Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance


{kind=link}
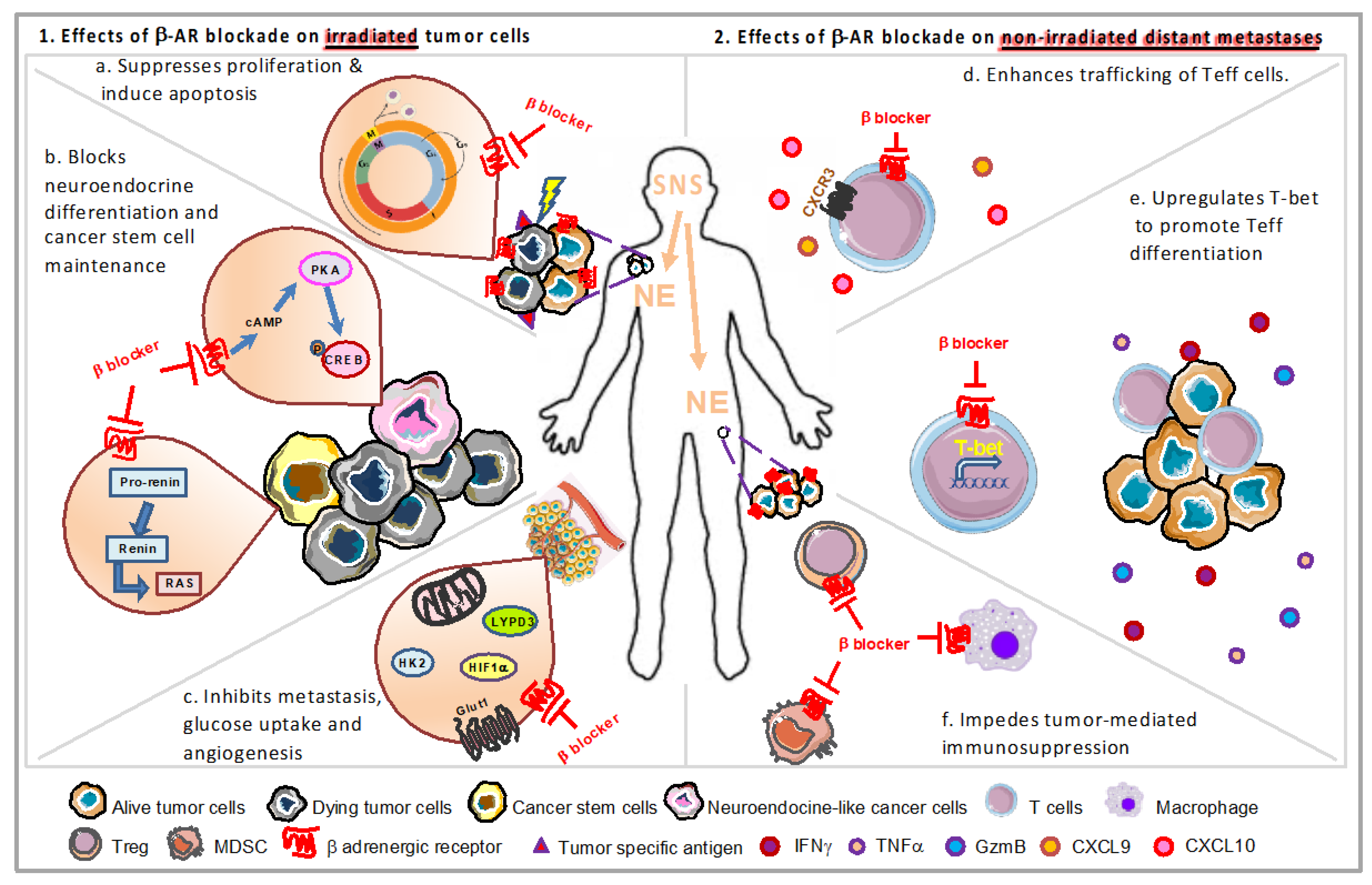{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Chronic Stress and Cancer
3. Modulation of the Tumor Microenvironment by Chronic Stress: Impact on Radiotherapy
3.1. Impact of Chronic Stress on Immune Cells
3.1.1. Adaptive Immune Response
CD8+ and CD4+ T-Cells
Regulatory T-Cells (Tregs)
3.1.2. Innate Immune Response
Dendritic Cells
NK Cells
Tumor-Associated Macrophages
Myeloid Derived Suppressor Cells (MDSCs)
3.2. Impact of Chronic Stress on Irradiated Tumors
3.2.1. Impact of Stress Signaling on Tumor Proliferation/Apoptosis through Cell Cycle
3.2.2. Stress Hormones Can Induce Neuroendocrine Differentiation
3.2.3. Cancer Stem Cells and Stress
3.2.4. Modulation of Hypoxia by Stress Signaling
3.2.5. Chronic Stress Can Promote Metastasis
3.2.6. Modulation of Metabolism in Cancer by Stress Hormones
4. Can Stress Affect the Frequency of the “Abscopal Effect” following Radiation?
4.1. Emerging Evidence of a Role for Stress in Regulating the Frequency of the Abscopal Effect
4.2. Variables That May Influence the Impact of Stress on the Abscopal Effect
5. Health-Related Quality of Life Depends upon Psychosocial Stress and Has the Potential to Influence Outcomes Following RT
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF | activating transcription factor |
| ATM APC | ataxia-telangiectasia mutated antigen-presenting cells |
| BAD | Bcl-2 antagonist of cell death |
| β-ARs | β-adrenergic receptors |
| cAMP | cyclic adenosine monophosphate |
| C/EBPβ | CCAAT-enhancer-binding protein β |
| cGAS | cyclic GMP-AMP synthase |
| COX-2 | cyclooxygenase-2 |
| CREB | cAMP response element-binding protein |
| CSF1 | colony-stimulating factor 1 |
| CTLA-4 | cytotoxic T-lymphocyte-associated antigen 4 |
| CXCL12 | CXC-motif chemokine ligand 12 |
| DCs | dendritic cells |
| DNA | deoxyribonucleic acid |
| EGFR | epidermal growth factor receptor |
| ERK | extracellular-signal-regulated kinase |
| FasL | Fas-Fas ligand |
| Foxp3 | forkhead box P3 |
| FT | financial toxicity |
| Glut | glucose transporter |
| GRK2 | G protein-coupled receptor kinase 2 |
| GSK3β | glycogen synthase kinase 3β |
| GTP | guanosine triphosphate |
| GzmB | granzyme B |
| HIF-1α | hypoxia-inducible factor-1α |
| HK2 | hexokinase 2 |
| HMGB1 | high mobility group box 1 |
| HPA axis | hypothalamic-pituitary-adrenal axis |
| HRQOL | health-related quality of life |
| HuR | human antigen R |
| ICAM-1 | intercellular adhesion molecule 1 |
| IFNs | interferons |
| IL-6 | interleukin-6 |
| iNOS | inducible nitric oxide synthase |
| IRF3 | interferon regulatory factor 3 |
| ISO | isoproterenol |
| JNK | c-Jun N-terminal kinase |
| KO | knockout |
| LDHA | lactate dehydrogenase A |
| LIMK1 | LIM kinase 1 |
| LNs | lymph nodes |
| LYPD3 | Ly6/PLAUR domain-containing protein 3 |
| MAPK | mitogenic activated protein kinase |
| MBSR | mindfulness-based stress reduction |
| MCL-1 | myeloid cell leukemia 1 |
| MDSCs | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MIP-1α | macrophage inflammatory protein 1α |
| MMP-2 | matrix metallopeptidase-2 |
| NE | norepinephrine |
| NF-κB | nuclear factor kappa B |
| NK | natural killer |
| NLRP3 | nucleotide-binding domain leucine-rich repeat family pyrin domain containing protein 3 |
| PAK1 | p21-activated kinase 1 |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed death ligand 1 |
| PGE2 | prostaglandin E2 |
| PI3K | phosphoinositide 3-kinase |
| PKA | cAMP-protein kinase A |
| RAA | renin-angiotensin-aldosterone |
| RAC1 | Ras related C3 botulinum toxin substrate 1 |
| RANTES | regulated on activation, normal T-cell expressed and secreted |
| RAS | Renin-angiotensin system |
| ROS | reactive oxygen species |
| RT | radiation therapy |
| SNS | sympathetic nervous system |
| ST | standard room temperatures |
| STAT3 | signal transducer and activator of transcription 3 |
| STING | stimulator of interferon genes |
| TAMs | tumor-associated macrophages |
| TBI | total body irradiation |
| TLR-4 | toll-like receptor 4 |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor alpha |
| Tregs | regulatory T-cells |
| TT | thermoneutral temperatures |
| VASP | vasodilator-stimulated phosphoprotein |
| VCAM-1 | vascular cell adhesion protein 1 |
| VEGF | vascular endothelial growth factor |
References
- Moreno-Smith, M.; Lutgendorf, S.K.; Sood, A.K. Impact of stress on cancer metastasis. Future Oncol. 2010, 6, 1863–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Panahi, Y.; Sahraei, H.; Johnston, T.P.; Sahebkar, A. The impact of stress on body function: A review. EXCLI J. 2017, 16, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Macht, V.A.; Reagan, L.P. Chronic stress from adolescence to aging in the prefrontal cortex: A neuroimmune perspective. Front. Neuroendocrinol. 2018, 49, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Geyer, S. The role of social and psychosocial factors in the development and course of cancer. Wien. Klin. Wochenschr. 2000, 112, 986–994. [Google Scholar]
- Ollonen, P.; Lehtonen, J.; Eskelinen, M. Stressful and adverse life experiences in patients with breast symptoms; a prospective case-control study in Kuopio, Finland. Anticancer Res. 2005, 25, 531–536. [Google Scholar]
- Kruk, J.; Aboul-Enein, B.H.; Bernstein, J.; Gronostaj, M. Psychological Stress and Cellular Aging in Cancer: A Meta-Analysis. Oxid. Med. Cell. Longev. 2019, 2019, 1270397. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Huang, J.; Wu, H.; Fu, C.; Li, Y.; Qiu, J. Impact of lifestyle and psychological stress on the development of early onset breast cancer. Medicine 2016, 95, e5529. [Google Scholar] [CrossRef]
- Song, H.; Saito, E.; Sawada, N.; Abe, S.K.; Hidaka, A.; Shimazu, T.; Yamaji, T.; Goto, A.; Iwasaki, M.; Sasazuki, S.; et al. Perceived stress level and risk of cancer incidence in a Japanese population: The Japan Public Health Center (JPHC)-based Prospective Study. Sci. Rep. 2017, 7, 12964. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, N.K.; Ozkan, M.; Ilgun, A.S.; Sarsenov, D.; Alco, G.; Aktepe, F.; Kalyoncu, N.; Izci, F.; Selamoglu, D.; Ordu, C.; et al. Possible role of stress, coping strategies, and life style in the development of breast cancer. Int. J. Psychiatry Med. 2018, 53, 207–220. [Google Scholar] [CrossRef]
- Yang, T.; Qiao, Y.; Xiang, S.; Li, W.; Gan, Y.; Chen, Y. Work stress and the risk of cancer: A meta-analysis of observational studies. Int. J. Cancer 2019, 144, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Lapierre, A.; Rousseau, M.C.; Parent, M.E. Perceived Workplace Stress Is Associated with an Increased Risk of Prostate Cancer before Age 65. Front. Oncol. 2017, 7, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc-Lapierre, A.; Rousseau, M.C.; Weiss, D.; El-Zein, M.; Siemiatycki, J.; Parent, M.E. Lifetime report of perceived stress at work and cancer among men: A case-control study in Montreal, Canada. Prev. Med. 2017, 96, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Barre, P.V.; Padmaja, G.; Rana, S.; Tiamongla. Stress and Quality of Life in Cancer Patients: Medical and Psychological Intervention. Indian J. Psychol. Med. 2018, 40, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedz, C.L.; Knifton, L.; Robb, K.A.; Katikireddi, S.V.; Smith, D.J. Depression and anxiety among people living with and beyond cancer: A growing clinical and research priority. BMC Cancer 2019, 19, 943. [Google Scholar] [CrossRef] [Green Version]
