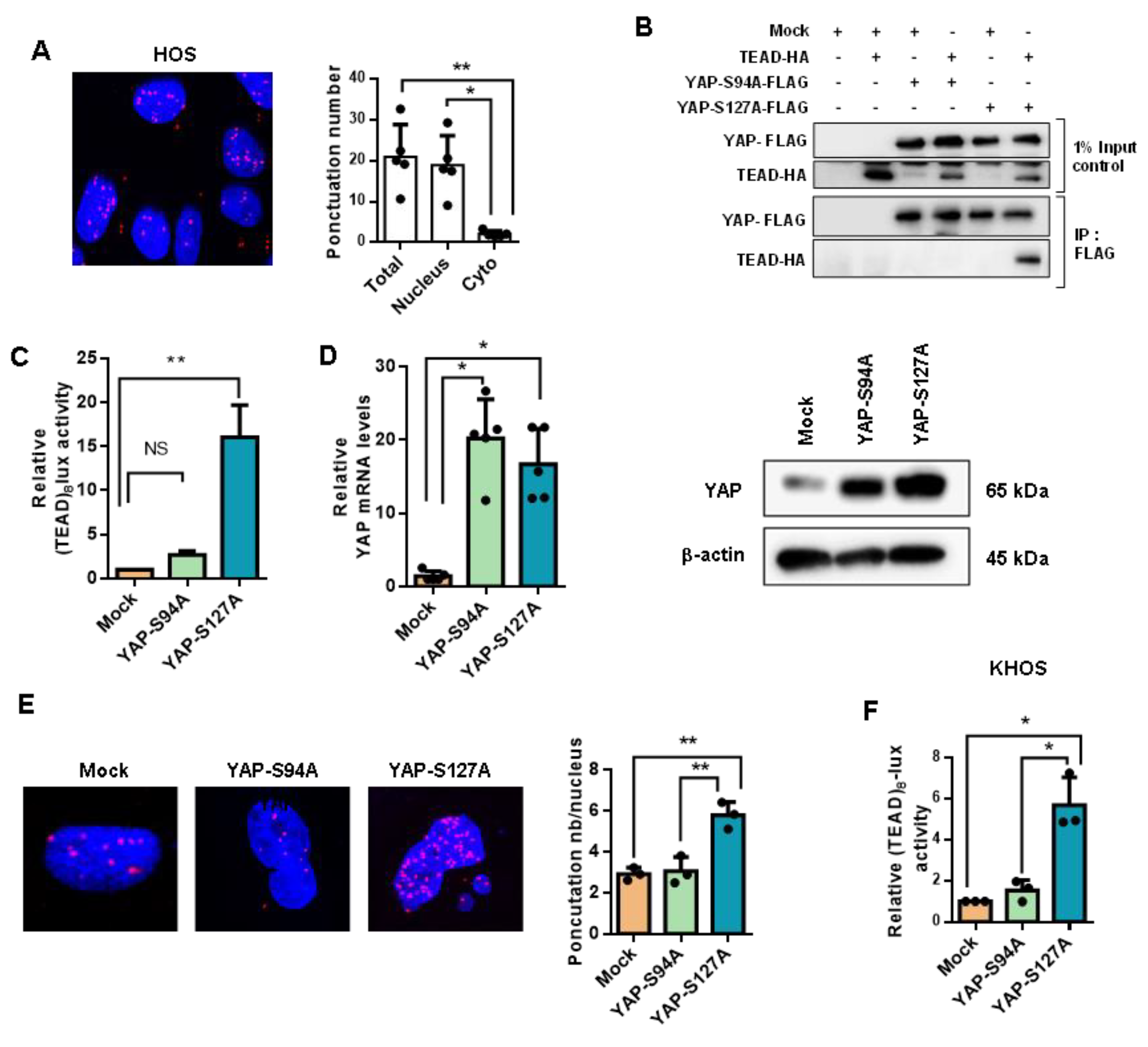
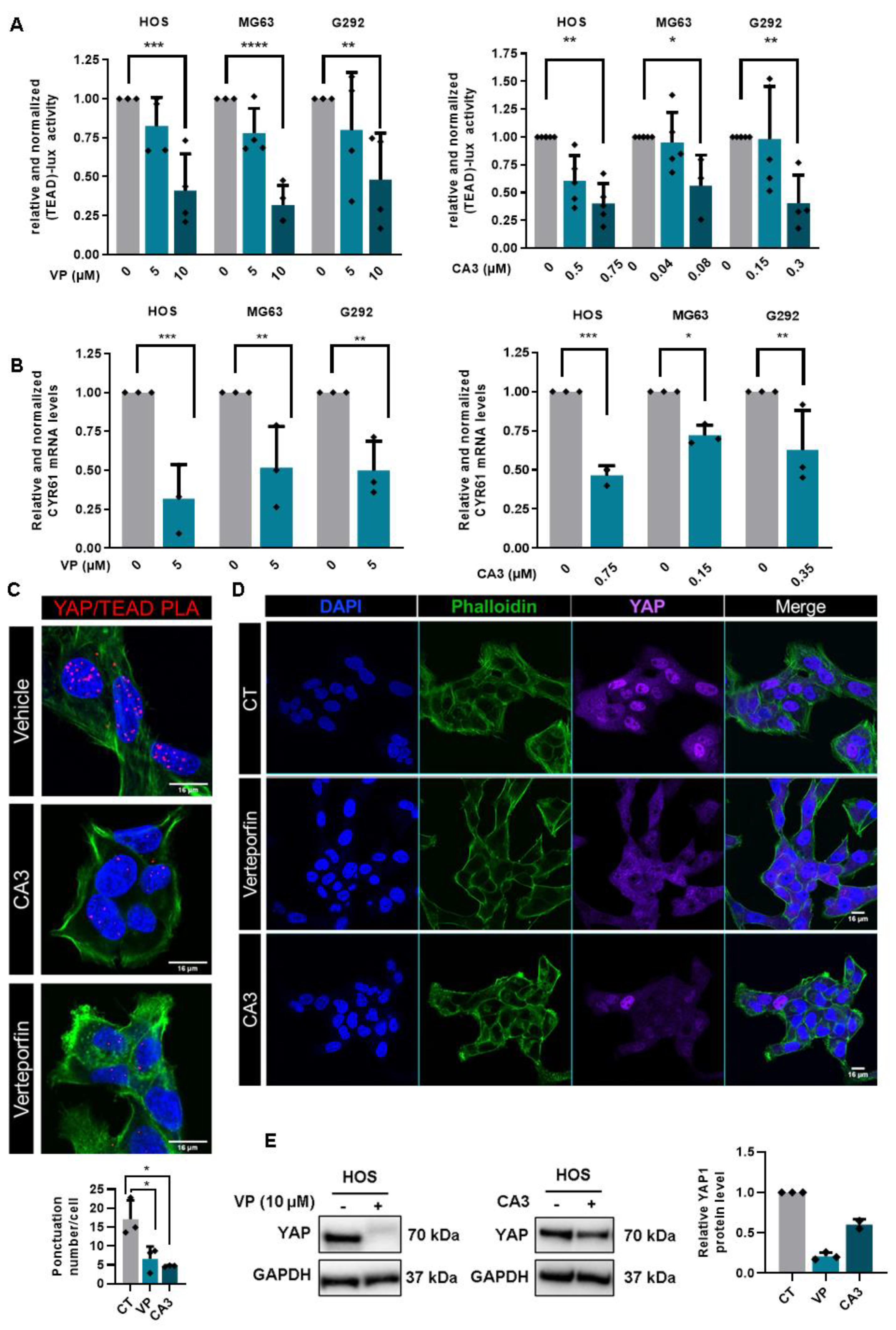
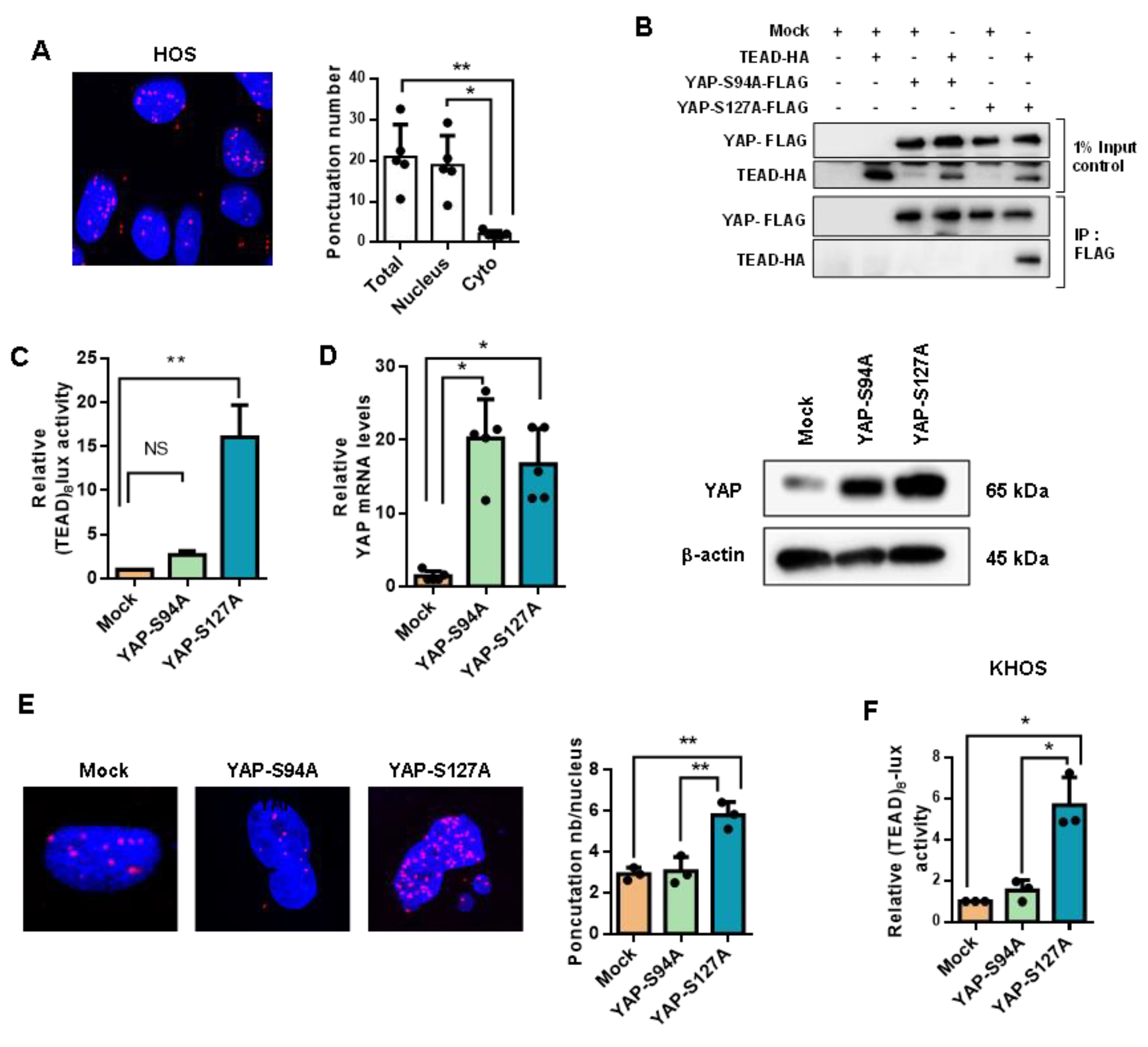
2.2. YAP/TEAD Interactions Are Crucial to Promote YAP-Driven TEAD Transcriptional Activity in OS Cells
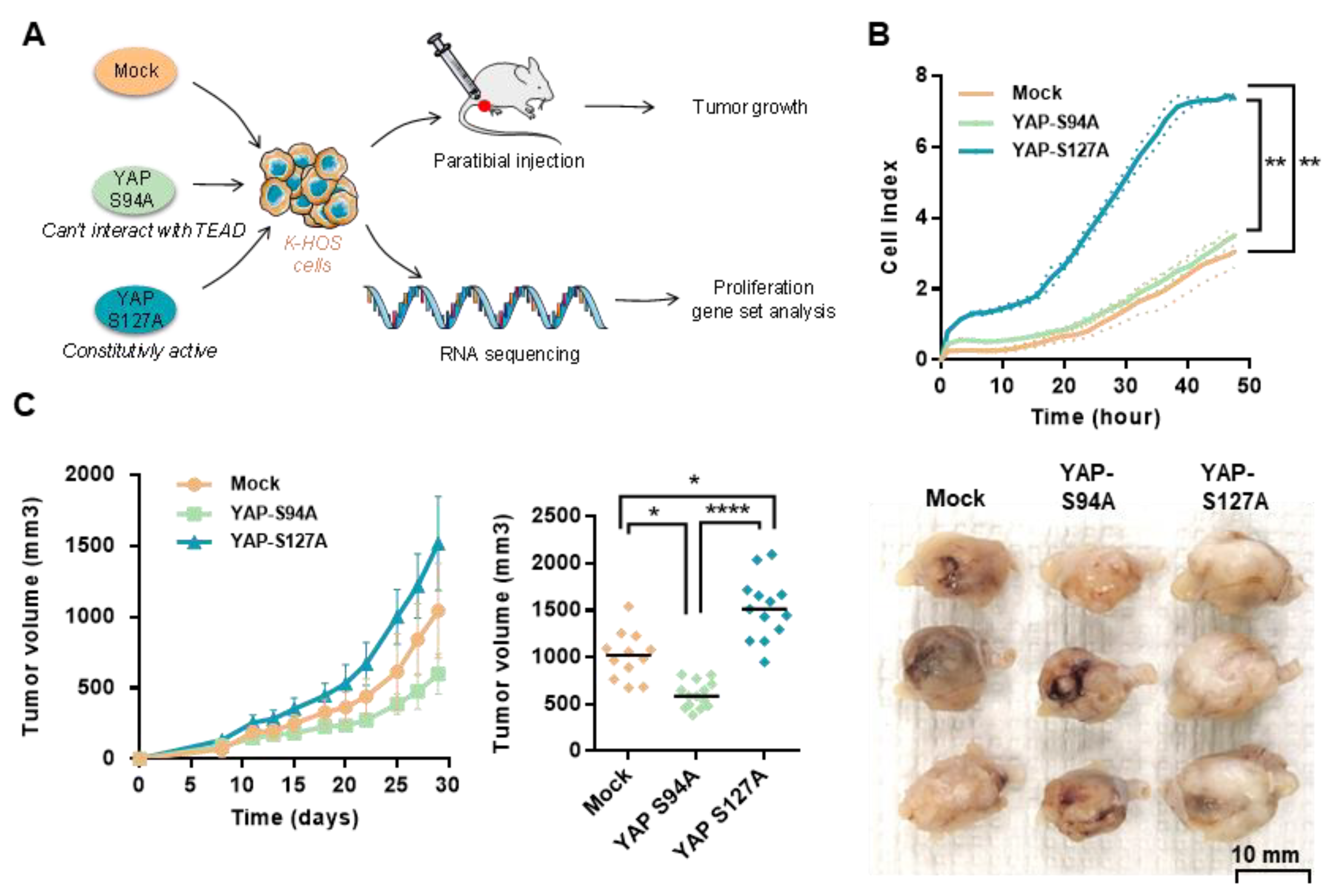
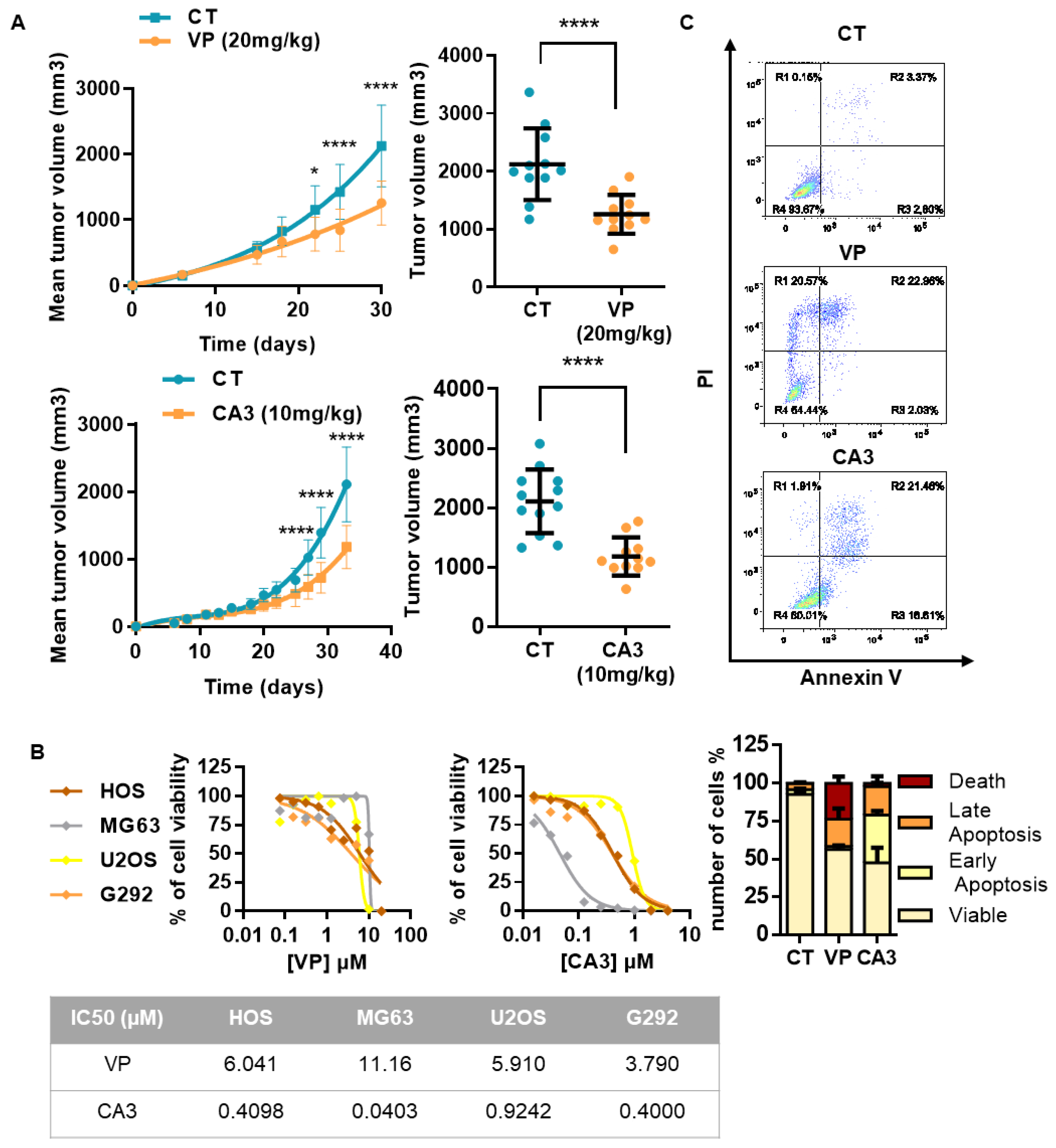
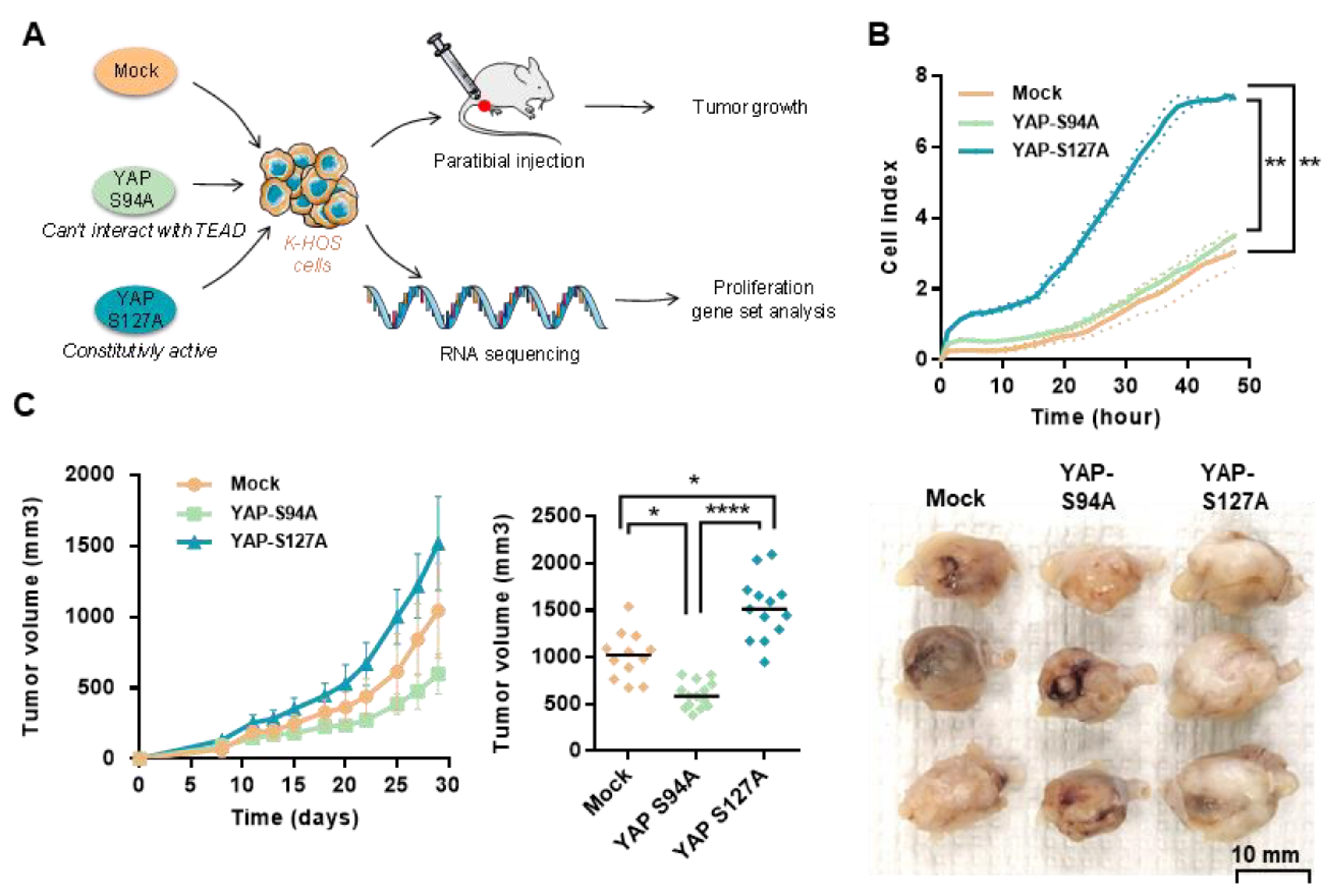
2.3. OS Cell Proliferation and In Vivo OS Tumor Growth Critically Depend on YAP-TEAD Interactions
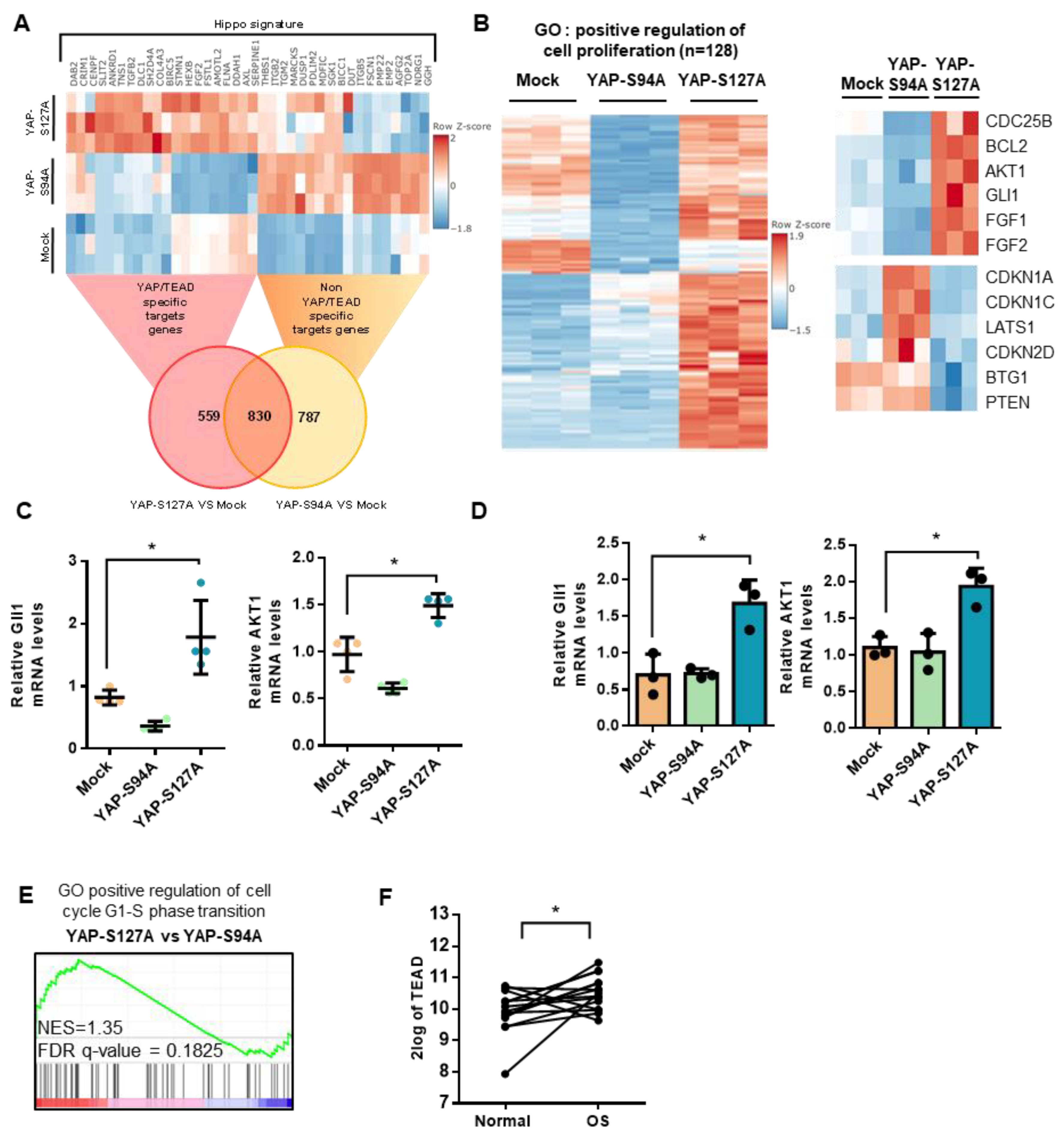
2.4. Role of TEAD in YAP-Driven Cell Cycle Genes Expression
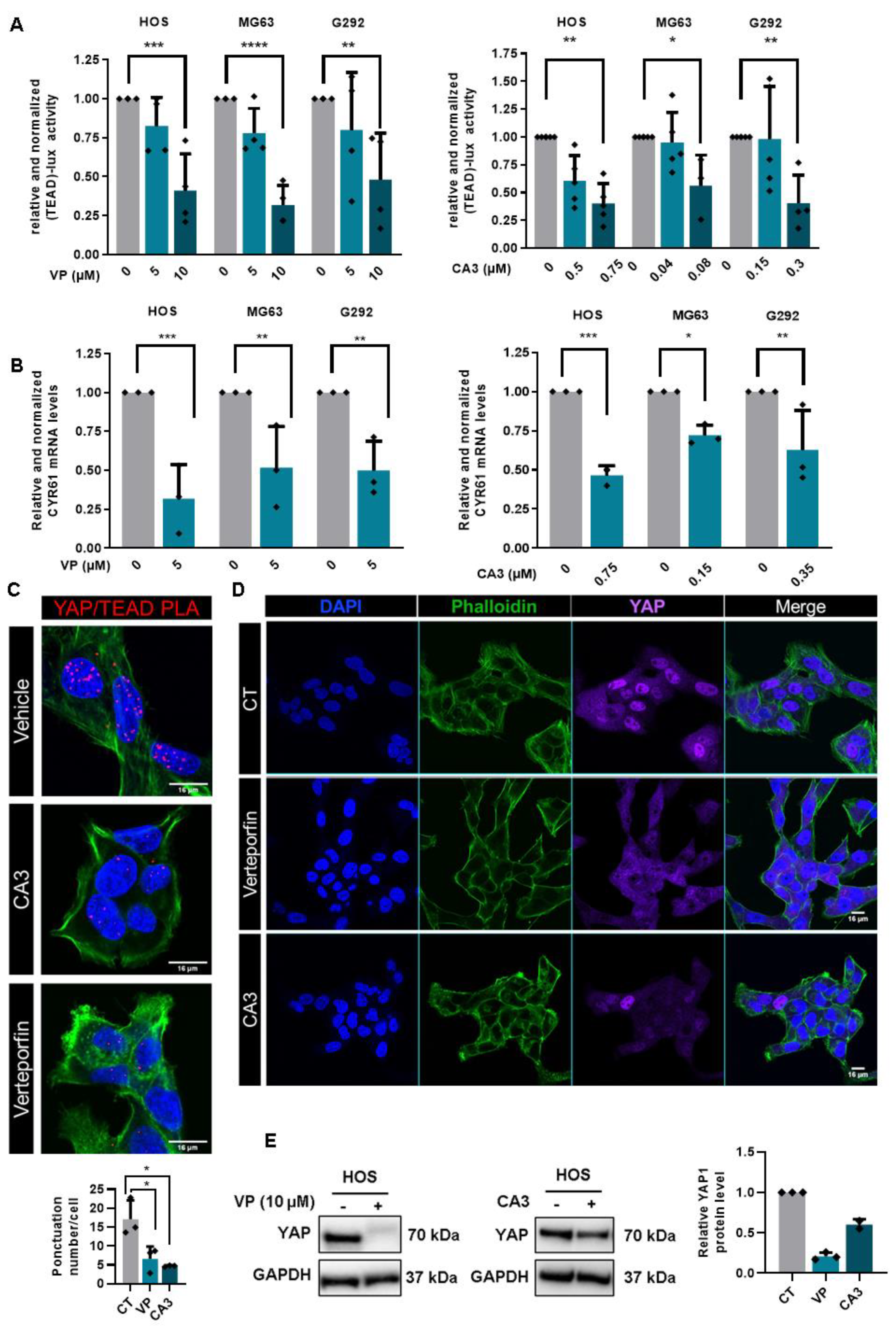
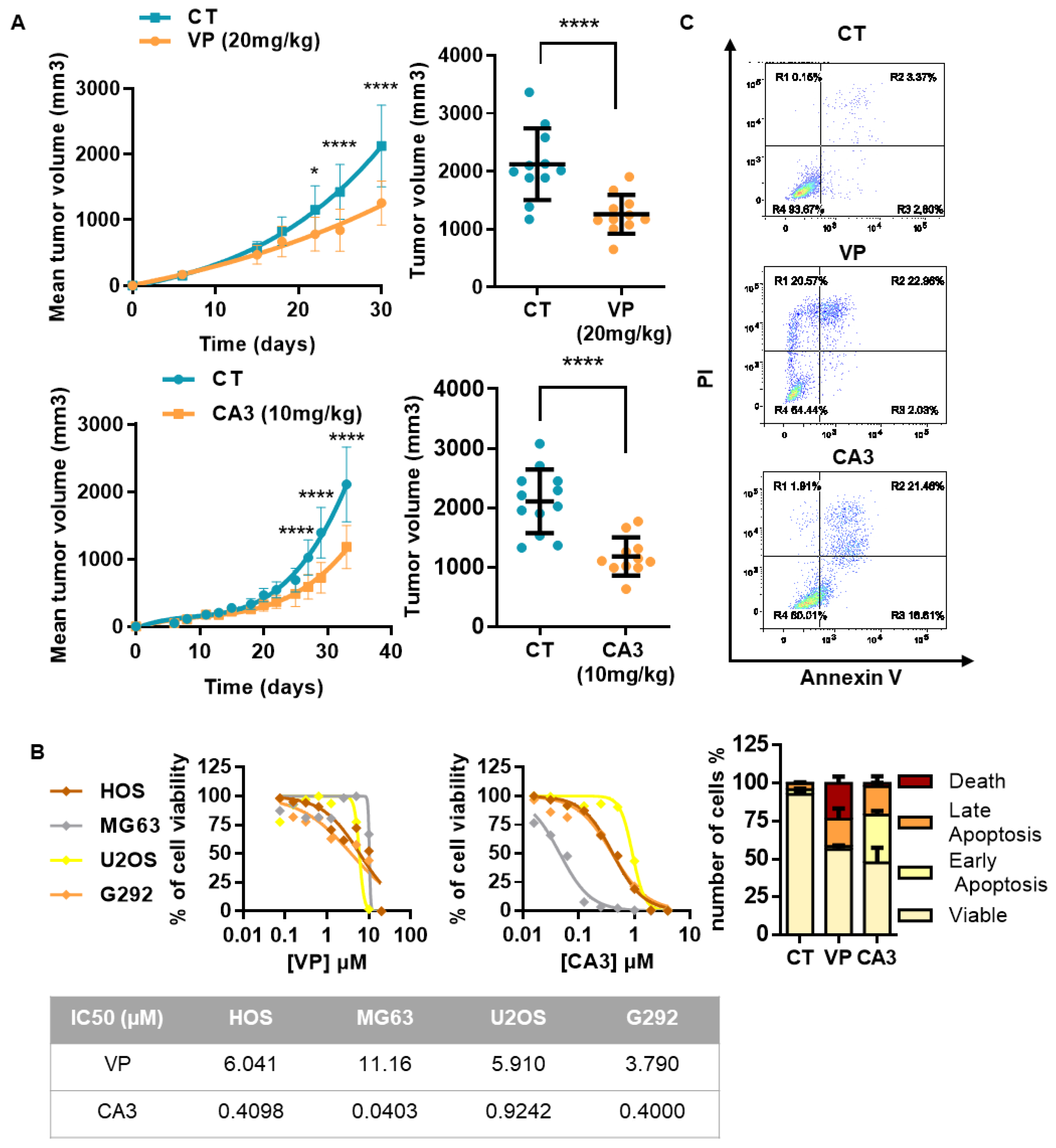
2.5. Verteporfin and CA3 Inhibit OS Primary Bone Tumor
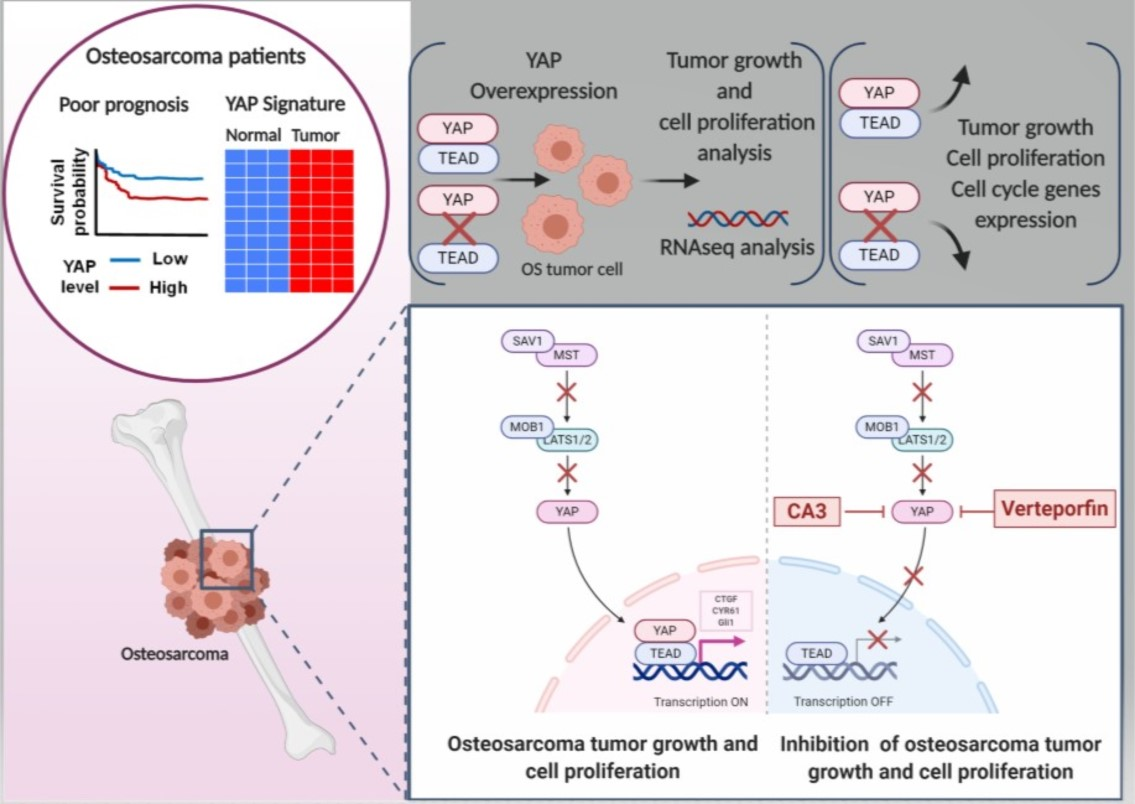
3. Discussion
3.1. YAP/TEAD Signaling as a Target Therapy against Primary Tumor Growth
3.2. Suppression of Primary Tumor Growth by YAP/TEAD Inhibitors
4. Materials and Methods
4.1. Osteosarcoma Mouse Model
4.2. Cell Culture and Reagents
4.3. Luciferase Reporter Assay and Plasmid Constructs
4.4. Real Time Proliferation and Annexin V Assays
4.5. Immunofluorescence
4.6. RNA Extraction and Real-Time Polymerase Chain Reaction
4.7. Western Blot Analysis
4.8. Immunoprecipitation
4.9. In Situ Proximity Ligation Assay (PLA), Immunofluorescence and Confocal Microscopy
4.10. RNA-seq Analysis
4.11. Statistical Analysis
4.12. Database
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simpson, E.; Brown, H.L. Understanding osteosarcomas. JAAPA 2018, 31, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The epidemiology of osteosarcoma. Cancer Treat. Res. 2009, 152, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Fletcher, C.D.M. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef]
- Moore, D.D.; Luu, H.H. Osteosarcoma. Cancer Treat. Res. 2014, 162, 65–92. [Google Scholar] [CrossRef]
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef] [Green Version]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- ESMO/European Sarcoma Network Working Group. Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii113–iii123. [Google Scholar] [CrossRef]
- Harrison, D.J.; Geller, D.S.; Gill, J.D.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 2018, 18, 39–50. [Google Scholar] [CrossRef]
- Duchman, K.R.; Gao, Y.; Miller, B.J. Prognostic factors for survival in patients with high-grade osteosarcoma using the Surveillance, Epidemiology, and End Results (SEER) Program database. Cancer Epidemiol. 2015, 39, 593–599. [Google Scholar] [CrossRef]
- Gibson, T.M.; Mostoufi-Moab, S.; Stratton, K.L.; Leisenring, W.M.; Barnea, D.; Chow, E.J.; Donaldson, S.S.; Howell, R.M.; Hudson, M.M.; Mahajan, A.; et al. Temporal patterns in the risk of chronic health conditions in survivors of childhood cancer diagnosed 1970-99: A report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2018, 19, 1590–1601. [Google Scholar] [CrossRef]
- Misra, J.R.; Irvine, K.D. The Hippo Signaling Network and Its Biological Functions. Annu. Rev. Genet. 2018, 52, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Gregorieff, A.; Wrana, J.L. Hippo signalling in intestinal regeneration and cancer. Curr. Opin. Cell Biol. 2017, 48, 17–25. [Google Scholar] [CrossRef]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.-X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in the Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Morice, S.; Danieau, G.; Rédini, F.; Brounais-Le-Royer, B.; Verrecchia, F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers 2020, 12, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-H.; Li, B.; Shen, L.; Shen, Y.; Chen, X.