- Fiorentino, L.; Ancoli-Israel, S. Sleep dysfunction in patients with cancer. Curr. Treat. Options Neurol. 2007, 9, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, R.L.; Falcinelli, M.; Flint, M.S. Stress and drug resistance in cancer. Cancer Drug Resist. 2019, 2, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, S.; Hartmann, F.; Papassotiropoulos, A.; de Quervain, D.J.; Rasch, B. Associations between basal cortisol levels and memory retrieval in healthy young individuals. J. Cogn. Neurosci. 2013, 25, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- Engert, V.; Kok, B.E.; Papassotiriou, I.; Chrousos, G.P.; Singer, T. Specific reduction in cortisol stress reactivity after social but not attention-based mental training. Sci. Adv. 2017, 3, e1700495. [Google Scholar] [CrossRef] [Green Version]
- Filipski, E.; Delaunay, F.; King, V.M.; Wu, M.W.; Claustrat, B.; Grechez-Cassiau, A.; Guettier, C.; Hastings, M.H.; Francis, L. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 2004, 64, 7879–7885. [Google Scholar] [CrossRef] [Green Version]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, K.P., Jr.; Drake, A.L.; Frey, D.J.; Fleshner, M.; Desouza, C.A.; Gronfier, C.; Czeisler, C.A. Influence of sleep deprivation and circadian misalignment on cortisol, inflammatory markers, and cytokine balance. Brain Behav. Immun. 2015, 47, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oster, H.; Challet, E.; Ott, V.; Arvat, E.; de Kloet, E.R.; Dijk, D.J.; Lightman, S.; Vgontzas, A.; Van Cauter, E. The Functional and Clinical Significance of the 24-Hour Rhythm of Circulating Glucocorticoids. Endocr. Rev. 2017, 38, 3–45. [Google Scholar] [CrossRef] [PubMed]
- Oshima, T.; Takenoshita, S.; Akaike, M.; Kunisaki, C.; Fujii, S.; Nozaki, A.; Numata, K.; Shiozawa, M.; Rino, Y.; Tanaka, K.; et al. Expression of circadian genes correlates with liver metastasis and outcomes in colorectal cancer. Oncol. Rep. 2011, 25, 1439–1446. [Google Scholar] [CrossRef] [Green Version]
- Kiessling, S.; Beaulieu-Laroche, L.; Blum, I.D.; Landgraf, D.; Welsh, D.K.; Storch, K.F.; Labrecque, N.; Cermakian, N. Enhancing circadian clock function in cancer cells inhibits tumor growth. BMC Biol. 2017, 15, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegrzyn, L.R.; Tamimi, R.M.; Rosner, B.A.; Brown, S.B.; Stevens, R.G.; Eliassen, A.H.; Laden, F.; Willett, W.C.; Hankinson, S.E.; Schernhammer, E.S. Rotating Night-Shift Work and the Risk of Breast Cancer in the Nurses’ Health Studies. Am. J. Epidemiol. 2017, 186, 532–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papantoniou, K.; Castano-Vinyals, G.; Espinosa, A.; Aragones, N.; Perez-Gomez, B.; Burgos, J.; Gomez-Acebo, I.; Llorca, J.; Peiro, R.; Jimenez-Moleon, J.J.; et al. Night shift work, chronotype and prostate cancer risk in the MCC-Spain case-control study. Int. J. Cancer 2015, 137, 1147–1157. [Google Scholar] [CrossRef]
- Nakane, T.; Szentendrei, T.; Stern, L.; Virmani, M.; Seely, J.; Kunos, G. Effects of IL-1 and cortisol on beta-adrenergic receptors, cell proliferation, and differentiation in cultured human A549 lung tumor cells. J. Immunol. 1990, 145, 260–266. [Google Scholar]
- Docherty, J.R. Subtypes of functional alpha1-adrenoceptor. Cell. Mol. Life Sci. 2010, 67, 405–417. [Google Scholar] [CrossRef]
- Kanagy, N.L. Alpha(2)-adrenergic receptor signalling in hypertension. Clin. Sci. 2005, 109, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.A.; Liggett, S.B. Cardiovascular pharmacogenomics of adrenergic receptor signaling: Clinical implications and future directions. Clin. Pharmacol. Ther. 2011, 89, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Hadi, T.; Douhard, R.; Dias, A.M.M.; Wendremaire, M.; Pezze, M.; Bardou, M.; Sagot, P.; Garrido, C.; Lirussi, F. Beta3 adrenergic receptor stimulation in human macrophages inhibits NADPHoxidase activity and induces catalase expression via PPARgamma activation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying beta-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, M.P. Sir James Black and propranolol. The role of the basic sciences in the history of cardiovascular pharmacology. Tex. Heart Inst. J. 1997, 24, 336–342. [Google Scholar]
- Vanhoutte, P.M.; Gao, Y. Beta blockers, nitric oxide, and cardiovascular disease. Curr. Opin. Pharmacol. 2013, 13, 265–273. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Sukhatme, V.; Meheus, L.; Rooman, I.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-Propranolol as an anti-cancer agent. Ecancermedicalscience 2016, 10, 680. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, E.; Street, J.; Pouchy, C.; Carre, M.; Gifford, A.J.; Murray, J.; Norris, M.D.; Trahair, T.; Andre, N.; Kavallaris, M. beta-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br. J. Cancer 2013, 108, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Z.; Bai, N.; Bi, J.H.; Liu, X.W.; Xu, G.Q.; Zhang, L.F.; Li, X.Q.; Huo, R. Propranolol inhibits the proliferation, migration and tube formation of hemangioma cells through HIF-1alpha dependent mechanisms. Braz. J. Med. Biol. Res. 2017, 50, e6138. [Google Scholar] [CrossRef] [Green Version]
- Brohee, L.; Peulen, O.; Nusgens, B.; Castronovo, V.; Thiry, M.; Colige, A.C.; Deroanne, C.F. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci. Rep. 2018, 8, 7050. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Ma, Q.; Wang, L.; Hu, H.; Li, J.; Zhang, D.; Zhang, M. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol. Rep. 2009, 22, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ma, Q.Y.; Hu, H.T.; Zhang, M. beta2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFkappaB and AP-1. Cancer Biol. Ther. 2010, 10, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamy, S.; Lachambre, M.P.; Lord-Dufour, S.; Beliveau, R. Propranolol suppresses angiogenesis in vitro: Inhibition of proliferation, migration, and differentiation of endothelial cells. Vasc. Pharmacol. 2010, 53, 200–208. [Google Scholar] [CrossRef]
- Kuang, X.; Qi, M.; Peng, C.; Zhou, C.; Su, J.; Zeng, W.; Liu, H.; Zhang, J.; Chen, M.; Shen, M.; et al. Propranolol enhanced the anti-tumor effect of sunitinib by inhibiting proliferation and inducing G0/G1/S phase arrest in malignant melanoma. Oncotarget 2018, 9, 802–811. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Che, X.; Zhao, W.; Zhang, D.; Long, H.; Chaudhary, P.; Li, H. Effects of propranolol in combination with radiation on apoptosis and survival of gastric cancer cells in vitro. Radiat. Oncol. 2010, 5, 98. [Google Scholar] [CrossRef] [Green Version]