-D. The role and clinical significance of YES-associated protein 1 in human osteosarcoma. Int. J. Immunopathol. Pharmacol. 2013, 26, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvier, C.; Macagno, N.; Nguyen, Q.; Loundou, A.; Jiguet-Jiglaire, C.; Gentet, J.-C.; Jouve, J.-L.; Rochwerger, A.; Mattei, J.-C.; Bouvard, D.; et al. Prognostic value of the Hippo pathway transcriptional coactivators YAP/TAZ and β1-integrin in conventional osteosarcoma. Oncotarget 2016, 7, 64702–64710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deel, M.D.; Li, J.J.; Crose, L.E.S.; Linardic, C.M. A Review: Molecular Aberrations within Hippo Signaling in Bone and Soft-Tissue Sarcomas. Front. Oncol. 2015, 5, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.; Xu, S.; Guo, F. TEAD1 mediates the oncogenic activities of Hippo-YAP1 signaling in osteosarcoma. Biochem. Biophys. Res. Commun. 2017, 488, 297–302. [Google Scholar] [CrossRef]
- Ho, X.D.; Phung, P.; Q Le, V.; H Nguyen, V.; Reimann, E.; Prans, E.; Kõks, G.; Maasalu, K.; Le, N.T.; H Trinh, L.; et al. Whole transcriptome analysis identifies differentially regulated networks between osteosarcoma and normal bone samples. Exp. Biol. Med. (Maywood) 2017, 242, 1802–1811. [Google Scholar] [CrossRef]
- Kuijjer, M.L.; Peterse, E.F.P.; van den Akker, B.E.W.M.; Briaire-de Bruijn, I.H.; Serra, M.; Meza-Zepeda, L.A.; Myklebost, O.; Hassan, A.B.; Hogendoorn, P.C.W.; Cleton-Jansen, A.-M. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer 2013, 13, 245. [Google Scholar] [CrossRef] [Green Version]
- Rosen, G.; Murphy, M.L.; Huvos, A.G.; Gutierrez, M.; Marcove, R.C. Chemotherapy, en bloc resection, and prosthetic bone replacement in the treatment of osteogenic sarcoma. Cancer 1976, 37, 1–11. [Google Scholar] [CrossRef]
- Kim, C.-L.; Choi, S.-H.; Mo, J.-S. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019, 8, 468. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Lézot, F.; Corre, I.; Morice, S.; Rédini, F.; Verrecchia, F. SHH Signaling Pathway Drives Pediatric Bone Sarcoma Progression. Cells 2020, 9, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisi, G.M.; Santucci, M.; D’Arca, D.; Lauriola, A.; Marverti, G.; Losi, L.; Scalvini, L.; Bolognesi, M.L.; Mor, M.; Costi, M.P. Repurposing of Drugs Targeting YAP-TEAD Functions. Cancers 2018, 10, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Al-Moujahed, A.; Brodowska, K.; Stryjewski, T.P.; Efstathiou, N.E.; Vasilikos, I.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci. Rep. 2017, 7, 7602. [Google Scholar] [CrossRef]
- Kandasamy, S.; Adhikary, G.; Rorke, E.A.; Friedberg, J.S.; Mickle, M.B.; Alexander, H.R.; Eckert, R.L. The YAP1 Signaling Inhibitors, Verteporfin and CA3, Suppress the Mesothelioma Cancer Stem Cell Phenotype. Mol. Cancer Res. 2020, 18, 343–351. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2016, 6, 27–37. [Google Scholar]
- Zucchini, C.; Manara, M.C.; Cristalli, C.; Carrabotta, M.; Greco, S.; Pinca, R.S.; Ferrari, C.; Landuzzi, L.; Pasello, M.; Lollini, P.-L.; et al. ROCK2 deprivation leads to the inhibition of tumor growth and metastatic potential in osteosarcoma cells through the modulation of YAP activity. J. Exp. Clin. Cancer Res. 2019, 38, 503. [Google Scholar] [CrossRef] [Green Version]