- Jean Wrobel, L.; Bod, L.; Lengagne, R.; Kato, M.; Prevost-Blondel, A.; Le Gal, F.A. Propranolol induces a favourable shift of anti-tumor immunity in a murine spontaneous model of melanoma. Oncotarget 2016, 7, 77825–77837. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Song, K.; Zhu, Z.; Liu, Z.; Zhang, W.; Li, W.; Hu, J.; Hu, Q.; Chen, C.; Chen, B.; et al. Propranolol Suppresses the Growth of Colorectal Cancer Through Simultaneously Activating Autologous CD8(+) T Cells and Inhibiting Tumor AKT/MAPK Pathway. Clin. Pharmacol. Ther. 2020, 108, 606–615. [Google Scholar] [CrossRef]
- Chen, M.; Qiao, G.; Hylander, B.L.; Mohammadpour, H.; Wang, X.Y.; Subjeck, J.R.; Singh, A.K.; Repasky, E.A. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat. Commun. 2020, 11, 1821. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.; Messmer, M.N.; et al. beta-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8(+) T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef] [Green Version]
- Mohammadpour, H.; MacDonald, C.R.; Qiao, G.; Chen, M.; Dong, B.; Hylander, B.L.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Investig. 2019, 129, 5537–5552. [Google Scholar] [CrossRef] [PubMed]
- Leigh, N.D.; Kokolus, K.M.; O’Neill, R.E.; Du, W.; Eng, J.W.; Qiu, J.; Chen, G.L.; McCarthy, P.L.; Farrar, J.D.; Cao, X.; et al. Housing Temperature-Induced Stress Is Suppressing Murine Graft-versus-Host Disease through beta2-Adrenergic Receptor Signaling. J. Immunol. 2015, 195, 5045–5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Katsube, T.; Begum, N.; Nenoi, M. Revisiting the health effects of psychological stress-its influence on susceptibility to ionizing radiation: A mini-review. J. Radiat. Res. 2016, 57, 325–335. [Google Scholar] [CrossRef]
- Andersen, B.L.; Farrar, W.B.; Golden-Kreutz, D.; Kutz, L.A.; MacCallum, R.; Courtney, M.E.; Glaser, R. Stress and immune responses after surgical treatment for regional breast cancer. J. Natl. Cancer Inst. 1998, 90, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zanos, P.; Jackson, I.L.; Zhang, X.; Zhu, X.; Gould, T.; Vujaskovic, Z. Psychological stress enhances tumor growth and diminishes radiation response in preclinical model of lung cancer. Radiother. Oncol. 2020, 146, 126–135. [Google Scholar] [CrossRef]
- Bautista, M.; Krishnan, A. The Autonomic Regulation of Tumor Growth and the Missing Links. Front. Oncol. 2020, 10, 744. [Google Scholar] [CrossRef]
- Won, E.; Kim, Y.K. Stress, the Autonomic Nervous System, and the Immune-kynurenine Pathway in the Etiology of Depression. Curr. Neuropharmacol. 2016, 14, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garelli, E.; Rittmeyer, A.; Putora, P.M.; Glatzer, M.; Dressel, R.; Andreas, S. Abscopal effect in lung cancer: Three case reports and a concise review. Immunotherapy 2019, 11, 1445–1461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol. 2008, 180, 3132–3139. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation 2009, 16, 300–317. [Google Scholar] [CrossRef] [Green Version]
- Kokolus, K.M.; Capitano, M.L.; Lee, C.T.; Eng, J.W.; Waight, J.D.; Hylander, B.L.; Sexton, S.; Hong, C.C.; Gordon, C.J.; Abrams, S.I.; et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc. Natl. Acad. Sci. USA 2013, 110, 20176–20181. [Google Scholar] [CrossRef] [Green Version]
- Eng, J.W.; Reed, C.B.; Kokolus, K.M.; Pitoniak, R.; Utley, A.; Bucsek, M.J.; Ma, W.W.; Repasky, E.A.; Hylander, B.L. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat. Commun. 2015, 6, 6426. [Google Scholar] [CrossRef] [Green Version]
- Hylander, B.L.; Repasky, E.A. Thermoneutrality, Mice, and Cancer: A Heated Opinion. Trends Cancer 2016, 2, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylander, B.L.; Gordon, C.J.; Repasky, E.A. Manipulation of Ambient Housing Temperature To Study the Impact of Chronic Stress on Immunity and Cancer in Mice. J. Immunol. 2019, 202, 631–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repasky, E.A.; Evans, S.S.; Dewhirst, M.W. Temperature matters! And why it should matter to tumor immunologists. Cancer Immunol. Res. 2013, 1, 210–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colon-Echevarria, C.B.; Lamboy-Caraballo, R.; Aquino-Acevedo, A.N.; Armaiz-Pena, G.N. Neuroendocrine Regulation of Tumor-Associated Immune Cells. Front. Oncol. 2019, 9, 1077. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Wang, Y.W.; Chen, R.; Zhou, B.; Ashwell, J.D.; Fornace, A.J., Jr. Ionizing Radiation Impairs T Cell Activation by Affecting Metabolic Reprogramming. Int. J. Biol. Sci. 2015, 11, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Nguy, S.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Daley, D.; Barilla, R.; et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology 2016, 150, 1659–1672. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Luo, Y.; Zhou, D. Hematopoietic stem cell injury induced by ionizing radiation. Antioxid. Redox Signal. 2014, 20, 1447–1462. [Google Scholar] [CrossRef] [Green Version]
- Spary, L.K.; Al-Taei, S.; Salimu, J.; Cook, A.D.; Ager, A.; Watson, H.A.; Clayton, A.; Staffurth, J.; Mason, M.D.; Tabi, Z. Enhancement of T cell responses as a result of synergy between lower doses of radiation and T cell stimulation. J. Immunol. 2014, 192, 3101–3110. [Google Scholar] [CrossRef]
- MacDonald, C.R.; Bucsek, M.J.; Qiao, G.; Chen, M.; Evans, L.; Greenberg, D.J.; Uccello, T.P.; Battaglia, N.G.; Hylander, B.L.; Singh, A.K.; et al. Adrenergic Receptor Signaling Regulates the Response of Tumors to Ionizing Radiation. Radiat. Res. 2019, 191, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Beauford, S.S.; Kumari, A.; Garnett-Benson, C. Ionizing radiation modulates the phenotype and function of human CD4+ induced regulatory T cells. BMC Immunol. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaue, D.; Comin-Anduix, B.; Ribas, A.; Zhang, L.; Goodglick, L.; Sayre, J.W.; Debucquoy, A.; Haustermans, K.; McBride, W.H. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin. Cancer Res. 2008, 14, 4883–4890. [Google Scholar] [CrossRef] [Green Version]
- Billiard, F.; Buard, V.; Benderitter, M.; Linard, C. Abdominal gamma-radiation induces an accumulation of function-impaired regulatory T cells in the small intestine. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 869–876. [Google Scholar] [CrossRef]
- Guereschi, M.G.; Araujo, L.P.; Maricato, J.T.; Takenaka, M.C.; Nascimento, V.M.; Vivanco, B.C.; Reis, V.O.; Keller, A.C.; Brum, P.C.; Basso, A.S. Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. Eur. J. Immunol. 2013, 43, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Li, Y.; Li, X.; Chen, G.; Liang, H.; Wu, Y.; Tong, J.; Ouyang, W. Propranolol Attenuates Surgical Stress-Induced Elevation of the Regulatory T Cell Response in Patients Undergoing Radical Mastectomy. J. Immunol. 2016, 196, 3460–3469. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Demaria, O.; Cornen, S.; Daeron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Dar, T.B.; Henson, R.M.; Shiao, S.L. Targeting Innate Immunity to Enhance the Efficacy of Radiation Therapy. Front. Immunol. 2018, 9, 3077. [Google Scholar] [CrossRef] [Green Version]