- Sanna, L.; Piredda, R.; Marchesi, I.; Bordoni, V.; Forcales, S.V.; Calvisi, D.F.; Bagella, L. Verteporfin exhibits anti-proliferative activity in embryonal and alveolar rhabdomyosarcoma cell lines. Chem. Biol. Interact. 2019, 312, 108813. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| AKT1 | TACGAGAAGAAGCTCAGCCC | TTGGTCAGGTGGTGTGATGG |
| Gli1 | CCAACTCCACAGGCATACAGGAT | CACAGATTCAGGCTCACGCTTC |
| GAPDH | TGGGTGTGAACCATGAGAAGTATG | GGTGCAGGAGGCATTGCT |
| YAP1 | TGACCCTCGTTTTGCCATGA | GTTGCTGCTGGTTGGAGTTG |
| Cyr61 | CCAGTGTACAGCAGCCTGAA | GGCCGGTATTTCTTCACACTC |
| CTGF | AGGAGTGGGTGTGTGACGAG | CGGGACAGTTGTAATGGCAG |
| Gene Set | Website Link |
|---|---|
| Hippo signature | http://software.broadinstitute.org/gsea/msigdb/geneset_page.jsp?geneSetName=CORDENONSI_YAP_CONSERVED_SIGNATURE&keywords=hippo |
| Positive regulation of cell proliferation (GO/0008284) | http://software.broadinstitute.org/gsea/msigdb/cards/GO_POSITIVE_REGULATION_OF_CELL_PROLIFERATION |