- Hiller, J.G.; Cole, S.W.; Crone, E.M.; Byrne, D.J.; Shackleford, D.M.; Pang, J.B.; Henderson, M.A.; Nightingale, S.S.; Ho, K.M.; Myles, P.S.; et al. Preoperative beta-Blockade with Propranolol Reduces Biomarkers of Metastasis in Breast Cancer: A Phase II Randomized Trial. Clin. Cancer Res. 2020, 26, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.H.; Park, G.Y.; Han, Y.K.; Kim, S.D.; Kim, J.S.; Lee, C.G.; Yang, K. Effect of low dose radiation on differentiation of bone marrow cells into dendritic cells. Dose Response 2012, 11, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Merrick, A.; Errington, F.; Milward, K.; O’Donnell, D.; Harrington, K.; Bateman, A.; Pandha, H.; Vile, R.; Morrison, E.; Selby, P.; et al. Immunosuppressive effects of radiation on human dendritic cells: Reduced IL-12 production on activation and impairment of naive T-cell priming. Br. J. Cancer 2005, 92, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, B.; Jia, X.; Ma, Y.; Gu, Y.; Zhang, P.; Wei, Q.; Cai, J.; Cui, J.; Gao, F.; et al. Radiation-induced decrease of CD8+ dendritic cells contributes to Th1/Th2 shift. Int. Immunopharmacol. 2017, 46, 178–185. [Google Scholar] [CrossRef]
- Wu, L.; Tai, Y.; Hu, S.; Zhang, M.; Wang, R.; Zhou, W.; Tao, J.; Han, Y.; Wang, Q.; Wei, W. Bidirectional Role of beta2-Adrenergic Receptor in Autoimmune Diseases. Front. Pharmacol. 2018, 9, 1313. [Google Scholar] [CrossRef]
- Nijhuis, L.E.; Olivier, B.J.; Dhawan, S.; Hilbers, F.W.; Boon, L.; Wolkers, M.C.; Samsom, J.N.; de Jonge, W.J. Adrenergic beta2 receptor activation stimulates anti-inflammatory properties of dendritic cells in vitro. PLoS ONE 2014, 9, e85086. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Chen, J.; Song, S.; Yuan, P.; Liu, L.; Zhang, Y.; Zhou, A.; Chang, Y.; Zhang, L.; Wei, W. beta2-adrenoceptor signaling reduction in dendritic cells is involved in the inflammatory response in adjuvant-induced arthritic rats. Sci. Rep. 2016, 6, 24548. [Google Scholar] [CrossRef] [Green Version]
- Giordani, L.; Cuzziol, N.; Del Pinto, T.; Sanchez, M.; Maccari, S.; Massimi, A.; Pietraforte, D.; Viora, M. β2-Agonist clenbuterol hinders human monocyte differentiation into dendritic cells. Exp. Cell Res. 2015, 339, 163–173. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; Quintana, F.J.; Basso, A.S. Norepinephrine Controls Effector T Cell Differentiation through beta2-Adrenergic Receptor-Mediated Inhibition of NF-kappaB and AP-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Herve, J.; Dubreil, L.; Tardif, V.; Terme, M.; Pogu, S.; Anegon, I.; Rozec, B.; Gauthier, C.; Bach, J.M.; Blancou, P. beta2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J. Immunol. 2013, 190, 3163–3171. [Google Scholar] [CrossRef] [Green Version]
- McGinnes, K.; Florence, J.; Penny, R. The effect of radiotherapy on the natural killer (NK)-cell activity of cancer patients. J. Clin. Immunol. 1987, 7, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, F.; Lindemalm, C.; Choudhury, A.; Granstam-Bjorneklett, H.; Helander, I.; Lekander, M.; Mikaelsson, E.; Nilsson, B.; Ojutkangas, M.L.; Osterborg, A.; et al. NK-cell and T-cell functions in patients with breast cancer: Effects of surgery and adjuvant chemo- and radiotherapy. Br. J. Cancer 2007, 97, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, B.; Benish, M.; Goldfarb, Y.; Sorski, L.; Melamed, R.; Rosenne, E.; Ben-Eliyahu, S. Continuous stress disrupts immunostimulatory effects of IL-12. Brain Behav. Immun. 2011, 25, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inbar, S.; Neeman, E.; Avraham, R.; Benish, M.; Rosenne, E.; Ben-Eliyahu, S. Do stress responses promote leukemia progression? An animal study suggesting a role for epinephrine and prostaglandin-E2 through reduced NK activity. PLoS ONE 2011, 6, e19246. [Google Scholar] [CrossRef]
- Ben-Eliyahu, S.; Shakhar, G.; Page, G.G.; Stefanski, V.; Shakhar, K. Suppression of NK cell activity and of resistance to metastasis by stress: A role for adrenal catecholamines and beta-adrenoceptors. Neuroimmunomodulation 2000, 8, 154–164. [Google Scholar] [CrossRef]
- Sun, Z.; Hou, D.; Liu, S.; Fu, W.; Wang, J.; Liang, Z. Norepinephrine inhibits the cytotoxicity of NK92MI cells via the beta2adrenoceptor/cAMP/PKA/pCREB signaling pathway. Mol. Med. Rep. 2018, 17, 8530–8535. [Google Scholar] [CrossRef] [Green Version]
- Kanemi, O.; Zhang, X.; Sakamoto, Y.; Ebina, M.; Nagatomi, R. Acute stress reduces intraparenchymal lung natural killer cells via beta-adrenergic stimulation. Clin. Exp. Immunol. 2005, 139, 25–34. [Google Scholar] [CrossRef]
- Avraham, R.; Benish, M.; Inbar, S.; Bartal, I.; Rosenne, E.; Ben-Eliyahu, S. Synergism between immunostimulation and prevention of surgery-induced immune suppression: An approach to reduce post-operative tumor progression. Brain Behav. Immun. 2010, 24, 952–958. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.F.; Lee, S.J.; Sood, A.K. Immunological and pleiotropic effects of individual beta-blockers and their relevance in cancer therapies. Expert Opin. Investig. Drugs 2016, 25, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Shakhar, G.; Ben-Eliyahu, S. In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J. Immunol. 1998, 160, 3251–3258. [Google Scholar]
- Ben-Eliyahu, S.; Shakhar, G.; Shakhar, K.; Melamed, R. Timing within the oestrous cycle modulates adrenergic suppression of NK activity and resistance to metastasis: Possible clinical implications. Br. J. Cancer 2000, 83, 1747–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, G.G.; Ben-Eliyahu, S. Natural killer cell activity and resistance to tumor metastasis in prepubescent rats: Deficient baselines, but invulnerability to stress and beta-adrenergic stimulation. Neuroimmunomodulation 2000, 7, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Page, G.G.; Fennelly, A.M.; Littleton-Kearney, M.T.; Ben-Eliyahu, S. Male--female differences in the impact of beta-adrenoceptor stimulation on resistance to experimental metastasis: Exploring the effects of age and gonadal hormone involvement. J. Neuroimmunol. 2008, 193, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.; Klein, E.; Kuten, A.; Fried, G.; Zinder, O.; Pollack, S. Increased emotional distress in daughters of breast cancer patients is associated with decreased natural cytotoxic activity, elevated levels of stress hormones and decreased secretion of Th1 cytokines. Int. J. Cancer 2002, 100, 347–354. [Google Scholar] [CrossRef]
- Fang, C.Y.; Reibel, D.K.; Longacre, M.L.; Rosenzweig, S.; Campbell, D.E.; Douglas, S.D. Enhanced psychosocial well-being following participation in a mindfulness-based stress reduction program is associated with increased natural killer cell activity. J. Altern. Complement. Med. 2010, 16, 531–538. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Vinogradov, S.; Warren, G.; Wei, X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine 2014, 9, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.S.; Brown, J.M. The irradiated tumor microenvironment: Role of tumor-associated macrophages in vascular recovery. Front. Physiol. 2013, 4, 157. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.S.; Chen, F.H.; Wang, C.C.; Huang, H.L.; Jung, S.M.; Wu, C.J.; Lee, C.C.; McBride, W.H.; Chiang, C.S.; Hong, J.H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Qin, J.F.; Jin, F.J.; Li, N.; Guan, H.T.; Lan, L.; Ni, H.; Wang, Y. Adrenergic receptor beta2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015, 48, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Lamkin, D.M.; Srivastava, S.; Bradshaw, K.P.; Betz, J.E.; Muy, K.B.; Wiese, A.M.; Yee, S.K.; Waggoner, R.M.; Arevalo, J.M.G.; Yoon, A.J.; et al. C/EBPbeta regulates the M2 transcriptome in beta-adrenergic-stimulated macrophages. Brain Behav. Immun. 2019, 80, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Lamkin, D.M.; Ho, H.Y.; Ong, T.H.; Kawanishi, C.K.; Stoffers, V.L.; Ahlawat, N.; Ma, J.C.Y.; Arevalo, J.M.G.; Cole, S.W.; Sloan, E.K. beta-Adrenergic-stimulated macrophages: Comprehensive localization in the M1-M2 spectrum. Brain Behav. Immun. 2016, 57, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.J.; Yang, Y.; Peng, W.T.; Sun, J.C.; Sun, W.Y.; Wei, W. G protein-coupled receptor kinase 2 regulating beta2-adrenergic receptor signaling in M2-polarized macrophages contributes to hepatocellular carcinoma progression. Onco Targets Ther. 2019, 12, 5499–5513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef] [Green Version]
- Leonard, W.; Dufait, I.; Schwarze, J.K.; Law, K.; Engels, B.; Jiang, H.; Van den Berge, D.; Gevaert, T.; Storme, G.; Verovski, V.; et al. Myeloid-derived suppressor cells reveal radioprotective properties through arginase-induced l-arginine depletion. Radiother. Oncol. 2016, 119, 291–299. [Google Scholar] [CrossRef]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [Green Version]
- Deorukhkar, A.; Krishnan, S. Targeting inflammatory pathways for tumor radiosensitization. Biochem. Pharmacol. 2010, 80, 1904–1914. [Google Scholar] [CrossRef] [Green Version]
- Tomic, S.; Joksimovic, B.; Bekic, M.; Vasiljevic, M.; Milanovic, M.; Colic, M.; Vucevic, D. Prostaglanin-E2 Potentiates the Suppressive Functions of Human Mononuclear Myeloid-Derived Suppressor Cells and Increases Their Capacity to Expand IL-10-Producing Regulatory T Cell Subsets. Front. Immunol. 2019, 10, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Tang, X.Y.; Li, Y.X.; Zhao, D.D.; Cao, Q.H.; Wu, H.X.; Yang, H.B.; Hao, K.; Yang, Y. Depression-Induced Neuropeptide Y Secretion Promotes Prostate Cancer Growth by Recruiting Myeloid Cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Gerweck, L.E.; Vijayappa, S.; Kurimasa, A.; Ogawa, K.; Chen, D.J. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer Res. 2006, 66, 8352–8355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Seshacharyulu, P.; Baine, M.J.; Souchek, J.J.; Menning, M.; Kaur, S.; Yan, Y.; Ouellette, M.M.; Jain, M.; Lin, C.; Batra, S.K. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Rains, S.L.; Amaya, C.N.; Bryan, B.A. Beta-adrenergic receptors are expressed across diverse cancers. Oncoscience 2017, 4, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Mo, Y.; Wang, Y.; Xiang, B.; Liao, Q.; Zhou, M.; Li, X.; Li, Y.; Xiong, W.; Li, G.; et al. Chronic Stress Promotes Cancer Development. Front. Oncol. 2020, 10, 1492. [Google Scholar] [CrossRef]
- Peixoto, R.; Pereira, M.L.; Oliveira, M. Beta-Blockers and Cancer: Where Are We? Pharmaceuticals 2020, 13, 105. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Montoya, A.; Varela-Ramirez, A.; Dickerson, E.; Pasquier, E.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef]
- Montoya, A.; Amaya, C.N.; Belmont, A.; Diab, N.; Trevino, R.; Villanueva, G.; Rains, S.; Sanchez, L.A.; Badri, N.; Otoukesh, S.; et al. Use of non-selective beta-blockers is associated with decreased tumor proliferative indices in early stage breast cancer. Oncotarget 2017, 8, 6446–6460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Chen, S.; Li, K.; Xiao, X.; Zheng, S.; Xu, T. The role of beta-adrenergic receptor signaling in the proliferation of hemangioma-derived endothelial cells. Cell Div. 2013, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Chen, X.; Zeng, W.; Peng, C.; Huang, G.; Li, X.; Ouyang, Z.; Luo, Y.; Xu, X.; Xu, B.; et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget 2016, 7, 68314–68327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fan, S.; Shi, Y.; Ren, H.; Hong, H.; Gao, X.; Zhang, M.; Qin, Q.; Li, H. Propranolol induced apoptosis and autophagy via the ROS/JNK signaling pathway in human ovarian cancer. J. Cancer 2020, 11, 5900–5910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ma, Q.; Wang, Z.; Zhang, M.; Guo, K.; Wang, F.; Wu, E. beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol. Cancer 2011, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective beta2-AR Blockage Suppresses Colorectal Cancer Growth Through Regulation of EGFR-Akt/ERK1/2 Signaling, G1-Phase Arrest, and Apoptosis. J. Cell. Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef]
- Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, D.; Abidi, W.; Guardavaccaro, D.; Zhou, M.; Ahearn, I.; Pagano, M.; Philips, M.R. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 2008, 181, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Yi, P.; Wang, H.; Xia, L.; Han, Y.; Wang, H.; Zeng, B.; Tang, L.; Pan, Q.; Tian, Y.; et al. RAC1 Involves in the Radioresistance by Mediating Epithelial-Mesenchymal Transition in Lung Cancer. Front. Oncol. 2020, 10, 649. [Google Scholar] [CrossRef]
- Bachmann, V.A.; Riml, A.; Huber, R.G.; Baillie, G.S.; Liedl, K.R.; Valovka, T.; Stefan, E. Reciprocal regulation of PKA and Rac signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8531–8536. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Elzey, B.D.; Poulson, J.M.; Morrison, W.B.; Ko, S.C.; Hahn, N.M.; Ratliff, T.L.; Hu, C.D. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro, in vivo and in prostate cancer patients. Am. J. Cancer Res. 2011, 1, 834–844. [Google Scholar] [PubMed]
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.D. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: Implications for disease progression. Cancer Res. 2008, 68, 9663–9670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, C.D.; Deng, X.; Hu, C.D. Targeting CREB inhibits radiation-induced neuroendocrine differentiation and increases radiation-induced cell death in prostate cancer cells. Am. J. Cancer Res. 2014, 4, 850–861. [Google Scholar] [PubMed]
- Cox, M.E.; Deeble, P.D.; Lakhani, S.; Parsons, S.J. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: Implications for prostate cancer progression. Cancer Res. 1999, 59, 3821–3830. [Google Scholar]