| GO positive regulation of cell cycle G1-S phase transition | https://www.gsea-msigdb.org/gsea/msigdb/cards/GO_POSITIVE_REGULATION_OF_CELL_CYCLE_G1_S_PHASE_TRANSITION.html |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morice, S.; Mullard, M.; Brion, R.; Dupuy, M.; Renault, S.; Tesfaye, R.; Brounais-Le Royer, B.; Ory, B.; Redini, F.; Verrecchia, F. The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers 2020, 12, 3847. https://doi.org/10.3390/cancers12123847
Morice S, Mullard M, Brion R, Dupuy M, Renault S, Tesfaye R, Brounais-Le Royer B, Ory B, Redini F, Verrecchia F. The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers. 2020; 12(12):3847. https://doi.org/10.3390/cancers12123847
Chicago/Turabian StyleMorice, Sarah, Mathilde Mullard, Regis Brion, Maryne Dupuy, Sarah Renault, Robel Tesfaye, Bénédicte Brounais-Le Royer, Benjamin Ory, Françoise Redini, and Franck Verrecchia. 2020. "The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth" Cancers 12, no. 12: 3847. https://doi.org/10.3390/cancers12123847
APA StyleMorice, S., Mullard, M., Brion, R., Dupuy, M., Renault, S., Tesfaye, R., Brounais-Le Royer, B., Ory, B., Redini, F., & Verrecchia, F. (2020). The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers, 12(12), 3847. https://doi.org/10.3390/cancers12123847