- Hu, C.D.; Choo, R.; Huang, J. Neuroendocrine differentiation in prostate cancer: A mechanism of radioresistance and treatment failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Zhu, R.; Yang, X.; Xue, X.; Shen, M.; Chen, F.; Chen, X.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; et al. Neuroendocrine differentiation contributes to radioresistance development and metastatic potential increase in non-small cell lung cancer. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1878–1890. [Google Scholar] [CrossRef]
- Volante, M.; Marci, V.; Andrejevic-Blant, S.; Tavaglione, V.; Sculli, M.C.; Tampellini, M.; Papotti, M. Increased neuroendocrine cells in resected metastases compared to primary colorectal adenocarcinomas. Virchows Arch. 2010, 457, 521–527. [Google Scholar] [CrossRef]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Tasken, K.A. beta-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2014, 4, 375. [Google Scholar] [CrossRef] [Green Version]
- Alberti, C. Molecularly targeted radiosensitization chances towards gene aberration-due organ confined/regionally advanced prostate cancer radioresistance. G. Chir. 2015, 36, 133–136. [Google Scholar] [CrossRef]
- Farias, V.A.; Tovar, I.; Del Moral, R.; O’Valle, F.; Exposito, J.; Oliver, F.J.; Ruiz de Almodovar, J.M. Enhancing the Bystander and Abscopal Effects to Improve Radiotherapy Outcomes. Front. Oncol. 2019, 9, 1381. [Google Scholar] [CrossRef] [Green Version]
- Roth, I.M.; Wickremesekera, A.C.; Wickremesekera, S.K.; Davis, P.F.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells via Modulation of the Renin-Angiotensin System. Front. Oncol. 2019, 9, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, D.C.; Roth, I.M.; Wickremesekera, A.C.; Davis, P.F.; Kaye, A.H.; Mantamadiotis, T.; Stylli, S.S.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells in Human Glioblastoma by Manipulating the Renin-Angiotensin System. Cells 2019, 8, 1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itinteang, T.; Brasch, H.D.; Tan, S.T.; Day, D.J. Expression of components of the renin-angiotensin system in proliferating infantile haemangioma may account for the propranolol-induced accelerated involution. J. Plast. Reconstr. Aesthet. Surg. 2011, 64, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Dornhoffer, J.R.; Wei, T.; Zhang, H.; Miller, E.; Cleves, M.A.; Richter, G.T. The expression of renin-angiotensin-aldosterone axis components in infantile hemangioma tissue and the impact of propranolol treatment. Pediatr. Res. 2017, 82, 155–163. [Google Scholar] [CrossRef]
- Leaute-Labreze, C.; Dumas de la Roque, E.; Hubiche, T.; Boralevi, F.; Thambo, J.B.; Taieb, A. Propranolol for severe hemangiomas of infancy. N. Engl. J. Med. 2008, 358, 2649–2651. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.I.; Asosingh, K.; Stephens, O.R.; Queisser, K.A.; Xu, W.; Willard, B.; Hu, B.; Dermawan, J.K.T.; Stark, G.R.; Naga Prasad, S.V.; et al. Hypoxia sensing through beta-adrenergic receptors. JCI Insight 2016, 1, e90240. [Google Scholar] [CrossRef]
- Shan, T.; Ma, J.; Ma, Q.; Guo, K.; Guo, J.; Li, X.; Li, W.; Liu, J.; Huang, C.; Wang, F.; et al. beta2-AR-HIF-1alpha: A novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr. Mol. Med. 2013, 13, 1023–1034. [Google Scholar] [CrossRef]
- Reglero, C.; Lafarga, V.; Rivas, V.; Albitre, A.; Ramos, P.; Berciano, S.R.; Tapia, O.; Martinez-Chantar, M.L.; Mayor, F., Jr.; Penela, P. GRK2-Dependent HuR Phosphorylation Regulates HIF1alpha Activation under Hypoxia or Adrenergic Stress. Cancers 2020, 12, 1216. [Google Scholar] [CrossRef]
- Hu, H.T.; Ma, Q.Y.; Zhang, D.; Shen, S.G.; Han, L.; Ma, Y.D.; Li, R.F.; Xie, K.P. HIF-1alpha links beta-adrenoceptor agonists and pancreatic cancer cells under normoxic condition. Acta Pharmacol. Sin. 2010, 31, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Guo, Z.; Gao, Y.; Pan, W. Propranolol represses infantile hemangioma cell growth through the beta2-adrenergic receptor in a HIF-1alpha-dependent manner. Oncol. Rep. 2015, 33, 3099–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chim, H.; Armijo, B.S.; Miller, E.; Gliniak, C.; Serret, M.A.; Gosain, A.K. Propranolol induces regression of hemangioma cells through HIF-1alpha-mediated inhibition of VEGF-A. Ann. Surg. 2012, 256, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Su, W.H.; Chuang, P.C.; Huang, E.Y.; Yang, K.D. Radiation-induced increase in cell migration and metastatic potential of cervical cancer cells operates via the K-Ras pathway. Am. J. Pathol. 2012, 180, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, Z.; Lu, L.; Cho, C.H. β-Adrenergic system, a backstage manipulator regulating tumour progression and drug target in cancer therapy. Semin. Cancer Biol. 2013, 23, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Gruet, M.; Cotton, D.; Coveney, C.; Boocock, D.J.; Wagner, S.; Komorowski, L.; Rees, R.C.; Pockley, A.G.; Garner, A.C.; Wallis, J.D.; et al. β2-Adrenergic Signalling Promotes Cell Migration by Upregulating Expression of the Metastasis-Associated Molecule LYPD3. Biology 2020, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Palm, D.; Lang, K.; Niggemann, B.; Drell, T.L.t.; Masur, K.; Zaenker, K.S.; Entschladen, F. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int. J. Cancer 2006, 118, 2744–2749. [Google Scholar] [CrossRef]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 87. [Google Scholar] [CrossRef]
- Cui, B.; Luo, Y.; Tian, P.; Peng, F.; Lu, J.; Yang, Y.; Su, Q.; Liu, B.; Yu, J.; Luo, X.; et al. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J. Clin. Investig. 2019, 129, 1030–1046. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.T.; Zhou, S.H. Warburg effect, hexokinase-II, and radioresistance of laryngeal carcinoma. Oncotarget 2017, 8, 14133–14146. [Google Scholar] [CrossRef] [Green Version]
- Kang, F.; Ma, W.; Ma, X.; Shao, Y.; Yang, W.; Chen, X.; Li, L.; Wang, J. Propranolol inhibits glucose metabolism and 18F-FDG uptake of breast cancer through posttranscriptional downregulation of hexokinase-2. J. Nucl. Med. 2014, 55, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Lucido, C.T.; Callejas-Valera, J.L.; Colbert, P.L.; Vermeer, D.W.; Miskimins, W.K.; Spanos, W.C.; Vermeer, P.D. beta2-Adrenergic receptor modulates mitochondrial metabolism and disease progression in recurrent/metastatic HPV(+) HNSCC. Oncogenesis 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Lucido, C.T.; Miskimins, W.K.; Vermeer, P.D. Propranolol Promotes Glucose Dependence and Synergizes with Dichloroacetate for Anti-Cancer Activity in HNSCC. Cancers 2018, 10, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico, M.; Baglioni, M.; Bondarenko, M.; Laluce, N.C.; Rozados, V.; Andre, N.; Carre, M.; Scharovsky, O.G.; Menacho Marquez, M. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget 2017, 8, 2874–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adra, J.; Lundstedt, D.; Killander, F.; Holmberg, E.; Haghanegi, M.; Kjellen, E.; Karlsson, P.; Alkner, S. Distribution of Locoregional Breast Cancer Recurrence in Relation to Postoperative Radiation Fields and Biological Subtypes. Int. J. Radiat. Oncol. Biol. Phys. 2019, 105, 285–295. [Google Scholar] [CrossRef]
- Vilalta, M.; Rafat, M.; Graves, E.E. Effects of radiation on metastasis and tumor cell migration. Cell. Mol. Life Sci. 2016, 73, 2999–3007. [Google Scholar] [CrossRef] [Green Version]
- Mole, R.H.; Whole body irradiation; radiobiology or medicine? Br., J. Whole body irradiation; radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Grass, G.D.; Krishna, N.; Kim, S. The immune mechanisms of abscopal effect in radiation therapy. Curr. Probl. Cancer 2016, 40, 10–24. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.I.; McArthur, H.L.; Ho, A.Y. The Abscopal Effect of Radiation Therapy: What Is It and How Can We Use It in Breast Cancer? Curr. Breast Cancer Rep. 2017, 9, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Vatner, R.E.; Formenti, S.C. Myeloid-derived cells in tumors: Effects of radiation. Semin. Radiat. Oncol. 2015, 25, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Qiao, G.; Bucsek, M.J.; Winder, N.M.; Chen, M.; Giridharan, T.; Olejniczak, S.H.; Hylander, B.L.; Repasky, E.A. β-Adrenergic signaling blocks murine CD8(+) T-cell metabolic reprogramming during activation: A mechanism for immunosuppression by adrenergic stress. Cancer Immunol. Immunother. 2019, 68, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Hayano, Y.; Nakai, A.; Furuta, F.; Noda, M. Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J. Exp. Med. 2016, 213, 2567–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, A.; Hayano, Y.; Furuta, F.; Noda, M.; Suzuki, K. Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. J. Exp. Med. 2014, 211, 2583–2598. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Meng, Y.; Blank, C.; Brown, I.; Kacha, A.; Kline, J.; Harlin, H. Immune resistance orchestrated by the tumor microenvironment. Immunol. Rev. 2006, 213, 131–145. [Google Scholar] [CrossRef]
- Kokolus, K.M.; Zhang, Y.; Sivik, J.M.; Schmeck, C.; Zhu, J.; Repasky, E.A.; Drabick, J.J.; Schell, T.D. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 2018, 7, e1405205. [Google Scholar] [CrossRef] [Green Version]
- Demaria, S.; Formenti, S.C. Can abscopal effects of local radiotherapy be predicted by modeling T cell trafficking? J. Immunother. Cancer 2016, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Dagoglu, N.; Karaman, S.; Caglar, H.B.; Oral, E.N. Abscopal Effect of Radiotherapy in the Immunotherapy Era: Systematic Review of Reported Cases. Cureus 2019, 11, e4103. [Google Scholar] [CrossRef] [Green Version]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Lemons, J.M.; Karrison, T.G.; Pitroda, S.P.; Melotek, J.M.; Zha, Y.; Al-Hallaq, H.A.; Arina, A.; Khodarev, N.N.; Janisch, L.; et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1611–1618. [Google Scholar] [CrossRef]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing tumor immunity with fractionated radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanguineti, G.; Giannarelli, D.; Petrongari, M.G.; Arcangeli, S.; Sangiovanni, A.; Saracino, B.; Farneti, A.; Faiella, A.; Conte, M.; Arcangeli, G. Leukotoxicity after moderately Hypofractionated radiotherapy versus conventionally fractionated dose escalated radiotherapy for localized prostate Cancer: A secondary analysis from a randomized study. Radiat. Oncol. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chin, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Zhang, W.; Gong, Y.; Xie, C. Radiotherapy activates autophagy to increase CD8(+) T cell infiltration by modulating major histocompatibility complex class-I expression in non-small cell lung cancer. J. Int. Med. Res. 2019, 47, 3818–3830. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Roger, A.; Finet, A.; Boru, B.; Beauchet, A.; Mazeron, J.J.; Otzmeguine, Y.; Blom, A.; Longvert, C.; de Maleissye, M.F.; Fort, M.; et al. Efficacy of combined hypo-fractionated radiotherapy and anti-PD-1 monotherapy in difficult-to-treat advanced melanoma patients. Oncoimmunology 2018, 7, e1442166. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [Green Version]
- Aboudaram, A.; Modesto, A.; Chaltiel, L.; Gomez-Roca, C.; Boulinguez, S.; Sibaud, V.; Delord, J.P.; Chira, C.; Delannes, M.; Moyal, E.; et al. Concurrent radiotherapy for patients with metastatic melanoma and receiving anti-programmed-death 1 therapy: A safe and effective combination. Melanoma Res. 2017, 27, 485–491. [Google Scholar] [CrossRef]
- Theurich, S.; Rothschild, S.I.; Hoffmann, M.; Fabri, M.; Sommer, A.; Garcia-Marquez, M.; Thelen, M.; Schill, C.; Merki, R.; Schmid, T.; et al. Local Tumor Treatment in Combination with Systemic Ipilimumab Immunotherapy Prolongs Overall Survival in Patients with Advanced Malignant Melanoma. Cancer Immunol. Res. 2016, 4, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Koller, K.M.; Mackley, H.B.; Liu, J.; Wagner, H.; Talamo, G.; Schell, T.D.; Pameijer, C.; Neves, R.I.; Anderson, B.; Kokolus, K.M.; et al. Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol. Ther. 2017, 18, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, M.A.; Callahan, M.K.; Barker, C.A.; Yamada, Y.; Yuan, J.; Kitano, S.; Mu, Z.; Rasalan, T.; Adamow, M.; Ritter, E.; et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 2012, 366, 925–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Chaudhary, P.; Qiu, G.; Che, X.; Fan, L. The role of propranolol as a radiosensitizer in gastric cancer treatment. Drug Des. Dev. Ther. 2018, 12, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.M.; Liao, Z.X.; Komaki, R.; Welsh, J.W.; O’Reilly, M.S.; Chang, J.Y.; Zhuang, Y.; Levy, L.B.; Lu, C.; Gomez, D.R. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann. Oncol. 2013, 24, 1312–1319. [Google Scholar] [CrossRef]
- Wang, H.; Liao, Z.; Zhuang, Y.; Liu, Y.; Levy, L.B.; Xu, T.; Yusuf, S.W.; Gomez, D.R. Incidental receipt of cardiac medications and survival outcomes among patients with stage III non-small-cell lung cancer after definitive radiotherapy. Clin. Lung Cancer 2015, 16, 128–136. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; Yan, S.X.; Heilbroner, S.P.; Sonett, J.R.; Stoopler, M.B.; Shu, C.; Halmos, B.; Wang, T.J.C.; Hei, T.K.; Cheng, S.K. Effects of beta-Adrenergic Antagonists on Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. J. Clin. Med. 2019, 8, 575. [Google Scholar] [CrossRef] [Green Version]
- Hilakivi-Clarke, L.; Rowland, J.; Clarke, R.; Lippman, M.E. Psychosocial factors in the development and progression of breast cancer. Breast Cancer Res. Treat. 1994, 29, 141–160. [Google Scholar] [CrossRef]
- Šoštarič, M.; Šprah, L. Psychological distress and intervention in cancer patients treated with radiotherapy. Radiol. Oncol. 2004, 38, 193–203. [Google Scholar]
- Guo, Z.; Tang, H.Y.; Li, H.; Tan, S.K.; Feng, K.H.; Huang, Y.C.; Bu, Q.; Jiang, W. The benefits of psychosocial interventions for cancer patients undergoing radiotherapy. Health Qual. Life Outcomes 2013, 11, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.P.; Gupta, D.; Grutsch, J.F.; Staren, E.D. Can changes in health related quality of life scores predict survival in stages III and IV colorectal cancer? Health Qual. Life Outcomes 2011, 9, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efficace, F.; Innominato, P.F.; Bjarnason, G.; Coens, C.; Humblet, Y.; Tumolo, S.; Genet, D.; Tampellini, M.; Bottomley, A.; Garufi, C.; et al. Validation of patient’s self-reported social functioning as an independent prognostic factor for survival in metastatic colorectal cancer patients: Results of an international study by the Chronotherapy Group of the European Organisation for Research and Treatment of Cancer. J. Clin. Oncol. 2008, 26, 2020–2026. [Google Scholar] [CrossRef] [PubMed]
- Gotay, C.C.; Kawamoto, C.T.; Bottomley, A.; Efficace, F. The prognostic significance of patient-reported outcomes in cancer clinical trials. J. Clin. Oncol. 2008, 26, 1355–1363. [Google Scholar] [CrossRef]
- Maisey, N.R.; Norman, A.; Watson, M.; Allen, M.J.; Hill, M.E.; Cunningham, D. Baseline quality of life predicts survival in patients with advanced colorectal cancer. Eur. J. Cancer 2002, 38, 1351–1357. [Google Scholar] [CrossRef]
- Mol, L.; Ottevanger, P.B.; Koopman, M.; Punt, C.J. The prognostic value of WHO performance status in relation to quality of life in advanced colorectal cancer patients. Eur. J. Cancer 2016, 66, 138–143. [Google Scholar] [CrossRef]
- Oskam, I.M.; Verdonck-de Leeuw, I.M.; Aaronson, N.K.; Kuik, D.J.; de Bree, R.; Doornaert, P.; Langendijk, J.A.; Leemans, C.R. Quality of life as predictor of survival: A prospective study on patients treated with combined surgery and radiotherapy for advanced oral and oropharyngeal cancer. Radiother. Oncol. 2010, 97, 258–262. [Google Scholar] [CrossRef]
- Osthus, A.A.; Aarstad, A.K.; Olofsson, J.; Aarstad, H.J. Prediction of survival by pretreatment health-related quality-of-life scores in a prospective cohort of patients with head and neck squamous cell carcinoma. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Quinten, C.; Martinelli, F.; Coens, C.; Sprangers, M.A.; Ringash, J.; Gotay, C.; Bjordal, K.; Greimel, E.; Reeve, B.B.; Maringwa, J.; et al. A global analysis of multitrial data investigating quality of life and symptoms as prognostic factors for survival in different tumor sites. Cancer 2014, 120, 302–311. [Google Scholar] [CrossRef]
- Vickers, M.M.; Lee, C.; Tu, D.; Wheatley-Price, P.; Parulekar, W.; Brundage, M.D.; Moore, M.J.; Au, H.; O’Callaghan, C.J.; Jonker, D.J.; et al. Significance of baseline and change in quality of life scores in predicting clinical outcomes in an international phase III trial of advanced pancreatic cancer: NCIC CTG PA.3. Pancreatology 2016, 16, 1106–1112. [Google Scholar] [CrossRef]
- Eldridge, R.C.; Pugh, S.L.; Trotti, A.; Hu, K.; Spencer, S.; Yom, S.S.; Rosenthal, D.; Read, N.; Desai, A.; Gore, E.; et al. Changing functional status within 6 months posttreatment is prognostic of overall survival in patients with head and neck cancer: NRG Oncology Study. Head Neck 2019, 41, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Jameson, M.J.; Karnell, L.H.; Christensen, A.J.; Funk, G.F. First-year trends in self-reported general health predict survival in patients with head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2008, 134, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.; Fortin, A.; Gelinas, M.; Nabid, A.; Brochet, F.; Tetu, B.; Bairati, I. Health-related quality of life as a survival predictor for patients with localized head and neck cancer treated with radiation therapy. J. Clin. Oncol. 2009, 27, 2970–2976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCI Dictionary of Cancer Terms: Financial Hardship. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/financial-toxicity (accessed on 18 December 2020).
- Ramsey, S.D.; Bansal, A.; Fedorenko, C.R.; Blough, D.K.; Overstreet, K.A.; Shankaran, V.; Newcomb, P. Financial Insolvency as a Risk Factor for Early Mortality Among Patients With Cancer. J. Clin. Oncol. 2016, 34, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Fayers, P.M.; Aaronson, N.K.; Bjordal, K.; Groenvold, M.; Curran, D.; Bottomley, A.; EORTC Quality of Life Group. The EORTC QLQ-C30 Scoring Manual (3rd Edition). Available online: https://www.eortc.org/app/uploads/sites/2/2018/02/SCmanual.pdf (accessed on 28 July 2020).
- Ma, S.J.; Iovoli, A.J.; Singh, A.K. Association of significant financial burden with survival for head and neck cancer patients treated with radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, e400. [Google Scholar] [CrossRef]
- Klein, J.; Bodner, W.; Garg, M.; Kalnicki, S.; Ohri, N. Pretreatment financial toxicity predicts progression-free survival following concurrent chemoradiotherapy for locally advanced non-small-cell lung cancer. Future Oncol. 2019, 15, 1697–1705. [Google Scholar] [CrossRef]
- Carrera, P.M.; Kantarjian, H.M.; Blinder, V.S. The financial burden and distress of patients with cancer: Understanding and stepping-up action on the financial toxicity of cancer treatment. CA Cancer J. Clin. 2018, 68, 153–165. [Google Scholar] [CrossRef]
- Hazell, S.Z.; Fu, W.; Hu, C.; Voong, K.R.; Lee, B.; Peterson, V.; Feliciano, J.L.; Nicholas, L.H.; McNutt, T.R.; Han, P.; et al. Financial toxicity in lung cancer: An assessment of magnitude, perception, and impact on quality of life. Ann. Oncol. 2020, 31, 96–102. [Google Scholar] [CrossRef]
- Jagsi, R.; Ward, K.C.; Abrahamse, P.H.; Wallner, L.P.; Kurian, A.W.; Hamilton, A.S.; Katz, S.J.; Hawley, S.T. Unmet need for clinician engagement regarding financial toxicity after diagnosis of breast cancer. Cancer 2018, 124, 3668–3676. [Google Scholar] [CrossRef]
- LaRocca, C.J.; Li, A.; Lafaro, K.; Clark, K.; Loscalzo, M.; Melstrom, L.G.; Warner, S.G. The impact of financial toxicity in gastrointestinal cancer patients. Surgery 2020, 168, 167–172. [Google Scholar] [CrossRef]
- Gandhi, S.; Pandey, M.R.; Attwood, K.; Ji, W.; Witkiewicz, A.K.; Knudsen, E.S.; Allen, C.; Tario, J.D.; Wallace, P.K.; Cedeno, C.D.; et al. Phase I Clinical Trial of Combination Propranolol and Pembrolizumab in Locally Advanced and Metastatic Melanoma: Safety, Tolerability, and Preliminary Evidence of Antitumor Activity. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Singh, A.K.; Repasky, E.A. Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance. Cancers 2020, 12, 3853. https://doi.org/10.3390/cancers12123853
Chen M, Singh AK, Repasky EA. Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance. Cancers. 2020; 12(12):3853. https://doi.org/10.3390/cancers12123853
Chicago/Turabian StyleChen, Minhui, Anurag K. Singh, and Elizabeth A. Repasky. 2020. "Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance" Cancers 12, no. 12: 3853. https://doi.org/10.3390/cancers12123853
APA StyleChen, M., Singh, A. K., & Repasky, E. A. (2020). Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance. Cancers, 12(12), 3853. https://doi.org/10.3390/cancers12123853